
2-(4-Methylsulfonylphenyl)pyrimidines as Prospective Radioligands for Imaging Cyclooxygenase-2 with PET—Synthesis, Triage, and Radiolabeling

 ,
,
Abstract
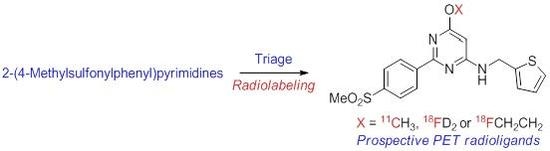
:
1. Introduction
2. Results and Discussion
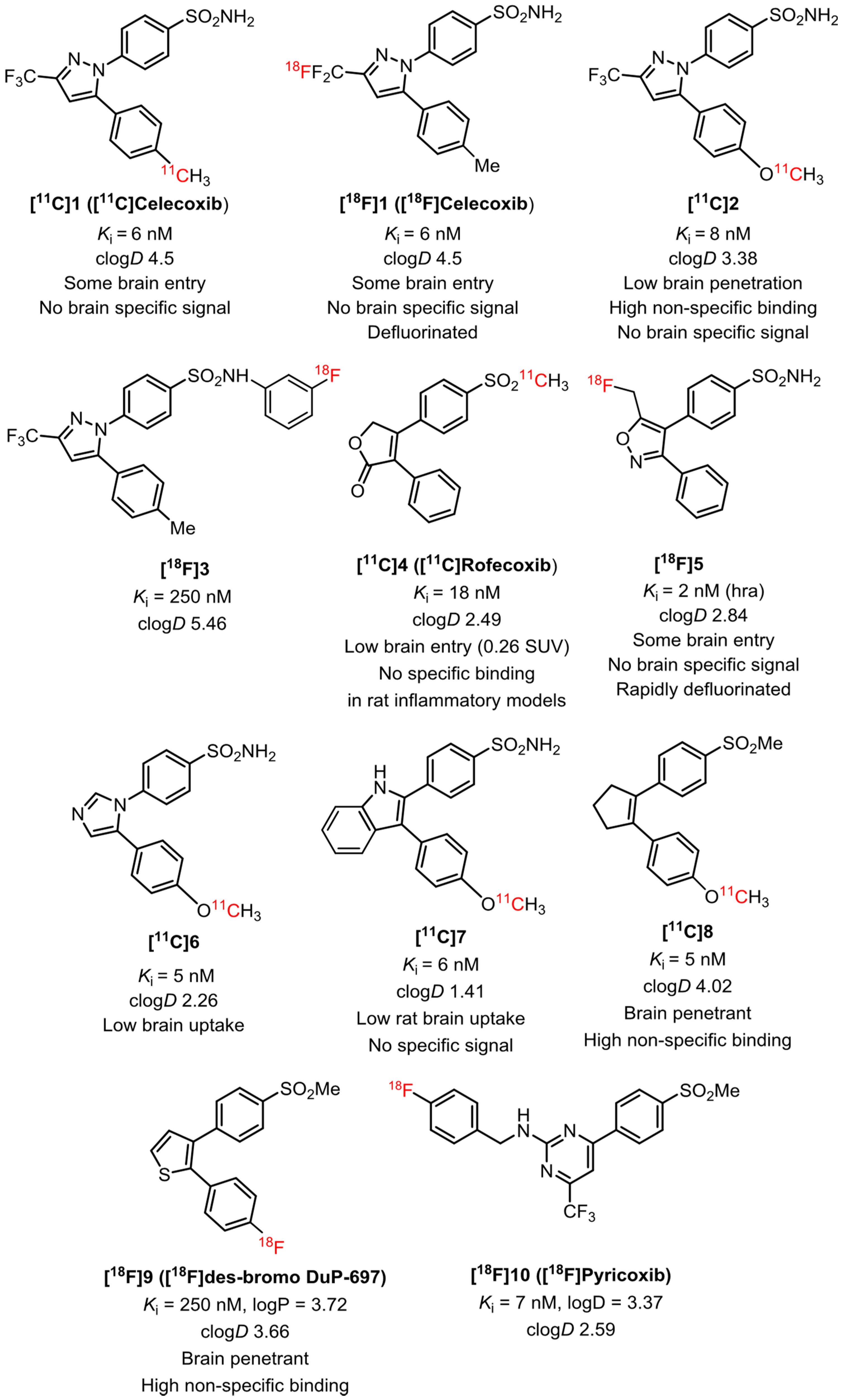
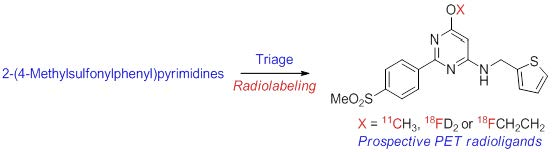
