
Amide Activation in Ground and Excited States


Abstract
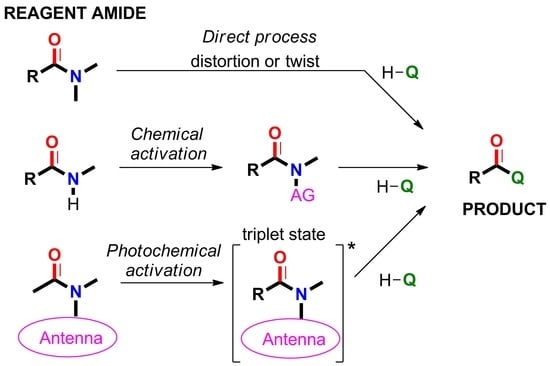
1. Introduction
2. Discussion
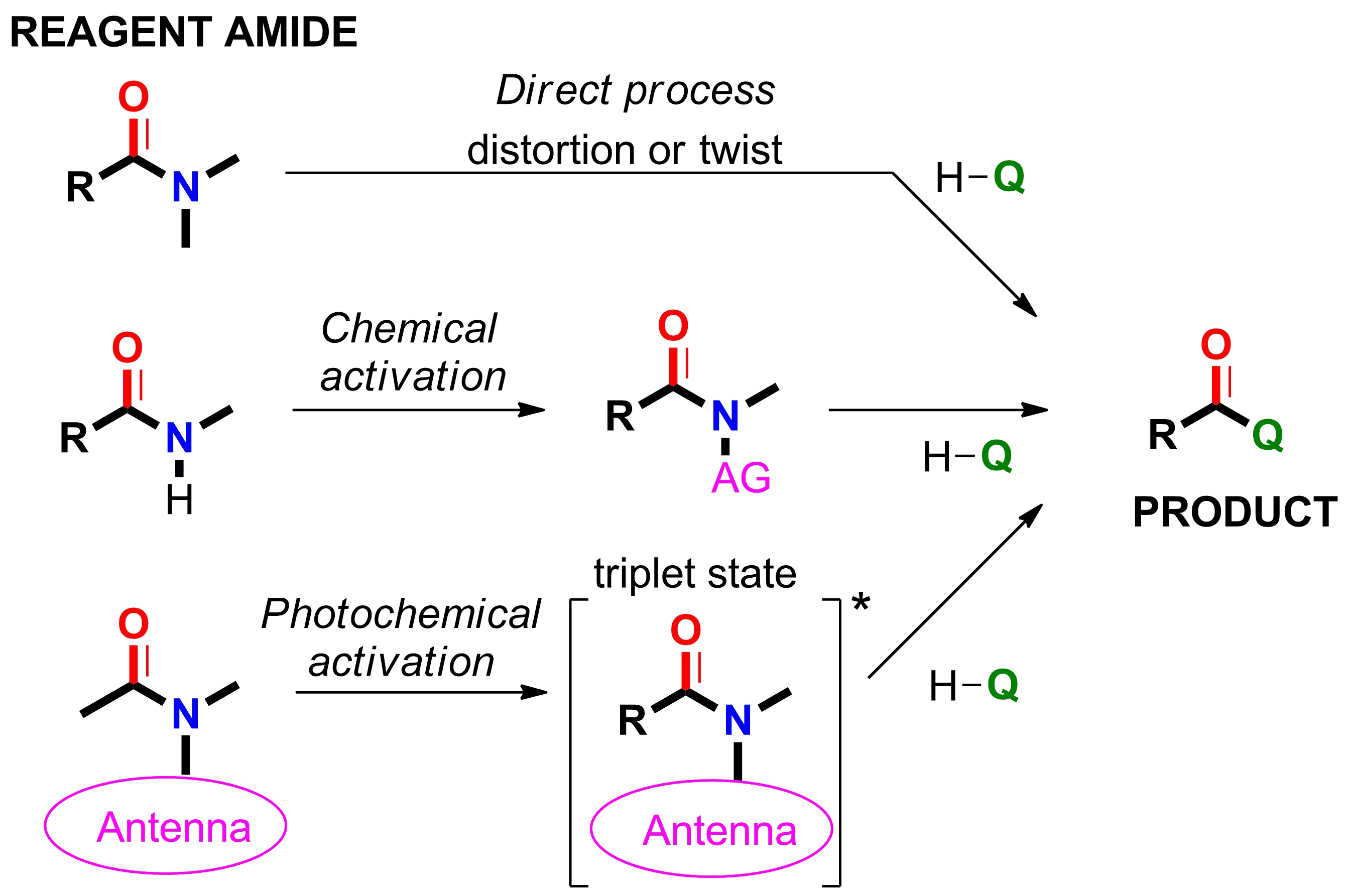
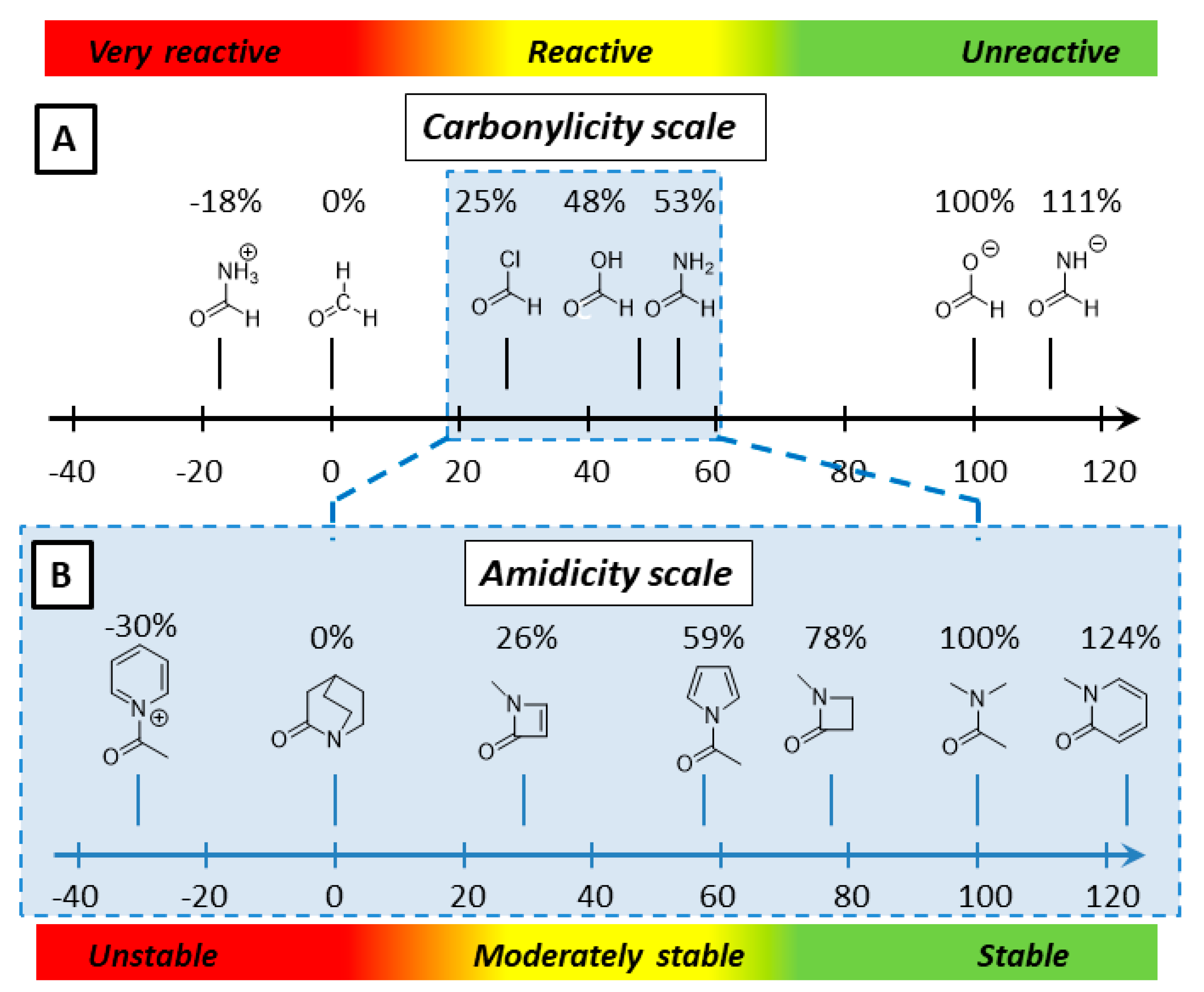
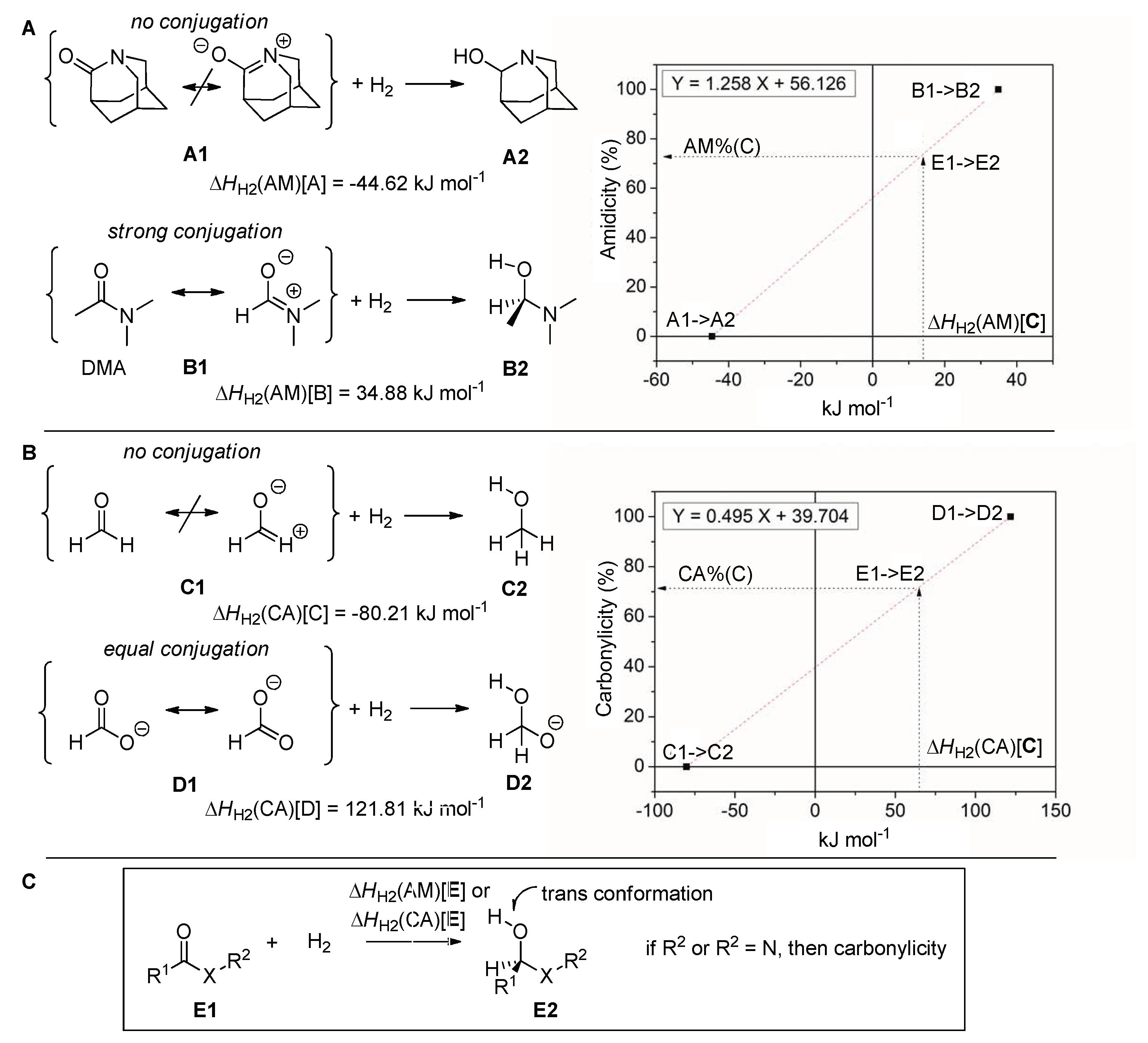
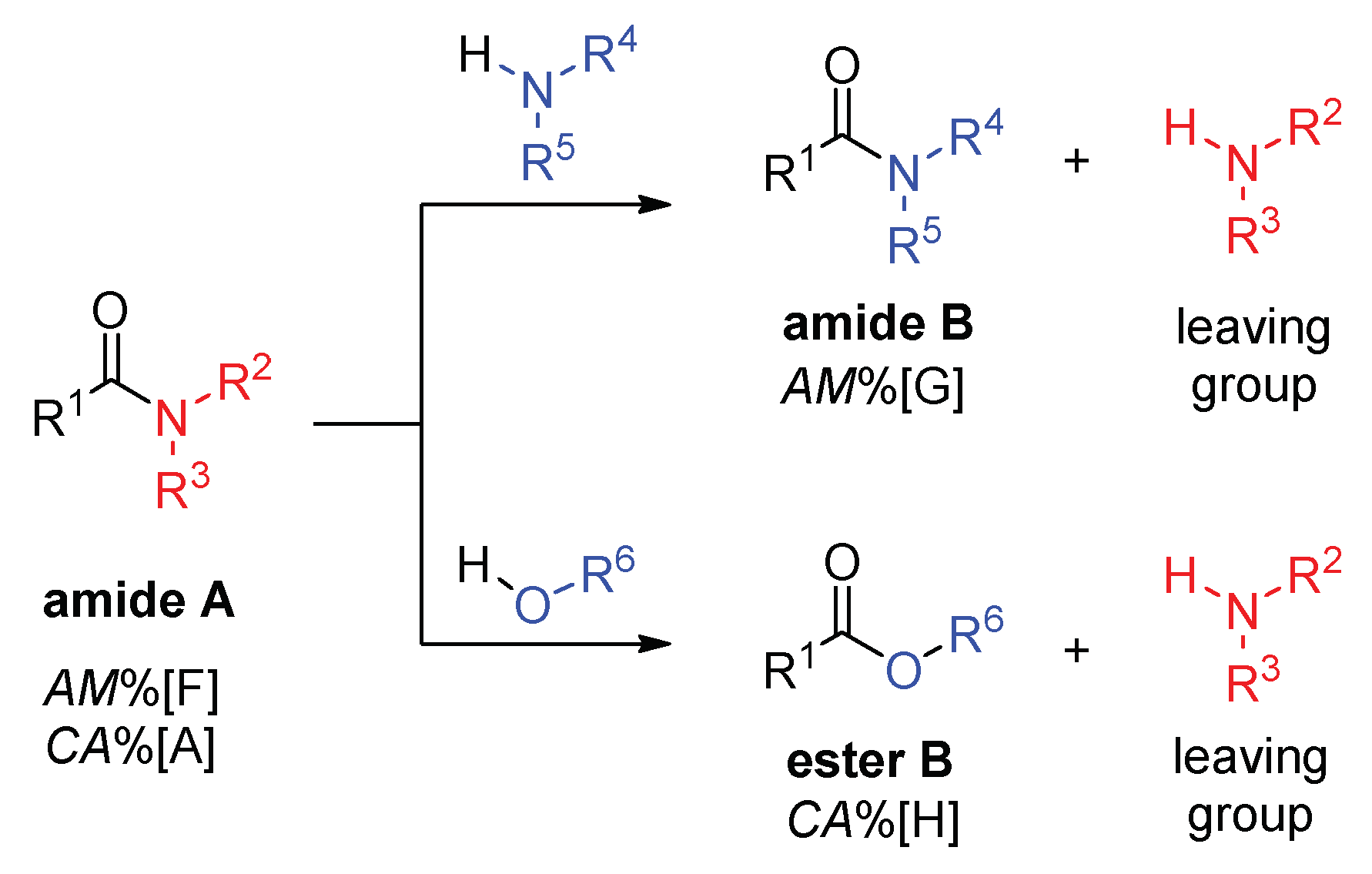
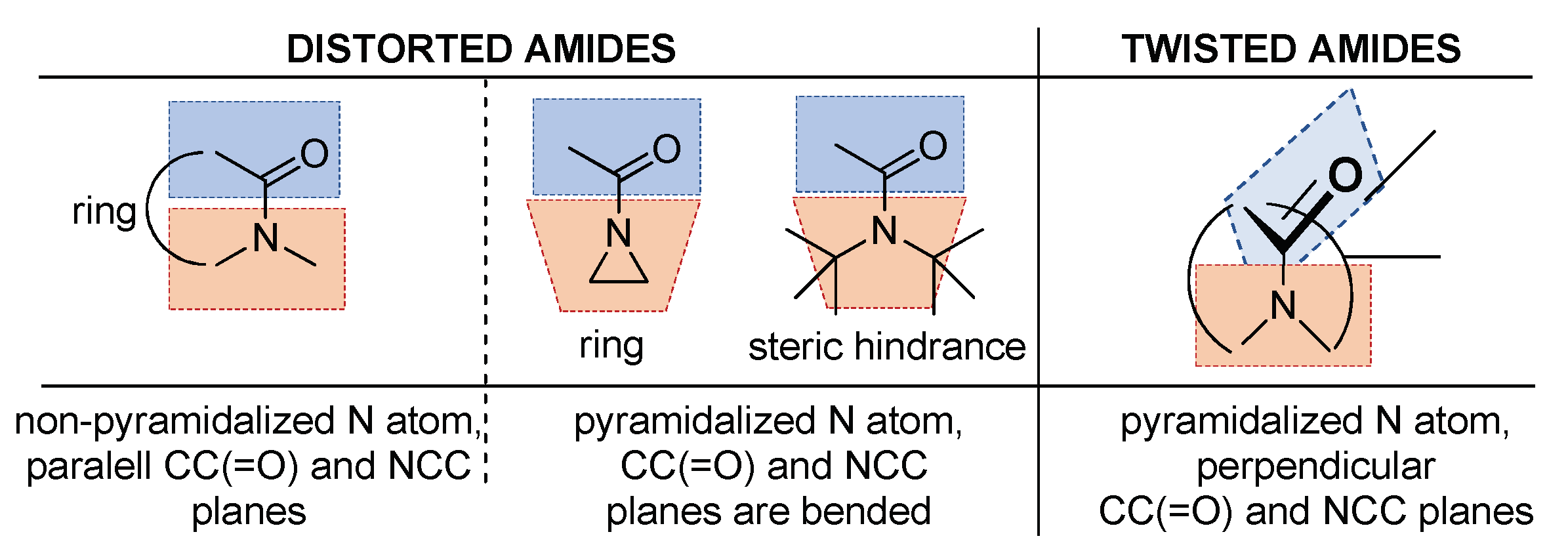
2.1. Amidicity Scale (AM%) and Carbonylicity Scale (CA%) and Their Resonance Enthalpies (HRE)
2.2. Transamidation and Transacylation Reactions in Synthetic Organic Chemistry
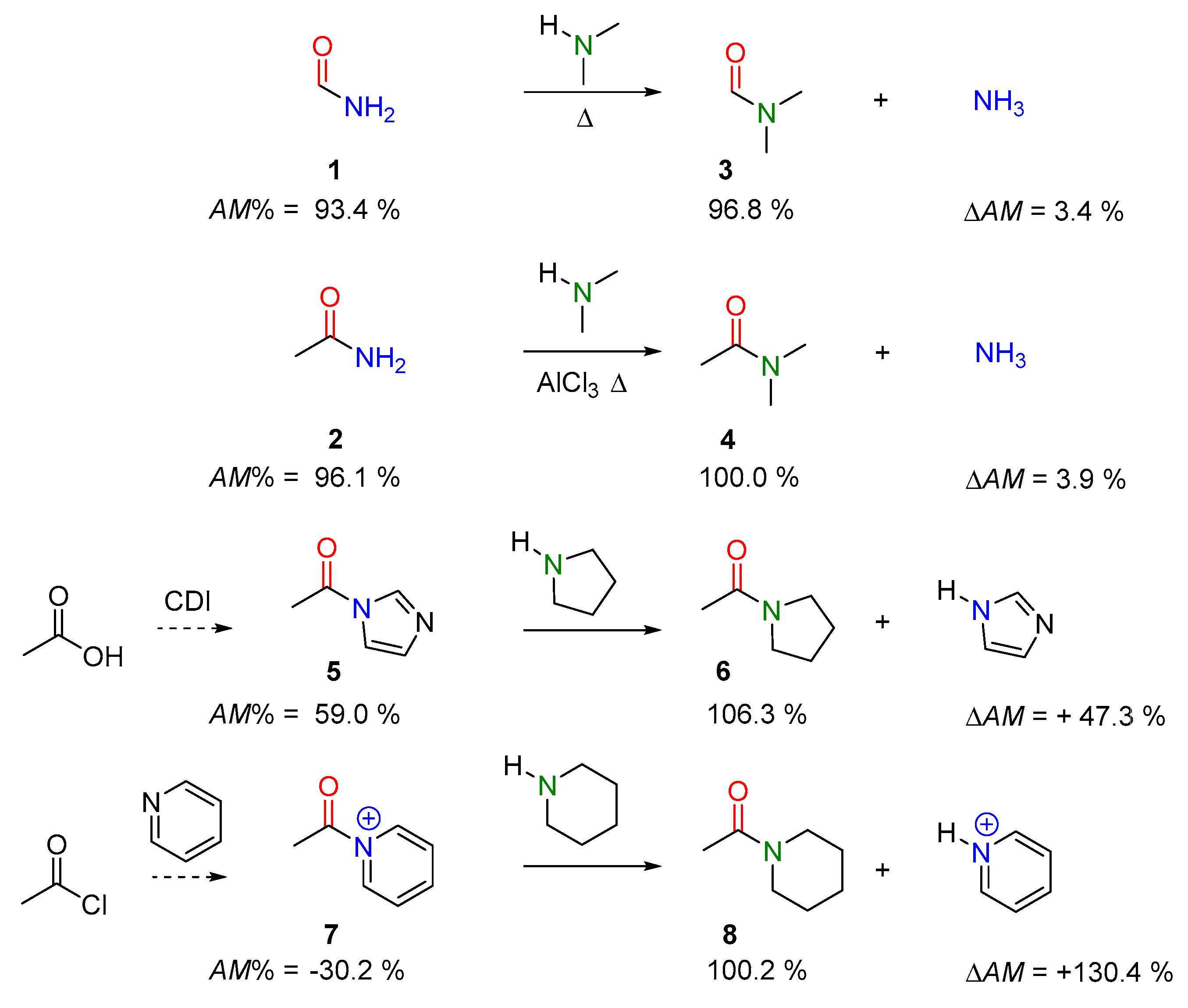
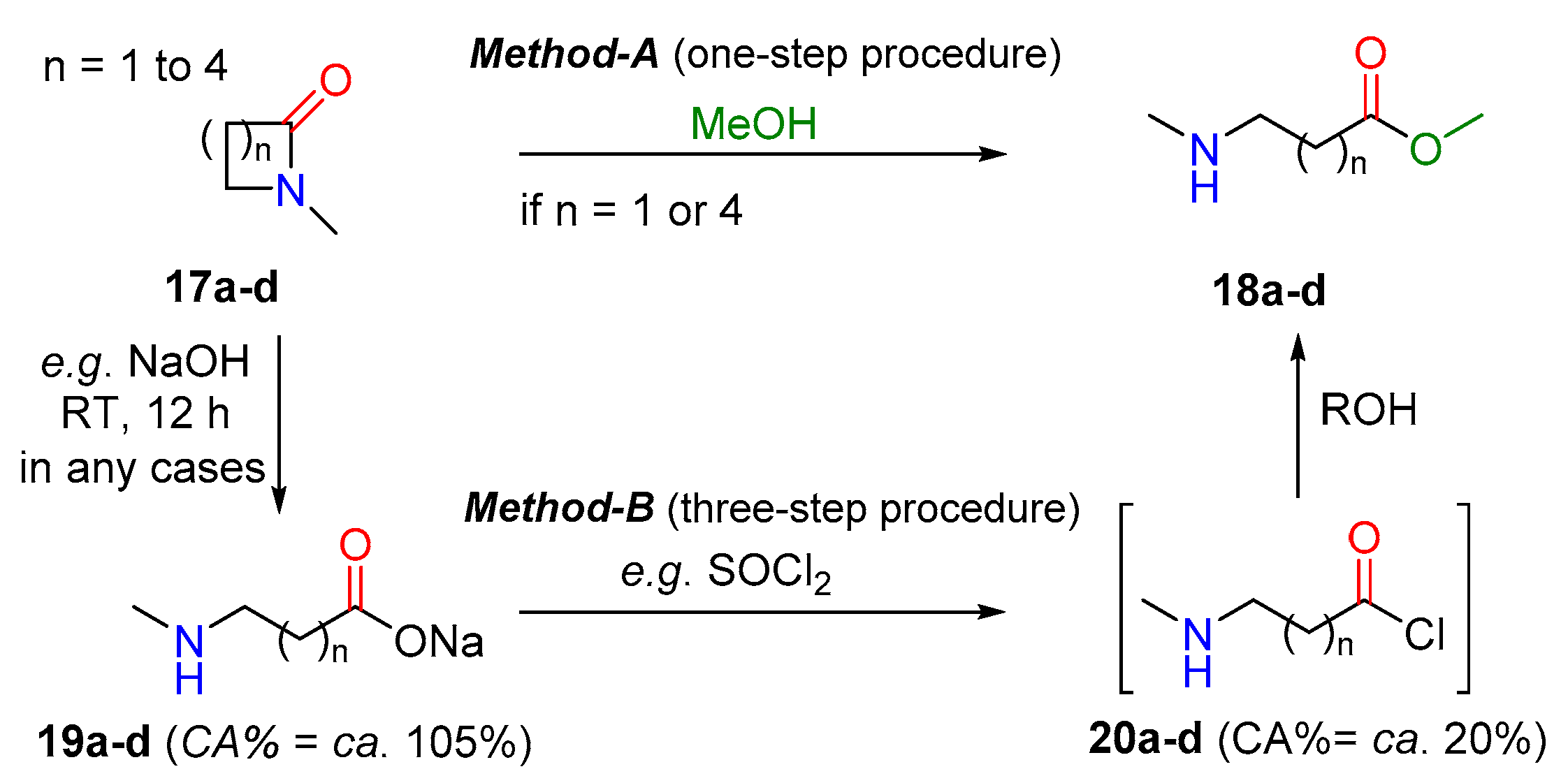
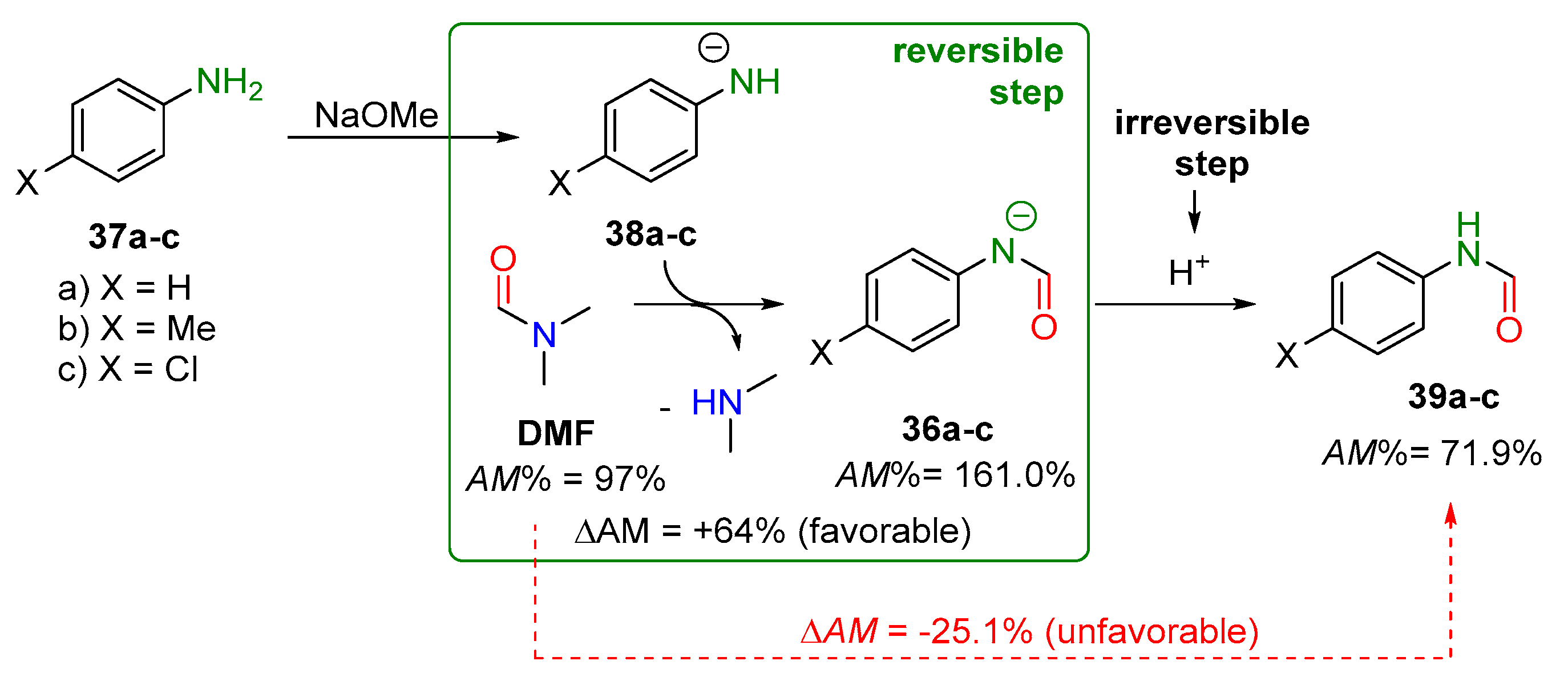
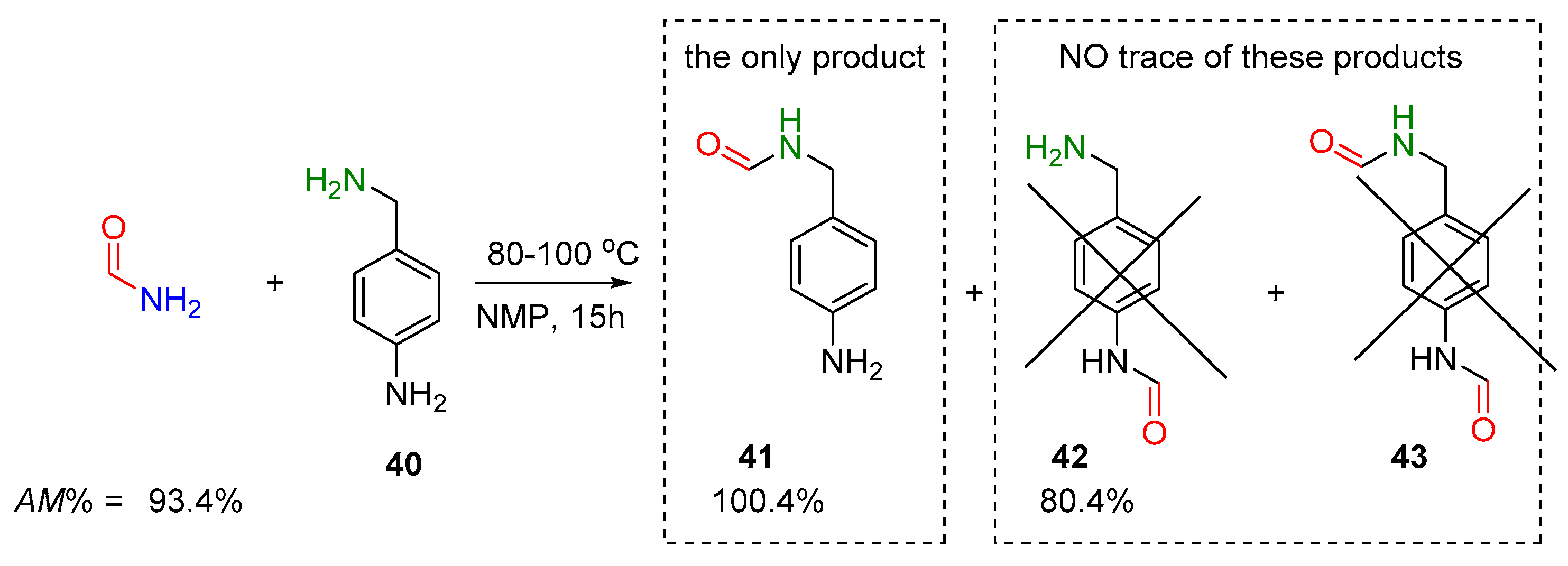
2.2.1. Examples of the Thermodynamically Allowed Transamidation Reactions (case I and II)
Transamidation Processes of Simple Amide Compounds
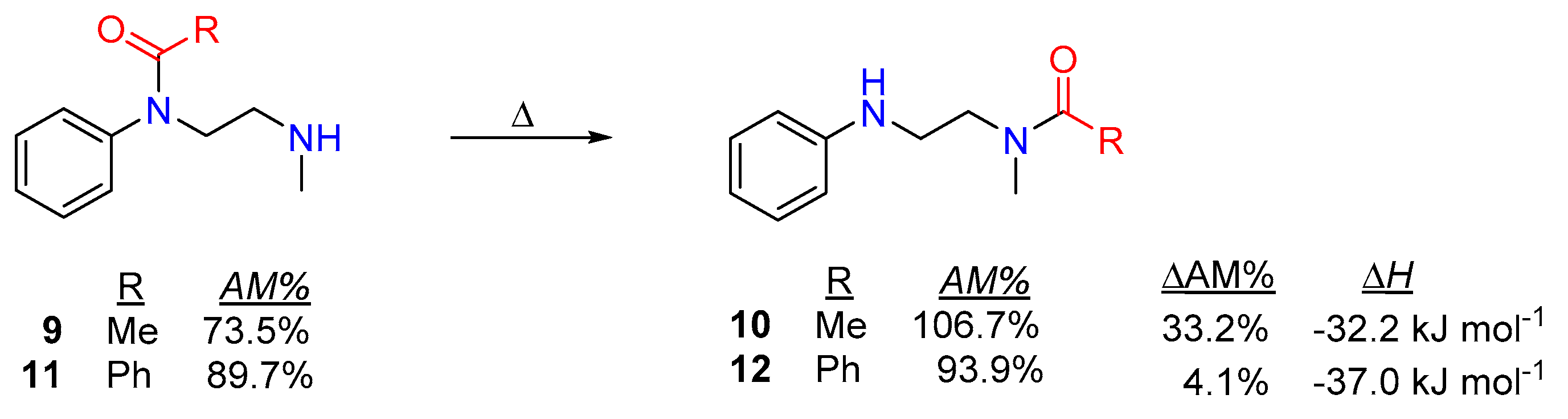
Transamidation and Acyl Transfer Processes of N-Phenylamides.
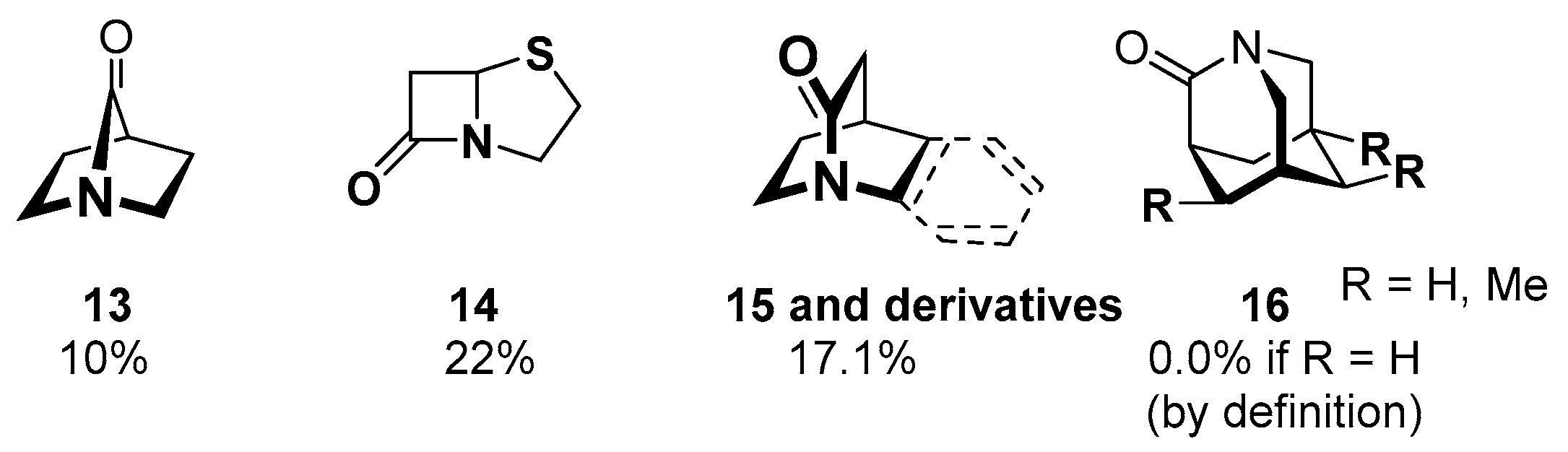
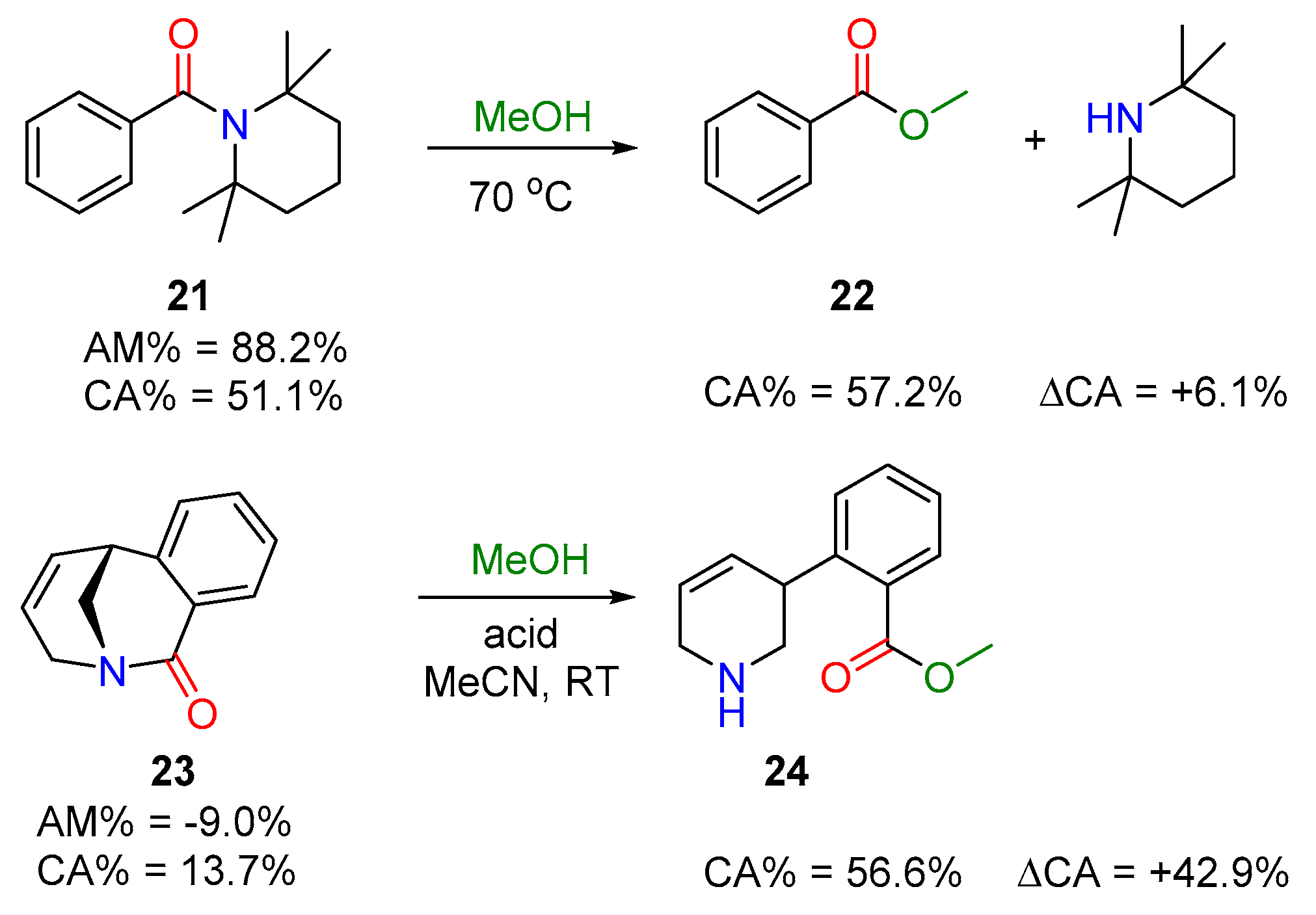
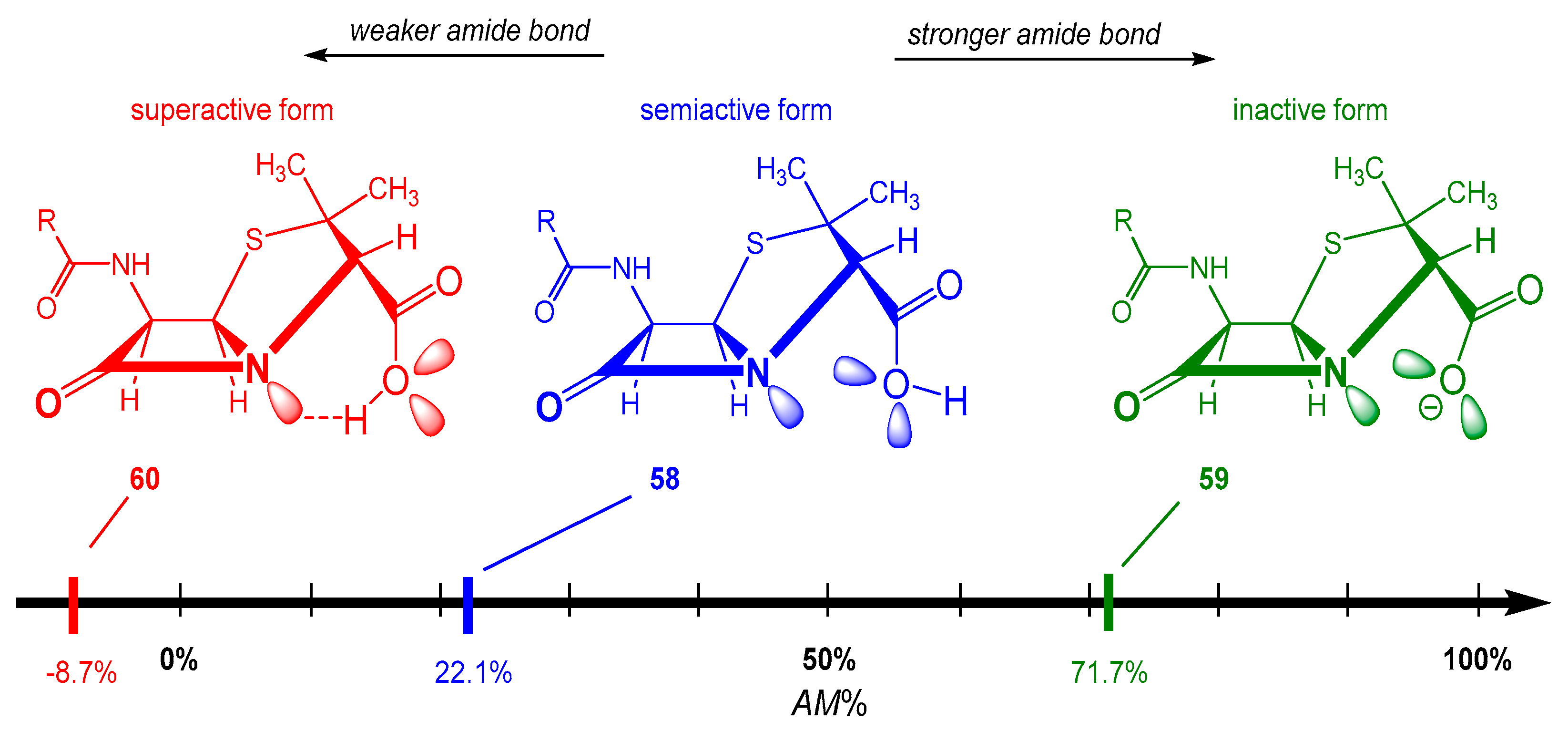
Acyl Transfer Reactions of Distorted or Twisted Amides
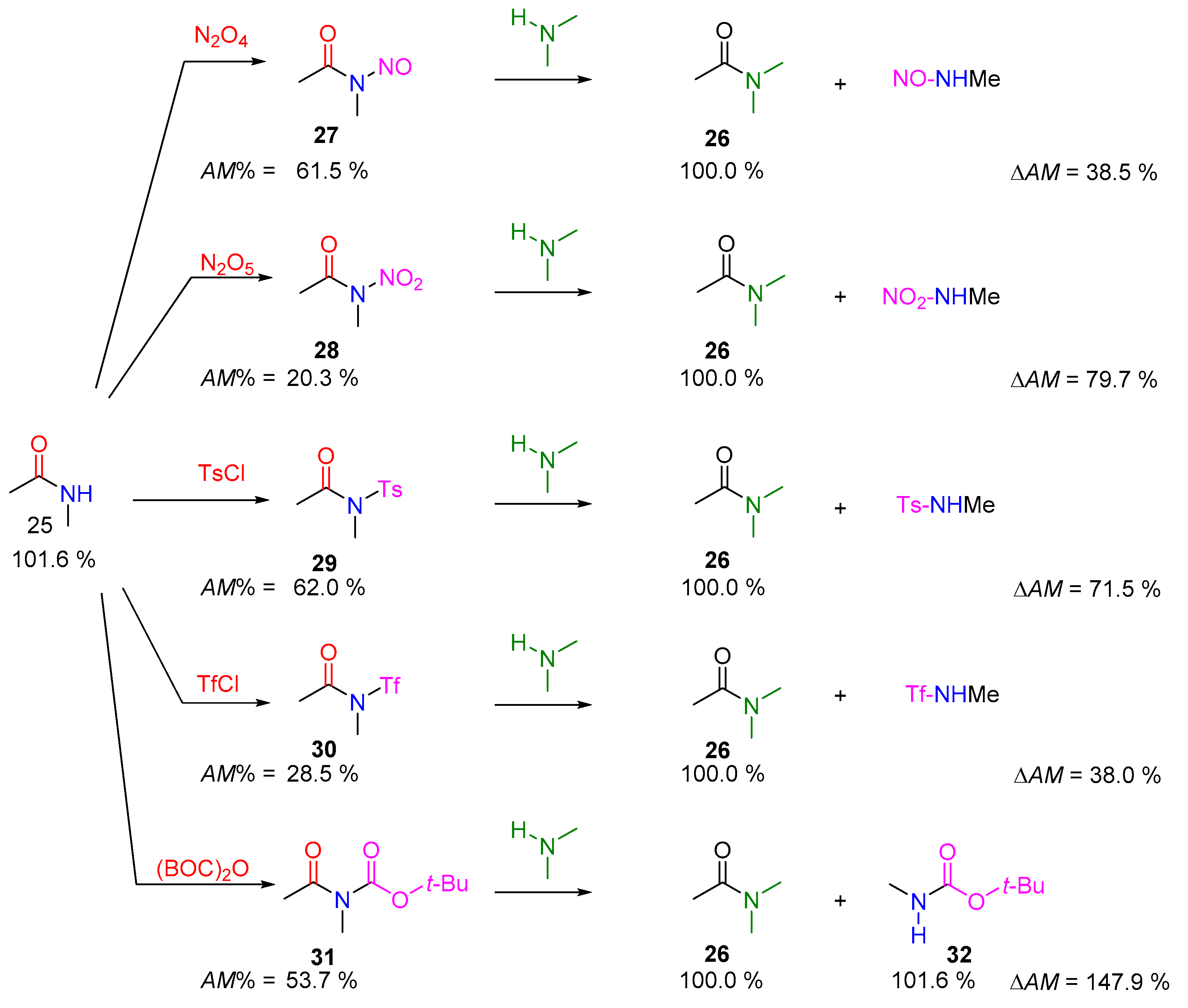
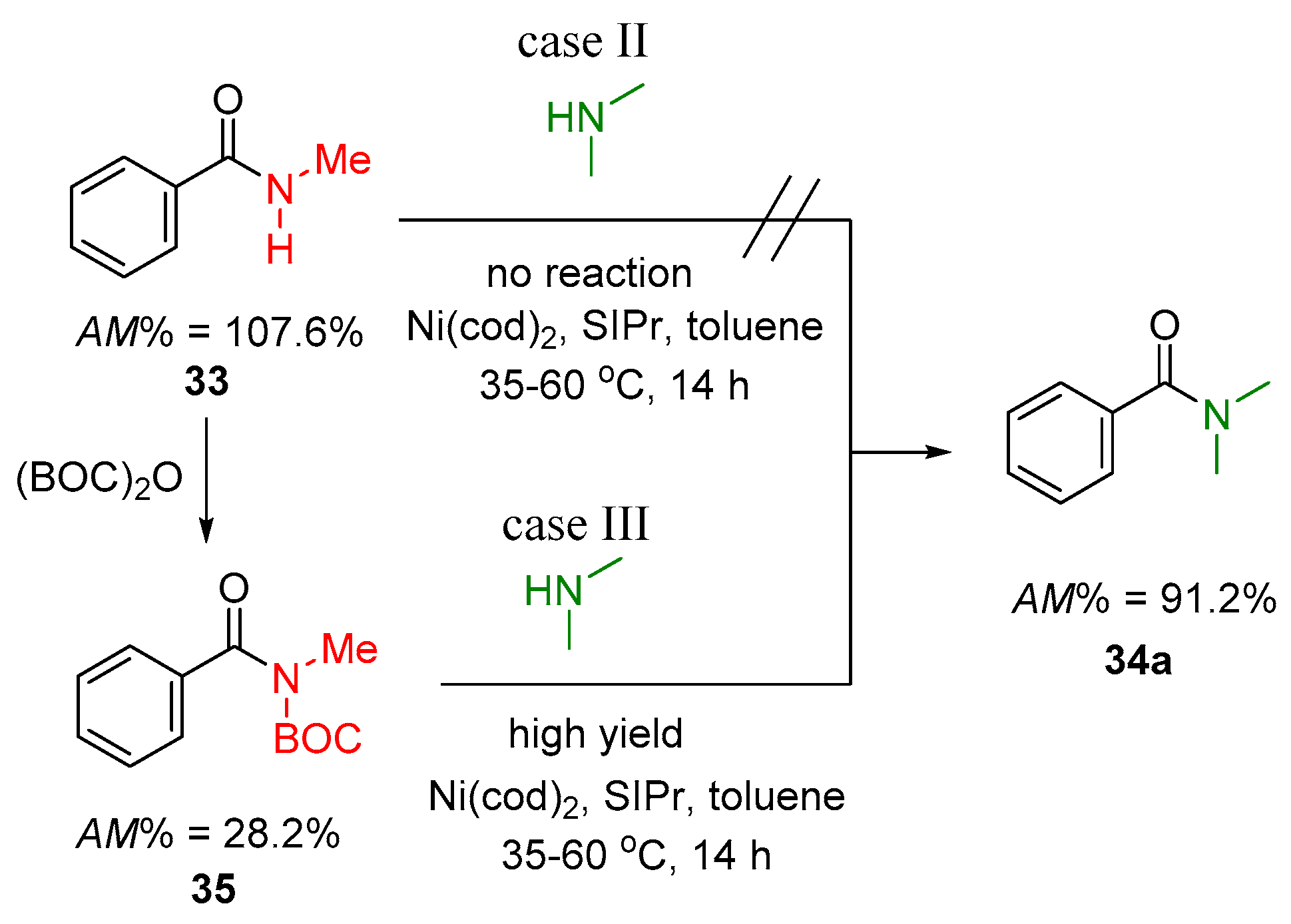
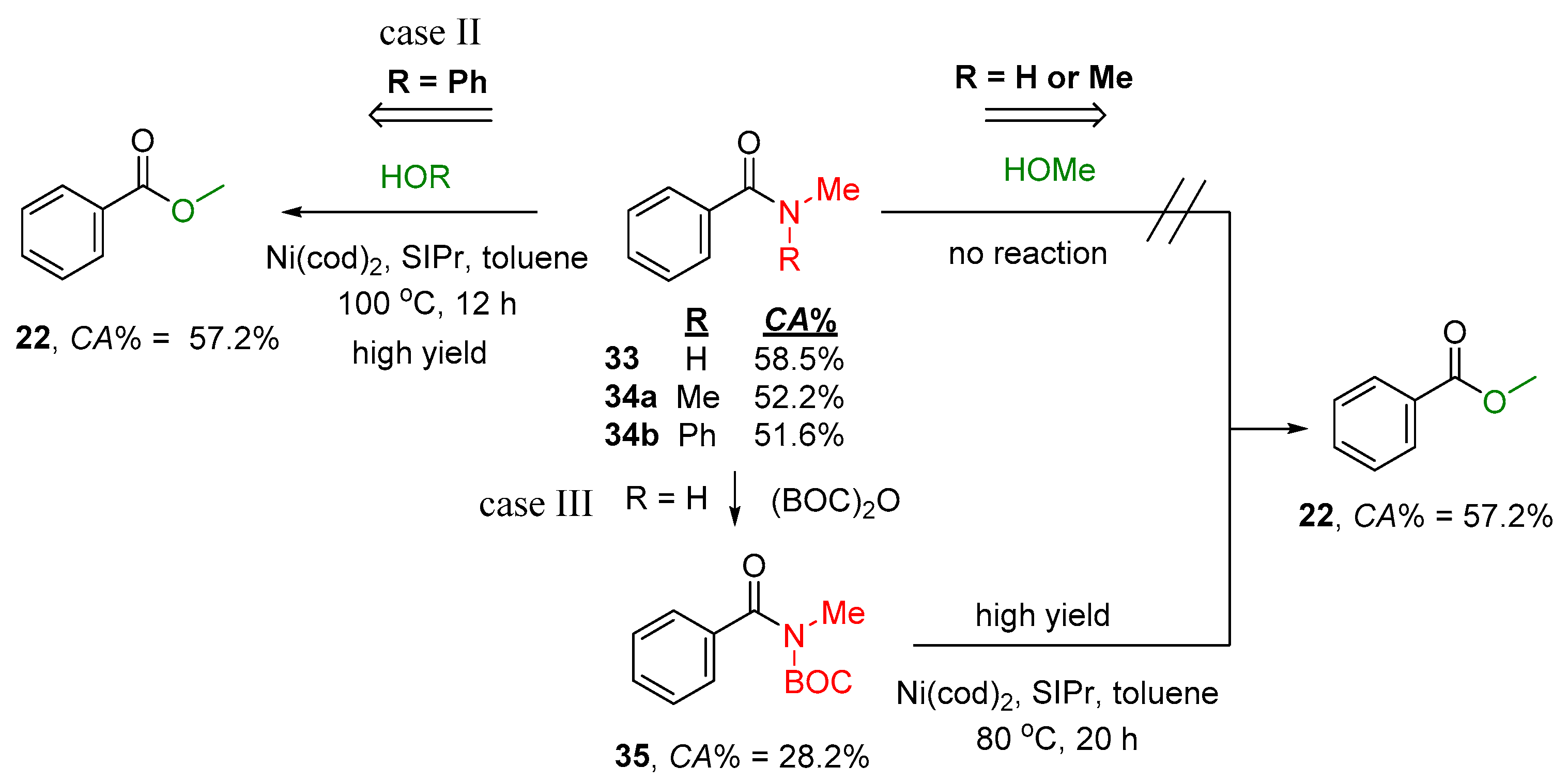
2.2.2. Transamidation Reaction via Activated Amides (case III)
2.2.3. Activated Transamidation Reaction via Product Stabilization [case IV]
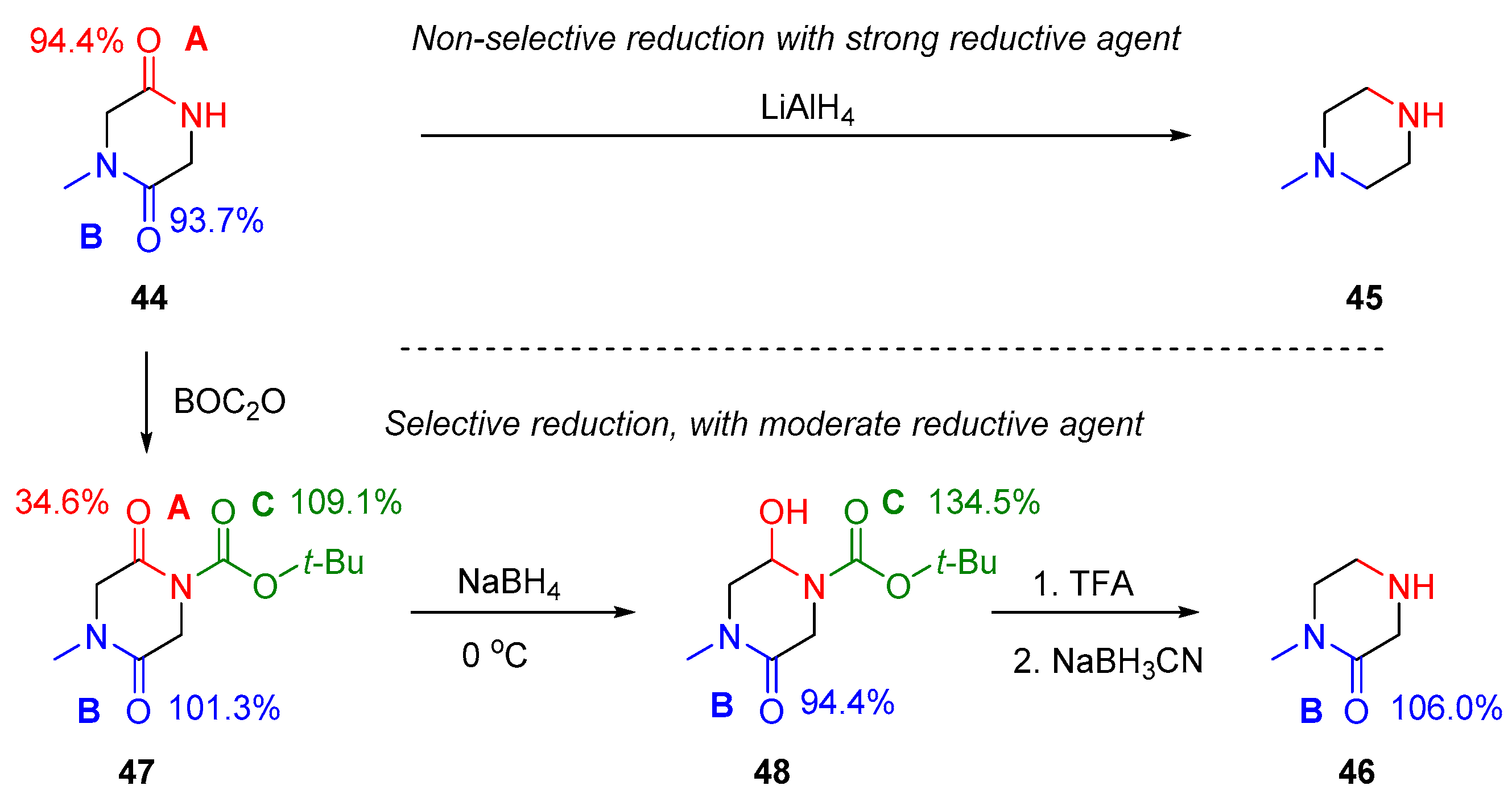
2.3. Amide Reduction via Amide Activation
2.4. Amide Reaction in the Biochemistry
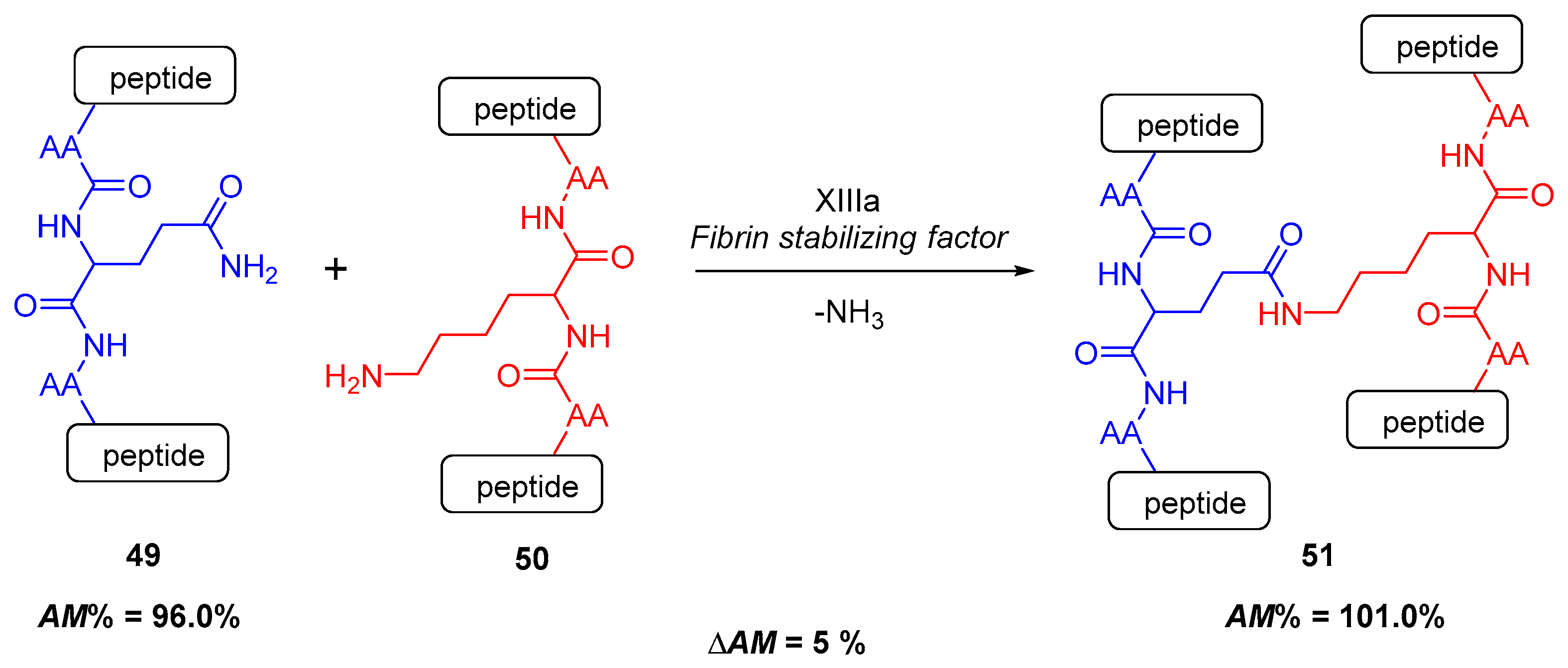
2.4.1. Cross-Linking in the Blood Clotting Process
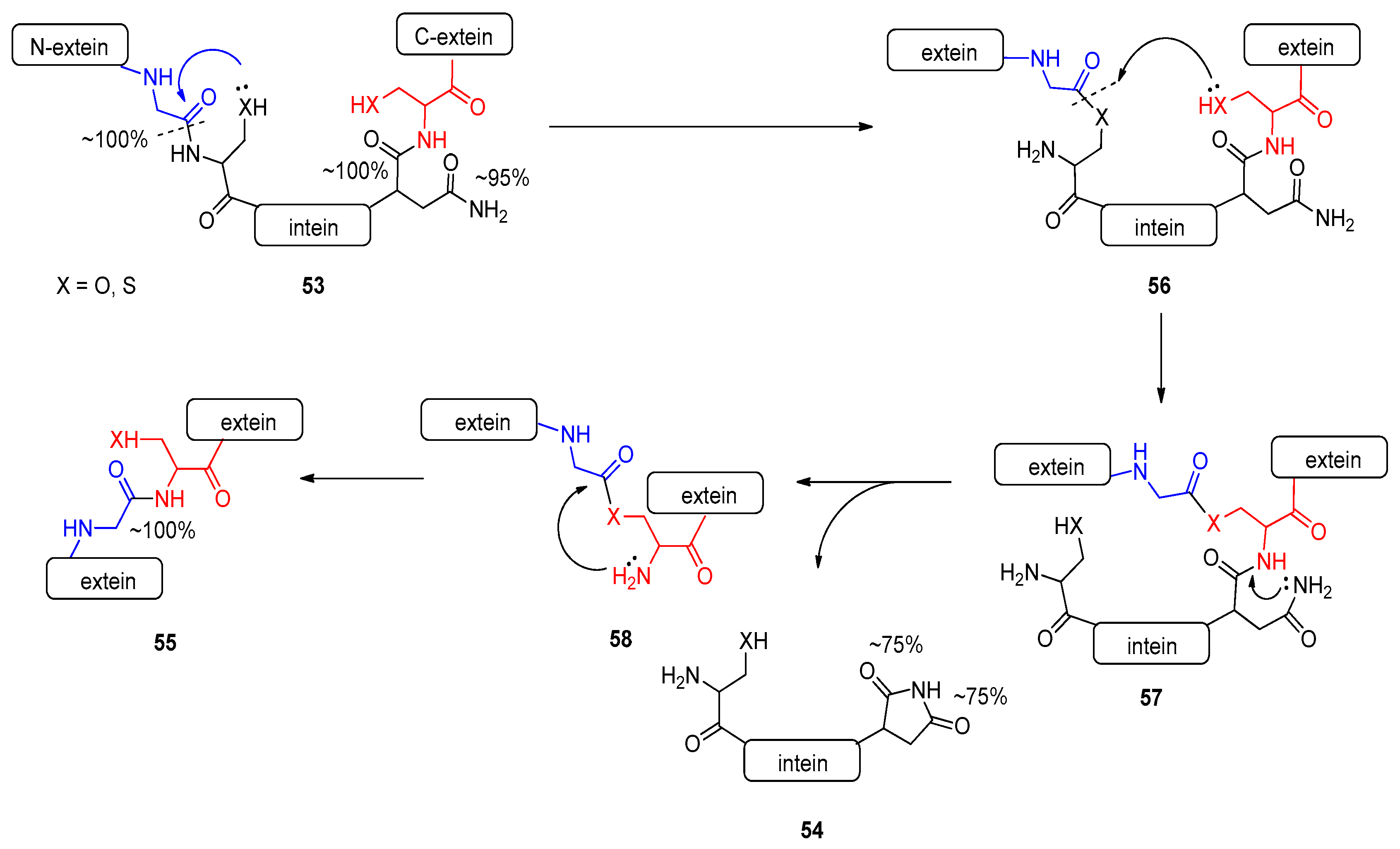
2.4.2. Intein-Mediated Protein Splicing
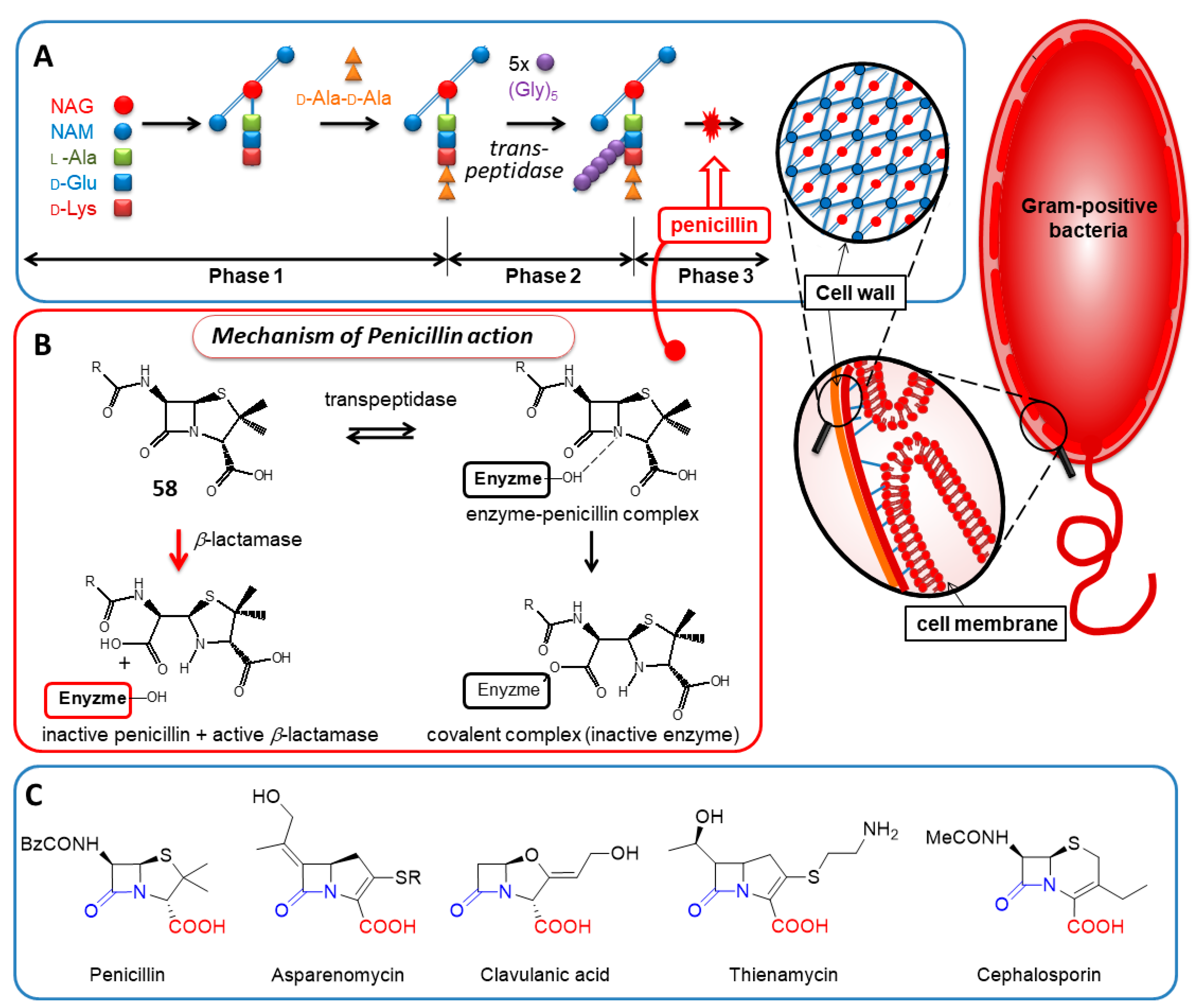
2.4.3. Penicillin
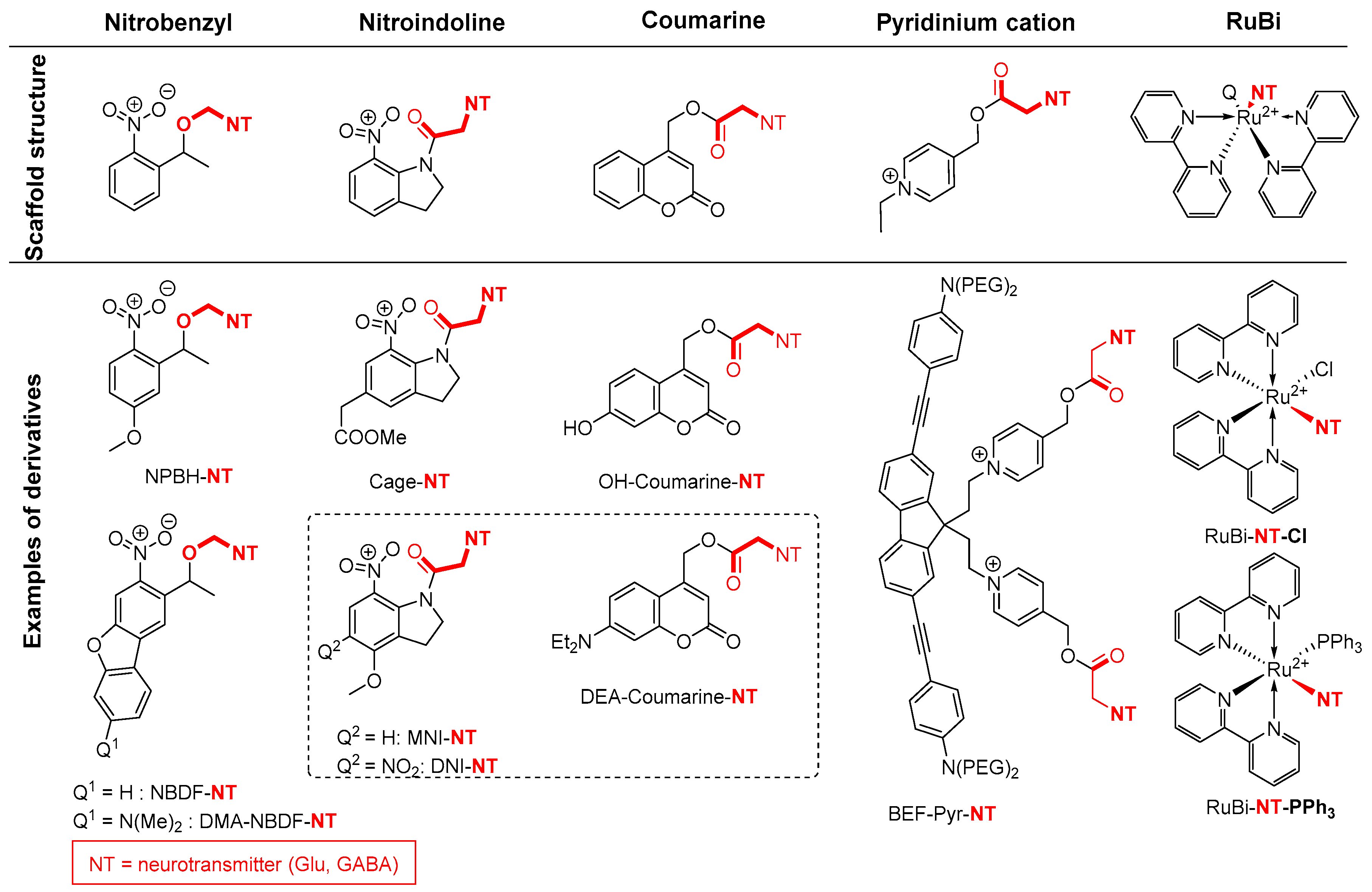
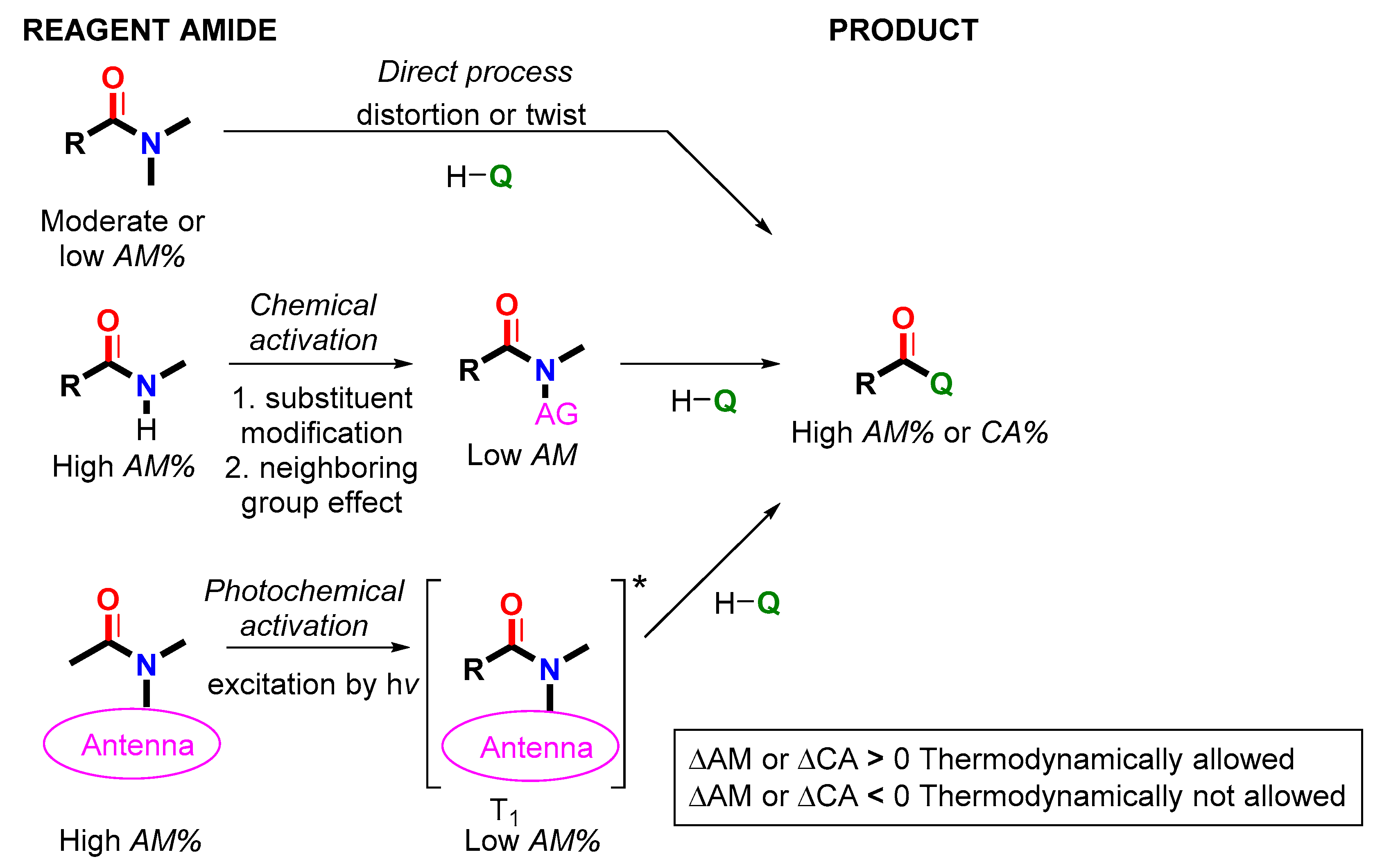
2.5. Amide Activation in Excited State.
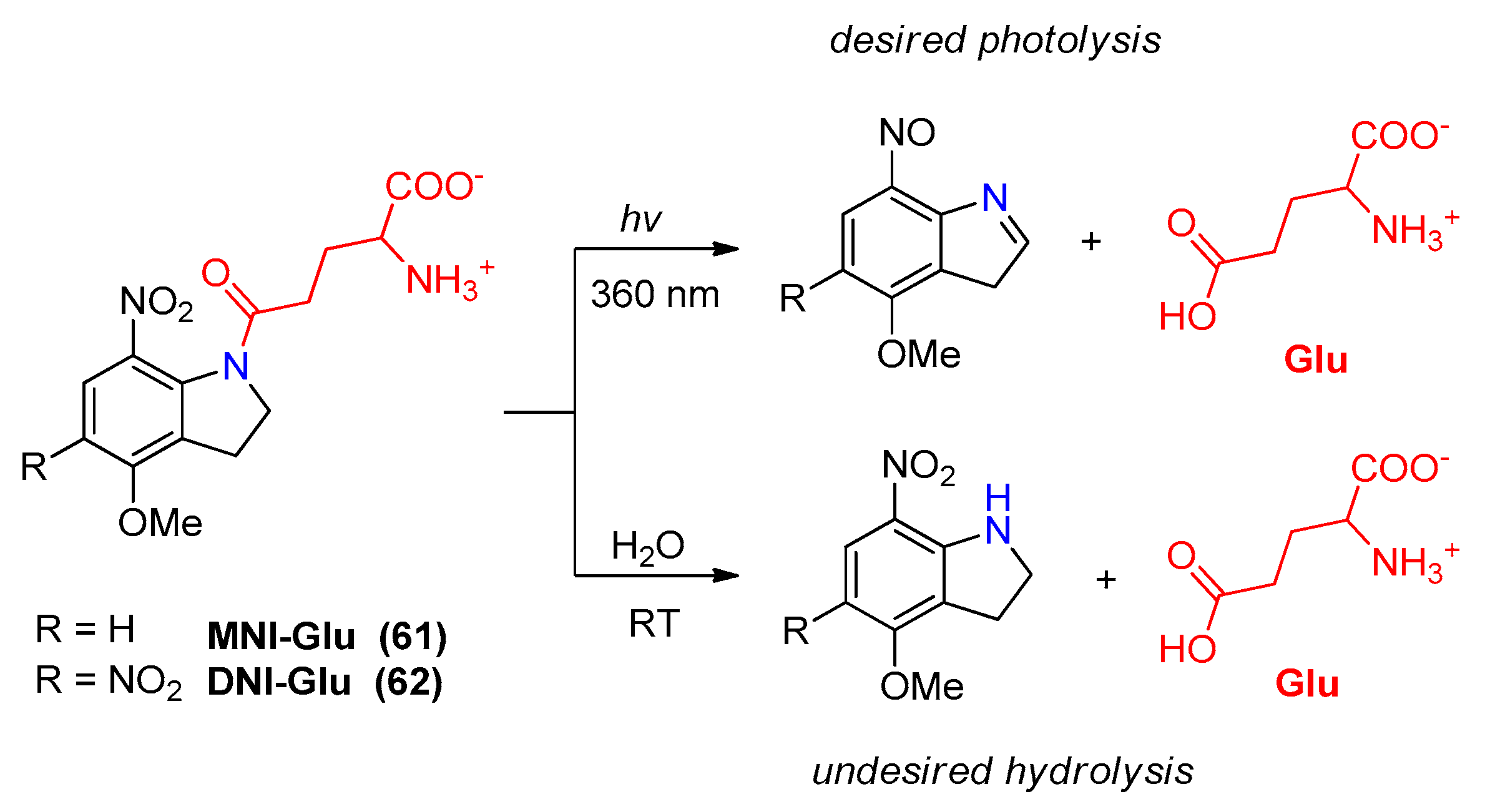
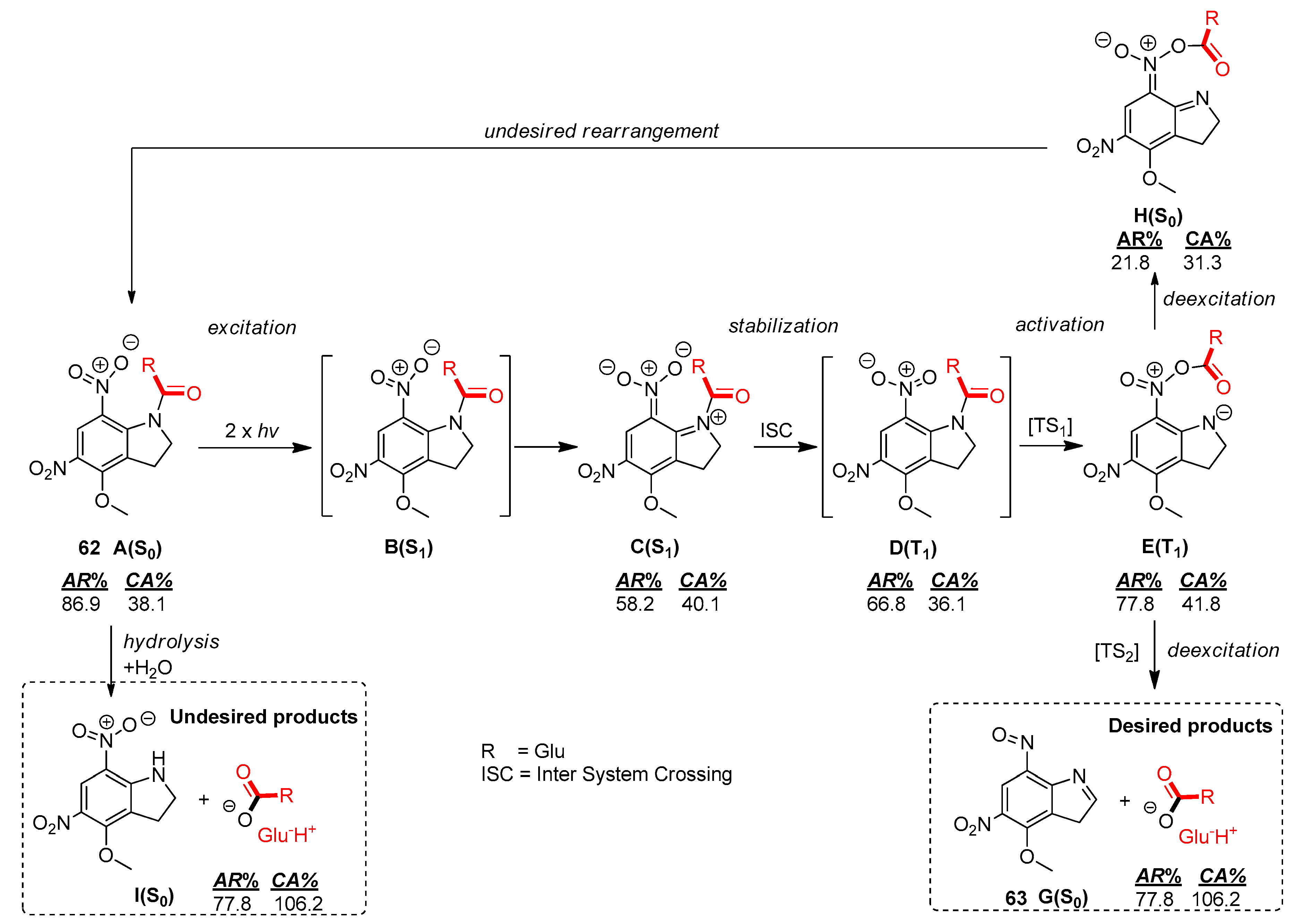
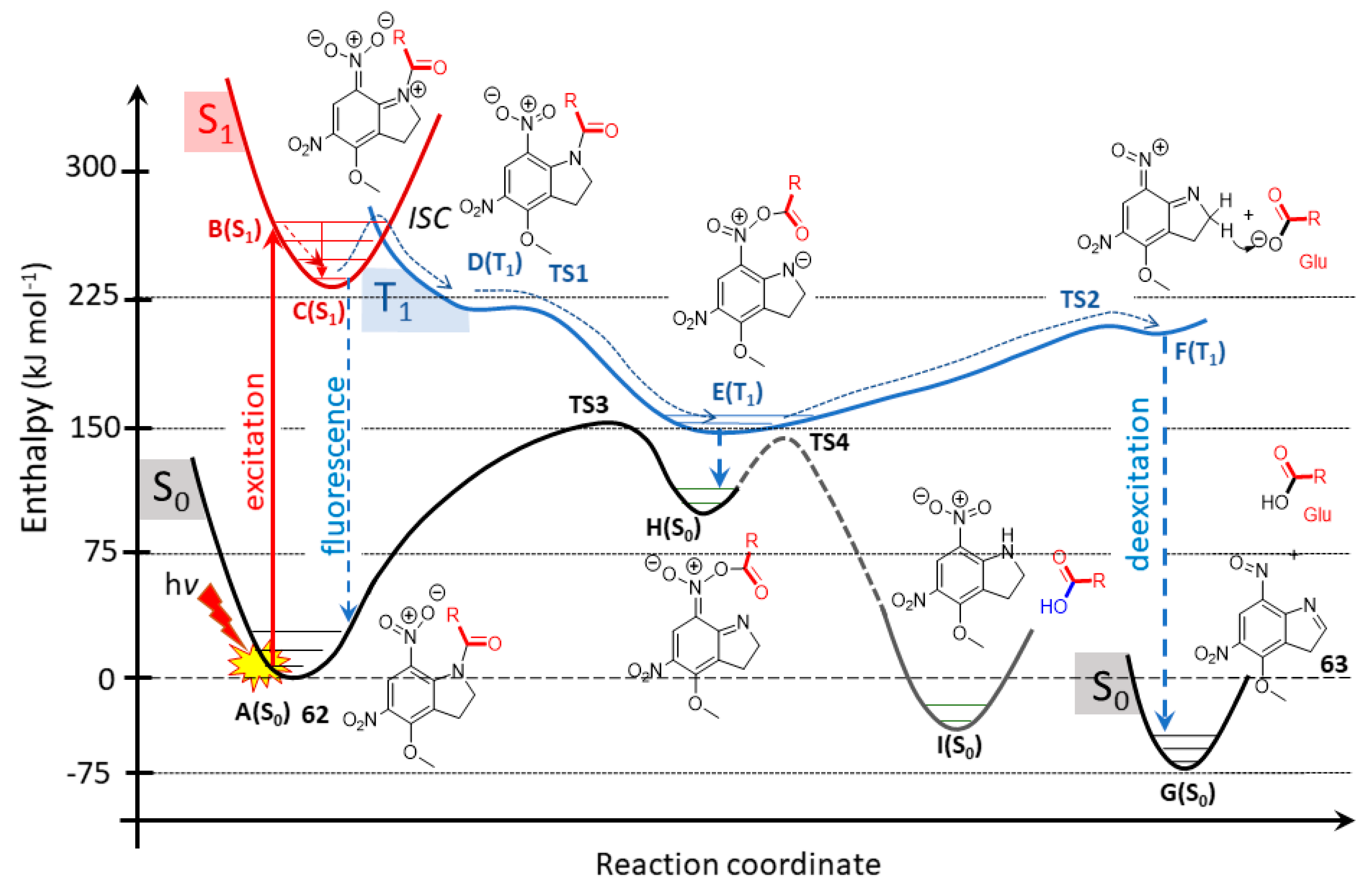
2.5.1. Photochemical Release of Glutamate from MNI-Glu and DNI-Glu at Excited State
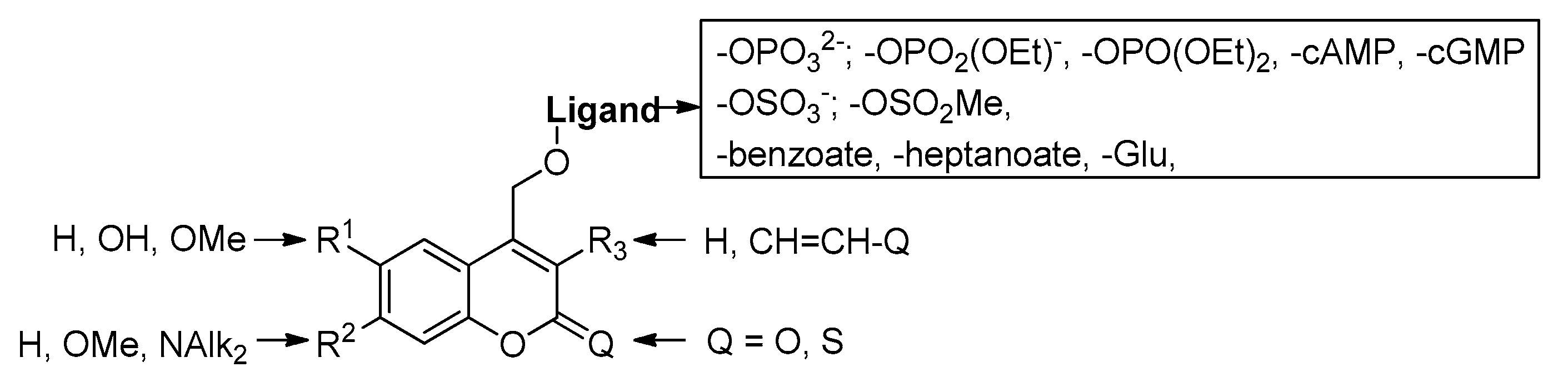
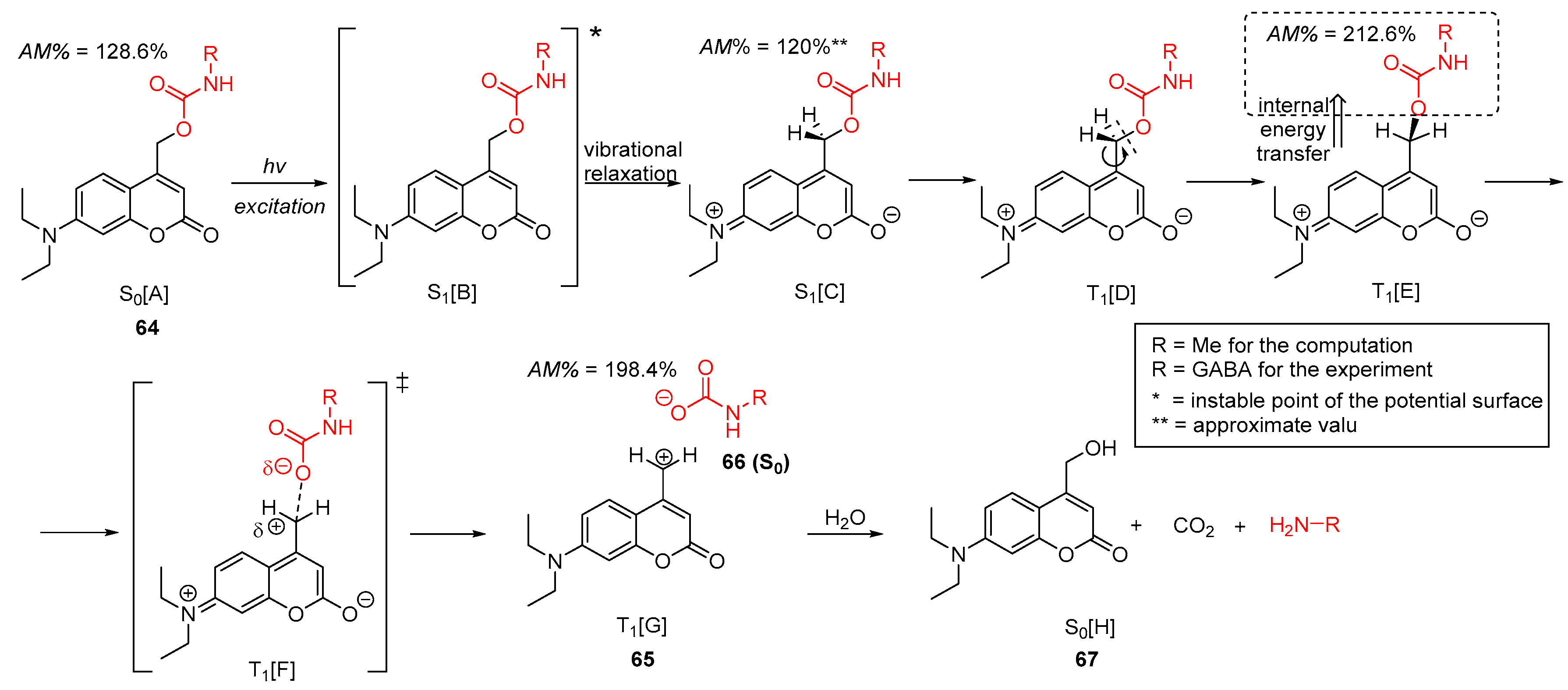
2.5.2. Photochemical Release of Glutamate from Coumarin-Caged Transmitters at Excited State
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, S.; Tang, K.; Raj, M. Amide Bond Activation of Biological Molecules Sriram. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.M.; Suppo, J.; Campagne, J. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Guzmán, P.; Mateus-Gómez, A.; Gamba-Sánchez, D. Direct Transamidation Reactions: Mechanism and Recent Advances. Molecules 2018, 23, 2382. [Google Scholar] [CrossRef] [PubMed]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley-VCH: New York, NY, USA, 2003; ISBN 978-0-471-35893-0. [Google Scholar]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.B. (Ed.) Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32102-5. [Google Scholar]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Oxford University Press: London, UK, 1940. [Google Scholar]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. (Eds.) Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Wiberg, K.B.; Breneman, C.M. Resonance Interactions in Acyclic Systems. 3. Formamide Internal Rotation Revisited. Charge and Energy Redistribution along the C-N Bond Rotational Pathway. J. Am. Chem. Soc. 1992, 114, 831–840. [Google Scholar] [CrossRef]
- Glover, S.A.; Rosser, A.A. Reliable determination of amidicity in acyclic amides and lactams. J. Org. Chem. 2012, 77, 5492–5502. [Google Scholar] [CrossRef] [PubMed]
- Van Vranken, D.; Weiss, G.A. Introduction to Bioorganic Chemistry and Chemical Biology; Garland Science (Taylor & Francis): Oxford, UK, 2013; ISBN 978-0815342144. [Google Scholar]
- Brix, K.; Stöcker, W. Proteases: Structure and Function; Brix, K., Stöcker, W., Eds.; Springer: Berlin, Germany, 2013; ISBN 978-3-7091-0885-7. [Google Scholar]
- Lukes, R. Sur une nouvelle application de la règle de bredt. Collect. Czechslovak Chem. Commun. 1938, 10, 148–152. [Google Scholar] [CrossRef]
- Woodward, R.B.; Neuberger, A.; Trenner, N.R. The Chemistry of Penicillin; Clarke, H.T., Johnson, J.R., Robinson, R., Eds.; Princeton University Press: New Jersey, NJ, USA, 1949. [Google Scholar]
- Wasserman, H.H. Chemistry: Synthesis with a twist. Nature 2006, 441, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Mucsi, Z.; Chass, G.A.; Viskolcz, B.; Csizmadia, I.G. Quantitative scale for the extent of conjugation of carbonyl groups: “Carbonylicity” percentage as a chemical driving force. J. Phys. Chem. A 2008, 112, 9153–9165. [Google Scholar] [CrossRef] [PubMed]
- Kemnitz, C.R.; Loewen, M.J. “Amide resonance” correlates with a breadth of C-N rotation barriers. J. Am. Chem. Soc. 2007, 129, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Mujika, J.I.; Matxain, J.M.; Eriksson, L.A.; Lopez, X. Resonance structures of the amide bond: The advantages of planarity. Chem.-Eur. J. 2006, 12, 7215–7224. [Google Scholar] [CrossRef] [PubMed]
- Bechara, W.S.; Pelletier, G.; Charette, A.B. Chemoselective synthesis of ketones and ketimines by addition of organometallic reagents to secondary amides. Nat. Chem. 2012, 4, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Mujika, J.I.; Mercero, J.M.; Lopez, X. Water-promoted hydrolysis of a highly twisted amide: Rate acceleration caused by the twist of the amide bond. J. Am. Chem. Soc. 2005, 127, 4445–4453. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, Z. Acid-catalyzed reactions of twisted amides in water solution: Competition between hydration and hydrolysis. Chem.-Eur. J. 2011, 17, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Cui, C.; Lee, J.Y.; Park, J.K.; Suh, S.B.; Park, J.; Kim, B.H.; Kim, K.S. N-Protonation vs O-Protonation in Strained Amides: Ab Initio Study. J. Org. Chem. 1997, 62, 4068–4071. [Google Scholar] [CrossRef]
- Mucsi, Z.; Tsai, A.; Szori, M.; Chass, G.A.; Viskolcz, B.; Csizmadia, I.G. A Quantitative Scale for the Extent of Conjugation of the Amide Bond. Amidity Percentage as a Chemical Driving Force. J. Phys. Chem. A 2007, 111, 13245–13254. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Greenberg, A.; Liebman, J.F. Paradigms and paradoxes: O- and N-protonated amides, stabilization energy, and resonance energy. Struct. Chem. 2012, 23, 197–199. [Google Scholar] [CrossRef]
- Morgan, J.; Greenberg, A. Novel bridgehead bicyclic lactams: (A) Molecules predicted to have O-protonated and N-protonated tautomers of comparable stability; (B) hyperstable lactams and their O-protonated tautomers. J. Chem. Thermodyn. 2014, 73, 206–212. [Google Scholar] [CrossRef]
- Tinnis, F.; Volkov, A.; Slagbrand, T.; Adolfsson, H. Chemoselective Reduction of Tertiary Amides under Thermal Control: Formation of either Aldehydes or Amines. Angew. Chem. Int. Ed. 2016, 55, 4562–4566. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.; Spain, M. Highly Chemoselective Reduction of Amides (Primary, Secondary, Tertiary) to Alcohols using SmI2/Amine/H2O under Mild Conditions. J. Am. Chem. Soc. 2014, 136, 2268–2271. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, B.; Svendsen, J.; Engh, R. Density Functional Studies on Secondary Amides: Role of Steric Factors in Cis/Trans Isomerization. Molecules 2018, 23, 2455. [Google Scholar] [CrossRef] [PubMed]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef] [PubMed]
- Ellis-Davies, G.C.R. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Hechel, A. Biologically active molecules with a “light switch”. Angew. Chem. Int. Ed. 2006, 45, 4900–4921. [Google Scholar] [CrossRef] [PubMed]
- Kotsuki, H.; Iwasaki, M.; Nishizawa, H. A new powerful method for the transformation of lactams into ω-amino-carboxamides under high pressure conditions. Tetrahedron Lett. 1992, 33, 4945–4948. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Greenberg, A.; Moore, D.T.; DuBois, T.D. Small and medium-sized bridgehead bicyclic lactams: A systematic ab initio molecular orbital study. J. Am. Chem. Soc. 1996, 118, 8658–8668. [Google Scholar] [CrossRef]
- Greenberg, A.; Venanzi, C.A. Structures and Energetics of Two Bridgehead Lactams and Their N- and O-Protonated Forms: An ab Initio Molecular Orbital Study. J. Am. Chem. Soc. 1993, 115, 6951–6957. [Google Scholar] [CrossRef]
- Mucsi, Z.; Chass, G.A.; Csizmadia, I.G. Amidicity change as a significant driving force and thermodynamic selection rule of transamidation reactions. A synergy between experiment and theory. J. Phys. Chem. B 2008, 112, 7885–7893. [Google Scholar] [CrossRef] [PubMed]
- Lukács, A.; Szabó, L.; Hazai, L.; Szántay, C.; Mák, M.; Gorka, Á.; Szántay, C. Seven-membered ring formation through Grewe-cyclization. Tetrahedron 2001, 57, 5843–5850. [Google Scholar] [CrossRef]
- Sibi, M.P.; Hasegawa, H.; Ghorpade, S.R. A convenient method for the conversion of N-acyloxazolidinones to hydroxamic acids. Org. Lett. 2002, 4, 3343–3346. [Google Scholar] [CrossRef] [PubMed]
- Galat, A.; Elion, G. The Interaction of Amides with Amines: A General Method of Acylation. J. Am. Chem. Soc. 1943, 65, 1566–1567. [Google Scholar] [CrossRef]
- Sowa, F.J.; Nieuwland, J.A. Organic Reactions with Boron Fluoride. XIV. The Reaction of Amides with Acids and Amines. J. Am. Chem. Soc. 1937, 59, 1202–1203. [Google Scholar] [CrossRef]
- Varga, T.R.; Nemes, P.; Mucsi, Z.; Scheiber, P. A concise synthetic pathway towards 5-substituted indolizidines. Tetrahedron Lett. 2007, 48, 1159–1161. [Google Scholar] [CrossRef]
- Wright, W.; Brabander, H.; Hardy, R. Notes- The Rearrangement of N-(Methylaminoalkyl)anilides. J. Org. Chem. 1961, 26, 2120–2123. [Google Scholar] [CrossRef]
- Kano, S.; Ebata, T.; Shibuya, S. Intra- and Intermolecular Nucleophilic Cleavage of the Amide Bond of b-Laactams. Chem. Pharm. Bull. 1979, 27, 2450–2455. [Google Scholar] [CrossRef]
- Wade, P.C.; Vogt, B.R.; Toeplitz, B.; Puar, M.S.; Gougoutas, J.Z. 1,2,4-Triazolo- and 1,2,5-triazino[4,3-d][1,4]benzodiazepinone ring systems: synthesis and barrier to ring inversion. J. Org. Chem. 1979, 44, 88–99. [Google Scholar] [CrossRef]
- Kano, S.; Ebata, T.; Denta, Y.; Hibino, S.; Shibuya, S. Formtion of Some Heterocycles through Ring Transformation of 1-Arylazetidin-2-ones. Heterocycles 1977, 8, 411–416. [Google Scholar] [CrossRef]
- Hie, L.; Fine Nathel, N.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of amides to esters by the nickel-catalysed activation of amide C-N bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Itai, A.; Toriumi, Y.; Tomioka, N.; Kagechika, H.; Azumaya, I.; Shudo, K. Stereochemistry of N-methylbenzanilide and benzanilide. Tetrahedron Lett. 1989, 30, 6177–6180. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, M.; Ge, S.; Hartwig, J.F. Palladium-Catalyzed, Enantioselective α-Arylation of α-Fluorooxindoles. Org. Lett. 2017, 19, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, G.; Mari, M.; Bartolucci, S.; Bartoccini, F.; Piersanti, G. Divergent reactions of oxindoles with amino alcohols: Via the borrowing hydrogen process: Oxindole ring opening vs. C3 alkylation. Org. Chem. Front. 2018, 5, 1622–1627. [Google Scholar] [CrossRef]
- Reverdito, A.M.; Orelli, L.; Dalmaso, M.; Perillo, I.; Fernández, B.M. Synthesis and hydrolysis of substituted tetrahydropyrimidinium salts. Behaviour of the degradation products on varying pH. J. Heterocycl. Chem. 1991, 28, 273–281. [Google Scholar] [CrossRef]
- Milos, S.; Hejtmánková, L.; Hanusek, J.; Machác, V. Synthesis and Ring Transformation of Substituted S-(1-Phenylpyrrolidine-2-ones-3-yl) isothiuronium Salts to Substituted. J. Heterocycl. Chem. 2002, 39, 1105–1107. [Google Scholar] [CrossRef]
- Schade, B.; Hagen, V.; Schmidt, R.; Herbrich, R.; Krause, E.; Eckardt, T.; Bendig, J. Deactivation Behavior and Excited-State Properties of (Coumarin-4-yl)methyl Derivatives. 1. Photocleavage of (7-Methoxycoumarin-4-yl)methyl-Caged Acids with Fluorescence Enhancement. J. Org. Chem. 1999, 64, 9109–9117. [Google Scholar] [CrossRef]
- Liu, C.; Szostak, M. Twisted Amides: From Obscurity to Broadly Useful Transition-Metal-Catalyzed Reactions by N–C Amide Bond Activation. Chem.-Eur. J. 2017, 23, 7157–7173. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D.; Davies, D.E.; Exall, A.M.; Livermore, D.G.; Sollis, S.L.; Nerozzi, F.; Allen, M.J.; Perren, M.; Shabbir, S.S.; Woollard, P.M.; et al. 2,5-Diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists. 2. Synthesis, chirality, and pharmacokinetics. J. Med. Chem. 2005, 48, 6956–6969. [Google Scholar] [CrossRef] [PubMed]
- Matulenko, M.A.; Lee, C.H.; Jiang, M.; Frey, R.R.; Cowart, M.D.; Bayburt, E.K.; DiDomenico, S.; Gfesser, G.A.; Gomtsyan, A.; Guo, Z.Z.; et al. 5-(3-Bromophenyl)-7-(6-morpholin-4-ylpyridin-3-yl)pyrido[2,3-d] pyrimidin-4-ylamine: Structure-activity relationships of 7-substituted heteroaryl analogs as non-nucleoside adenosine kinase inhibitors. Bioorg. Med. Chem. 2005, 13, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Mucsi, Z.; Chass, G.A.; Ábrányi-Balogh, P.; Jójárt, B.; Fang, D.C.; Ramirez-Cuesta, A.J.; Viskolcz, B.; Csizmadia, I.G. Penicillin’s catalytic mechanism revealed by inelastic neutrons and quantum chemical theory. Phys. Chem. Chem. Phys. 2013, 15, 20447–20455. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, G.M.; Skaife, C.J.; Kay, I.T. Strain effects in acyl transfer reactions. J. Chem. Res. Synopses 1980, 9, 294–295. [Google Scholar]
- Wang, Q.P.; Bennet, A.J.; Brown, R.S.; Santarsiero, B.D. Distorted amides as models for activated peptide N-C=O units. 2. The synthesis, hydrolytic profile, and molecular structure of 3,4-dihydro-2-oxo-1,4-propanoquinoline. Can. J. Chem. 1990, 68, 1732–1739. [Google Scholar] [CrossRef]
- Wang, Q.P.; Bennet, A.J.; Brown, R.S.; Santarsiero, B.D. Distorted Amides as Models for Activated Peptide N-C(O) Units. 3. Synthesis, Hydrolytic Profile, and Molecular Structure of 2,3,4,5-Tetrahydro-2-oxo-l,5-propanobenzazepine. J. Am. Chem. Soc. 1991, 113, 5757–5765. [Google Scholar] [CrossRef]
- Somayaji, V.; Brown, R.S. Distorted Amides as Models for Activated Peptide N—C=0 Units Produced during Enzyme-Catalyzed Acyl Transfer Reactions. 1. The Mechanism of Hydrolysis of 3,4-Dihydro-2-oxo-l,4-ethanoquinoline and 2,3,4,5-Tetrahydro-2-oxo-l,5-ethanobenzazepine. J. Org. Chem. 1986, 51, 2676–2686. [Google Scholar] [CrossRef]
- Tani, K.; Stoltz, B.M. Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate. Nature 2006, 441, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Synthesis, structure and reactions of the most twisted amide. J. Chem. Soc. Perkin Trans. 2 2001, 522–529. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Spontaneous, millisecond formation of a twisted amide from the amino acid, and the crystal structure of a tetrahedral intermediate [1]. J. Am. Chem. Soc. 1998, 120, 7101–7102. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Wothers, P.D.; Feeder, N. The Most Twisted Amide: Structure and Reactions. Angew. Chem. Int. Ed. 1998, 37, 785–786. [Google Scholar] [CrossRef]
- Andersson, P.G.; Munslow, I.J. (Eds.) Modern Reduction Methods; Wiley-VCH: New York, NY, USA, 2008; ISBN 978-3-527-31862-9. [Google Scholar]
- Levkoeva, E.I.; Nikitskaya, E.S.; Yakhontov, L.N. Reaction of 2-quinuclidones with and without C-N bond breaking. Dokl. Akad. Nauk 1970, 192, 342. [Google Scholar]
- Von Pracejus, H.; Kehlen, M.; Kehlen, H.; Matschiner, H. Neues zur sterischen mesomeriehinderung bei lactamen vom typ des α-chinuclidons. Tetrahedron 1965, 21, 2257–2270. [Google Scholar] [CrossRef]
- Werstiuk, N.H.; Brown, R.S.; Wang, Q. An AM1 calculational study of the protonation and reactions of 3,4-dihydro-2-oxo-1,4-ethanoquinoline, 3,4-dihydro-2-oxo-1,4-propanoquinoline, 3,3,4,5-tetrahydro-2-oxo-1,5-ethanobenzazepine, 3,3,4,5-tetrahydro-2-oxo-1,5-propanobenzazepine, and N-methyl-4-b. Can. J. Chem. 1996, 74, 524–532. [Google Scholar] [CrossRef]
- Hu, F.; Lalancette, R.; Szostak, M. Structural Characterization of N-Alkylated Twisted Amides: Consequences for Amide Bond Resonance and N-C Cleavage. Angew. Chem. Int. Ed. 2016, 55, 5062–5066. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Gair Ford, J.; Tyler, S.N.G.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered ureas as masked isocyanates: Facile carbamoylation of nucleophiles under neutral conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Haddow, M.F.; Tyler, S.N.G.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Switching pathways: Room-temperature neutral solvolysis and substitution of amides. Angew. Chem. Int. Ed. 2012, 51, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, V.; Skorey, K.I.; Brown, R.S. Molecular Structure of and Its Reaction with Alcohols as a Model for the Acylation Step of the Serine Proteases. J. Org. Chem. 1986, 51, 4866–4872. [Google Scholar] [CrossRef]
- Garcia, J.; González, J.; Segura, R.; Urpí, F.; Vilarrasa, J. Reaction of N-Nitroso- and N-Nitro-N-alkylamides with Amines. J. Org. Chem. 1984, 49, 3322–3327. [Google Scholar] [CrossRef]
- Garcia, J.; Vilarrasa, J. New synthetic “tricks” using old reagents. A mild method for the conversion of RCONHR′ to RCONHR″. Tetrahedron Lett. 1982, 23, 1127–1128. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Bergeron, R. Trifumides: New Acylating Ard Triflating Reagents. Tetrahedron Lett. 1973, 14, 4607–4610. [Google Scholar] [CrossRef]
- Bon, E.; Bigg, D.C.H.; Bertrand, G. Aluminum Chloride-Promoted Transamidation Reactions. J. Org. Chem. 1994, 59, 4035–4036. [Google Scholar] [CrossRef]
- Shah, K.R.; Blanton, C.D.W. Reaction of Maleimides and Ethyl 3-Aminocrotonates. A Reinvestigation Leading to an Improved Synthesis of Pyrrolo[3,4-c]pyridines. J. Org. Chem. 1982, 47, 502–508. [Google Scholar] [CrossRef]
- Davidsen, S.K.; May, P.D.; Summers, J.B. Di-tert-butyl N-Acylimidodicarbonates as Isolable Acylating Agents: Mild Conversion of Primary Carboxamides to Substituted Amides. J. Org. Chem. 1991, 56, 5482–5485. [Google Scholar] [CrossRef]
- Shapiro, G.; Marzi, M. Facile and Selective O-Alkyl Transesterification of Primary Carbamates with Titanium(IV) Alkoxides. J. Org. Chem. 1997, 62, 7096–7097. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Eagles, J.B.; Rudahindwa, L.; Hamaker, C.G.; Hitchcock, S.R. Synthesis, X-ray crystallography, and reactions of N-acyl and N-carbamoyl succinimides. Synth. Commun. 2013, 43, 2155–2164. [Google Scholar] [CrossRef]
- Goodman, C.A.; Hamaker, C.G.; Hitchcock, S.R. Synthesis and evaluation of some variants of the Nefkens’ reagent. Tetrahedron Lett. 2013, 54, 6012–6014. [Google Scholar] [CrossRef]
- Ji, C.-L.; Xie, P.-P.; Hong, X. Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters. Molecules 2018, 23, 2681. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, S.L.; Fu, Y.; Guo, Q.X.; Liu, L. Mechanism of Ni-catalyzed selective C-O bond activation in cross-coupling of aryl esters. J. Am. Chem. Soc. 2009, 131, 8815–8823. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Fu, Y. Mechanistic origin of cross-coupling selectivity in Ni-catalysed Tishchenko reactions. Chem.-Eur. J. 2012, 18, 16765–16773. [Google Scholar] [CrossRef] [PubMed]
- Stach, H.; Hesse, M. Synthesis of macrocyclic compounds by ring enlargement. Tetrahedron 1988, 44, 1573–1590. [Google Scholar] [CrossRef]
- Süsse, M.; Hájícek, J.; Hesse, M. Studies on the Carbon Zip Reaction of 2-OxocycIoalkane-l-carbonitriles. Helv. Chim. Acta 1985, 68, 1986–1997. [Google Scholar] [CrossRef]
- Askitoglu, E.; Guggisberg, A.; Hesse, M. N-substituted 3-aminopropanenitriles and 2-aminoacetonitriles asSchiff-base equivalents. Helv. Chim. Acta 1985, 68, 750–759. [Google Scholar] [CrossRef]
- Seyden-Penne, J. Reductions by the Alumino and Borohydrides in Organic Synthesis; Wiley-VCH: New York, NY, USA, 1997; ISBN 978-0-471-19036-3. [Google Scholar]
- Hudlicky, M. (Ed.) Reductions in Organic Chemistry; John Wiley & Sons: New York, NY, USA, 1984. [Google Scholar]
- Trost, B.M.; Fleming, I. (Eds.) Comprehensive Organic Synthesis; Pergamon: Oxford, UK, 1991. [Google Scholar]
- Gribble, G.W. Sodium borohydride in carboxylic acid media: A phenomenal reduction system. Chem. Soc. Rev. 1998, 27, 395. [Google Scholar] [CrossRef]
- Reeves, J.T.; Tan, Z.; Marsini, M.A.; Han, Z.S.; Xu, Y.; Reeves, D.C.; Lee, H.; Lu, B.Z.; Senanayake, C.H. A practical procedure for reduction of primary, secondary and tertiary amides to amines. Adv. Synth. Catal. 2013, 355, 47–52. [Google Scholar] [CrossRef]
- Simmons, B.J.; Hoffmann, M.; Hwang, J.; Jackl, M.K.; Garg, N.K. Nickel-Catalyzed Reduction of Secondary and Tertiary Amides. Org. Lett. 2017, 19, 1910–1913. [Google Scholar] [CrossRef] [PubMed]
- Ortín, I.; González, J.F.; de la Cuesta, E.; Avendaño, C. Synthesis of a novel tetrahydroisoquinoline pentacyclic framework. Tetrahedron 2009, 65, 2201–2211. [Google Scholar] [CrossRef]
- Ortín, I.; González, J.F.; de la Cuesta, E.; Manguan-García, C.; Perona, R.; Avendaño, C. Pyrazino[1,2-b]isoquinolines: Synthesis and study of their cytostatic and cytotoxic properties. Bioorganic Med. Chem. 2008, 16, 9065–9078. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.W.N.; Williams, R.M. The Concise and Versatile Synthesis of Epi-malbrancheamide and Structurally Related Analogs. Heterocycles 2006, 70, 249–259. [Google Scholar] [CrossRef]
- Lee, B.H.; Clothier, M.F. Selective reduction of secondary amides to amines in the presence of tertiary amides. Tetrahedron Lett. 1999, 40, 643–644. [Google Scholar] [CrossRef]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.; Poncin, M.; Nilakantan, R.; Ellestad, G. Calicheamicin gamma 1I and DNA: Molecular recognition process responsible for site-specificity. Science 1989, 244, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Garbaccio, R.M. Shape-dependent catalysis: Insights into the source of catalysis for the CC-1065 and duocarmycin DNA alkylation reaction. Acc. Chem. Res. 1999, 32, 1043–1052. [Google Scholar] [CrossRef]
- Boger, D.L.; Johnson, D.S. CC-1065 and the duocarmycins: Understanding their biological function through mechanistic studies. Angew. Chem. Int. Ed. 1996, 35, 1438–1474. [Google Scholar] [CrossRef]
- Boger, D.L.; Boyce, C.W.; Garbaccio, R.M.; Goldberg, J.A. CC-1065 and the duocarmycins: Synthetic studies. Chem. Rev. 1997, 97, 787–828. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Schellenberg, B.; Bachmann, A.S.; Archer, C.R.; Huber, R.; Powell, T.K.; Lindow, S.; Kaiser, M.; Dudler, R. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 2008, 452, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Thomas, P.M.; Scheerer, J.R.; Vagstad, A.L.; Kelleher, N.L.; Townsend, C.A. Deconstruction of iterative multidomain polyketide synthase function. Science 2008, 320, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Bud, R. The fall of a wonder drug. Nature 2007, 446, 981. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.C.; Bird, R.J. Review article: Coagulation cascade and therapeutics update: Relevance to nephrology. Part 1: Overview of coagulation, thrombophilias and history of anticoagulants. Nephrology 2009, 14, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.; Benz, E.; Shattil, S.; Furie, B.; Cohen, H. (Eds.) Hematology: Basic Principles and Practice, 4th ed.; Elsevier: Philadelphia, PA, USA, 2005; ISBN 978-0443079542. [Google Scholar]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Kane, P.; Yamashiro, C.; Wolczyk, D.; Neff, N.; Goebi, M.; Stevens, T. Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science 1990, 250, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Anraku, Y.; Mizutani, R.; Satow, Y. Protein splicing: Its discovery and structural insight into novel chemical mechanisms. IUBMB Life 2005, 57, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Hirata, R.; Ohsumi, Y.; Nakano, A.; Kawasaki, H.; Suzuki, K.; Anraku, Y. Molecular Structure of a Gene, VMA1, Encoding the Catalytic Subunit of H + -Translocating Adenosine Triphosphatase from Vacuolar Membranes of Saccharomyces cerevisiae. J. Biol.Chem. 1990, 265, 6726–6733. [Google Scholar] [PubMed]
- Gimble, F.S.; Thorner, J. Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature 1992, 357, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Makino, S.I.; Matsuzawa, H.; Satow, Y.; Ohya, Y.; Anraku, Y. Folding-dependent in vitro protein splicing of the Saccharomyces cerevisiae VMA1 protozyme. Biochem. Biophys. Res. Commun. 1996, 222, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Nogami, S.; Satow, Y.; Ohya, Y.; Anraku, Y. Identification of three core regions essential for protein splicing of the yeast Vma1 protozyme. A random mutagenesis study of the entire VMA1- derived endonuclease sequence. J. Biol. Chem. 1997, 272, 15668–15674. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Satow, Y.; Ohya, Y.; Anraku, Y. Protein splicing in the yeast Vma1 protozyme: Evidence for an intramolecular reaction. FEBS Lett. 1997, 412, 518–520. [Google Scholar] [CrossRef]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775. [Google Scholar] [CrossRef] [PubMed]
- Dennis, C. The bugs of war. Nature 2001, 411, 232. [Google Scholar] [CrossRef] [PubMed]
- Theilgaard, H.B.A.; Kristiansen, K.N.; Henriksen, C.M.; Nielsen, J. Purification and characterization of delta-(l-alpha-aminoadipyl)-l-cysteinyl-d-valine synthetase from Penicillium chrysogenum. Biochem. J. 1997, 327, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Huffman, G.W.; Gesellchen, P.D.; Turner, J.R.; Rothenberger, R.B.; Osborne, H.E.; Miller, F.D.; Chapman, J.L.; Queener, S.W. Substrate Specificity of Isopenicillin N Synthase. J. Med. Chem. 1992, 35, 1897–1914. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.L.; Clifton, I.J.; Fülöp, V.; Harlos, K.; Barton, G.J.; Hajdu, J.; Andersson, I.; Schofield, C.J.; Baldwin, J.E. Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature 1995, 375, 700. [Google Scholar] [CrossRef] [PubMed]
- Voet, D.; Voet, J.G. Biochemistry, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Cohen, M.L. Changing patterns of infectious disease. Nature 2000, 406, 762. [Google Scholar] [CrossRef] [PubMed]
- Smaglik, P. Antibiotic resistance must be monitored, US Senate is told. Nature 2000, 407, 437. [Google Scholar] [CrossRef] [PubMed]
- Nogrady, T. Medicinal Chemistry A Biochemical Approach; Oxford University Press: New York, NY, USA, 1985. [Google Scholar]
- Sheehan, J.C. The enchanted Ring: The Untold Story of Penicillin; The MIT Press: Cambridge, MA, USA, 1982; ISBN 9780262192040. [Google Scholar]
- Johnson, J.R.; Woodward, R.B.; Robinson, R. The Chemistry of Penicillin; Clarke, H.T., Ed.; Princton University Press: Princton, NJ, USA, 1949; ISBN 9780691627489. [Google Scholar]
- Woodward, R.B. Recent advances in the chemistry of natural products. Science 1966, 153, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.; Abraham, E.P. Cephalosporin C, a New Antibiotic containing Sulphur and d-α-Aminoadipic Acid. Nature 1955, 175, 548. [Google Scholar] [CrossRef] [PubMed]
- Tally, F.P.; Jacobus, N.V.; Gorbach, S.L. In vitro activity of thienamycin. Antimicrob. Agents Chemother. 1978, 14, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.B.R.; Schmitt, S.M.; Bouffard, F.A.; Christensen, B.G. Total Synthesis of (±)-Thienamycin. J. Am. Chem. Soc. 1978, 100, 313–315. [Google Scholar] [CrossRef]
- Araya, R. Input transformation by dendritic spines of pyramidal neurons. Front. Neuroanat. 2014, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Granger, A.J.; Nicoll, R.A. Expression mechanisms underlying long-term potentiation: A postsynaptic view, 10 years on. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 1–6. [Google Scholar] [CrossRef]
- Pálfi, D.; Chiovini, B.; Szalay, G.; Kaszás, A.; Turi, G.F.; Katona, G.; Ábrányi-Balogh, P.; Szori, M.; Potor, A.; Frigyesi, O.; et al. High efficiency two-photon uncaging coupled by the correction of spontaneous hydrolysis. Org. Biomol. Chem. 2018, 16, 1958–1970. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, G.; Ogden, D.; Kelly, G.; Corrie, J.E.T. Synthetic and photochemical studies of substituted 1-acyl-7-nitroindolines. Photochem. Photobiol. Sci. 2005, 4, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Warther, D.; Gug, S.; Specht, A.; Bolze, F.; Nicoud, J.F.; Mourot, A.; Goeldner, M. Two-photon uncaging: New prospects in neuroscience and cellular biology. Bioorg. Med. Chem. 2010, 18, 7753–7758. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Kovács, E.; Sághy, P.; Turczel, G.; Tóth, I.; Lendvay, G.; Domján, A.; Anastas, P.T.; Tuba, R. Metathesis of renewable polyene feedstocks–Indirect evidences of the formation of catalytically active ruthenium allylidene species. J. Organomet. Chem. 2017, 847, 213–217. [Google Scholar] [CrossRef]
- Lu, M.; Fedoryak, O.D.; Moister, B.R.; Dore, T.M. Bhc-diol as a photolabile protecting group for aldehydes and ketones. Org. Lett. 2003, 5, 2119–2122. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, H.J.; Perdicakis, B.; Fishlock, D.; Lajoie, G.A.; Jervis, E.; Guy Guillemette, J. Photo-control of nitric oxide synthase activity using a caged isoform specific inhibitor. Bioorg. Med. Chem. 2002, 10, 1919–1927. [Google Scholar] [CrossRef]
- Takaoka, K.; Tatsu, Y.; Yumoto, N.; Nakajima, T.; Shimamoto, K. Synthesis and photoreactivity of caged blockers for glutamate transporters. Bioorg. Med. Chem. Lett. 2003, 13, 965–970. [Google Scholar] [CrossRef]
- Takaoka, K.; Tatsu, Y.; Yumoto, N.; Nakajima, T.; Shimamoto, K. Synthesis of carbamate-type caged derivatives of a novel glutamate transporter blocker. Bioorg. Med. Chem. 2004, 12, 3687–3694. [Google Scholar] [CrossRef] [PubMed]
- Givens, R.S.; Matuszewski, B. Photochemistry of Phosphate Esters: An Efficient Method for the Generation of Electrophiles1. J. Am. Chem. Soc. 1984, 106, 6860–6861. [Google Scholar] [CrossRef]
- Furata, T.; Torigai, H.; Sugimoto, M.; Iwamura, M. Photochemical Properties of New Photolabile cAMP Derivatives in a Physiological Saline Solution. J. Org. Chem. 1995, 60, 3953–3956. [Google Scholar] [CrossRef]
- Hagen, V.; Bendig, J.; Frings, S.; Wiesner, B.; Schade, B.; Helm, S.; Lorenz, D.; Benjamin Kaupp, U. Synthesis, photochemistry and application of (7-methoxycoumarin-4-yl)methyl-caged 8-bromoadenosine cyclic 3’,5’-monophosphate and 8-bromoguanosine cyclic 3′,5′-monophosphate photolyzed in the nanosecond time region. J. Photochem. Photobiol. B Biol. 1999, 53, 91–102. [Google Scholar] [CrossRef]
- Eckardt, T.; Hagen, V.; Schade, B.; Schmidt, R.; Schweitzer, C.; Bendig, J. Deactivation behavior and excited-state properties of (coumarin-4-yl)methyl derivatives. 2. Photocleavage of selected (coumarin-4-yl)methyl-caged adenosine cyclic 3′,5′-monophosphates with fluorescence enhancement. J. Org. Chem. 2002, 67, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Geißler, D.; Kresse, W.; Wiesner, B.; Bendig, J.; Kettenmann, H.; Hagen, V. DMACM-caged adenosine nucleotides: Ultrafast phototriggers for ATP, ADP, and AMP activated by long-wavelength irradiation. ChemBioChem 2003, 4, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Hagen, V.; Frings, S.; Wiesner, B.; Helm, S.; Kaupp, U.B.; Bendig, J. [7-(Dialkylamino)coumarin-4-yl]methyl-caged compounds as ultrafast and effective long-wavelength phototriggers of 8-bromo-substituted cyclic nucleotides. ChemBioChem 2003, 4, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Takeuchi, H.; Isozaki, M.; Takahashi, Y.; Kanehara, M.; Sugimoto, M.; Watanabe, T.; Noguchi, K.; Dore, T.M.; Kurahashi, T.; et al. Bhc-cNMPs as either water-soluble or membrane-permeant photoreleasable cyclic nucleotides for both one- and two-photon excitation. ChemBioChem 2004, 5, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Geißler, D.; Antonenko, Y.N.; Schmidt, R.; Keller, S.; Krylova, O.O.; Wiesner, B.; Bendig, J.; Pohl, P.; Hagen, V. (Coumarin-4-yl)methyl esters as highly efficient, ultrafast phototriggers for protons and their application to acidifying membrane surfaces. Angew. Chem. Int. Ed. 2005, 44, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Hagen, V.; Dekowski, B.; Nache, V.; Schmidt, R.; Geißler, D.; Lorenz, D.; Eichhorst, J.; Keller, S.; Kaneko, H.; Benndorf, K.; Wiesner, B. Coumarinylmethyl esters for ultrafast release of high concentrations of cyclic nucleotides upon one- and two-photon photolysis. Angew. Chem. Int. Ed. 2005, 44, 7887–7891. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Wang, S.S.-H.; Dantzker, J.L.; Dore, T.M.; Bybee, W.J.; Callaway, E.M.; Denk, W.; Tsien, R.Y. Brominated 7-hydroxycoumarin-4-ylmethyls: Photolabile protecting groups with biologically useful cross-sections for two photon photolysis. Proc. Natl. Acad. Sci. USA 1999, 96, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lawrence, D.S. A strategy for the construction of caged diols using a photolabile protecting group. J. Org. Chem. 2002, 67, 2723–2726. [Google Scholar] [CrossRef] [PubMed]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | n = 1 (a) | n = 2 (a) | n = 3 (a) | n = 4 (a) |
|---|---|---|---|---|
| Start (CA%) 17 | 46.5% | 60.3% | 55.2% | 53.4% |
| Product (CA%) 18 | 56.2% | 56.1% | 56.1% | 56.2% |
| ΔCA | +10.7% | −4.2% | +0.9% | +2.8% |
| Method | A or B | only B | A 1 or B | A or B |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, E.; Rózsa, B.; Csomos, A.; Csizmadia, I.G.; Mucsi, Z. Amide Activation in Ground and Excited States. Molecules 2018, 23, 2859. https://doi.org/10.3390/molecules23112859
Kovács E, Rózsa B, Csomos A, Csizmadia IG, Mucsi Z. Amide Activation in Ground and Excited States. Molecules. 2018; 23(11):2859. https://doi.org/10.3390/molecules23112859
Chicago/Turabian StyleKovács, Ervin, Balázs Rózsa, Attila Csomos, Imre G. Csizmadia, and Zoltán Mucsi. 2018. "Amide Activation in Ground and Excited States" Molecules 23, no. 11: 2859. https://doi.org/10.3390/molecules23112859
APA StyleKovács, E., Rózsa, B., Csomos, A., Csizmadia, I. G., & Mucsi, Z. (2018). Amide Activation in Ground and Excited States. Molecules, 23(11), 2859. https://doi.org/10.3390/molecules23112859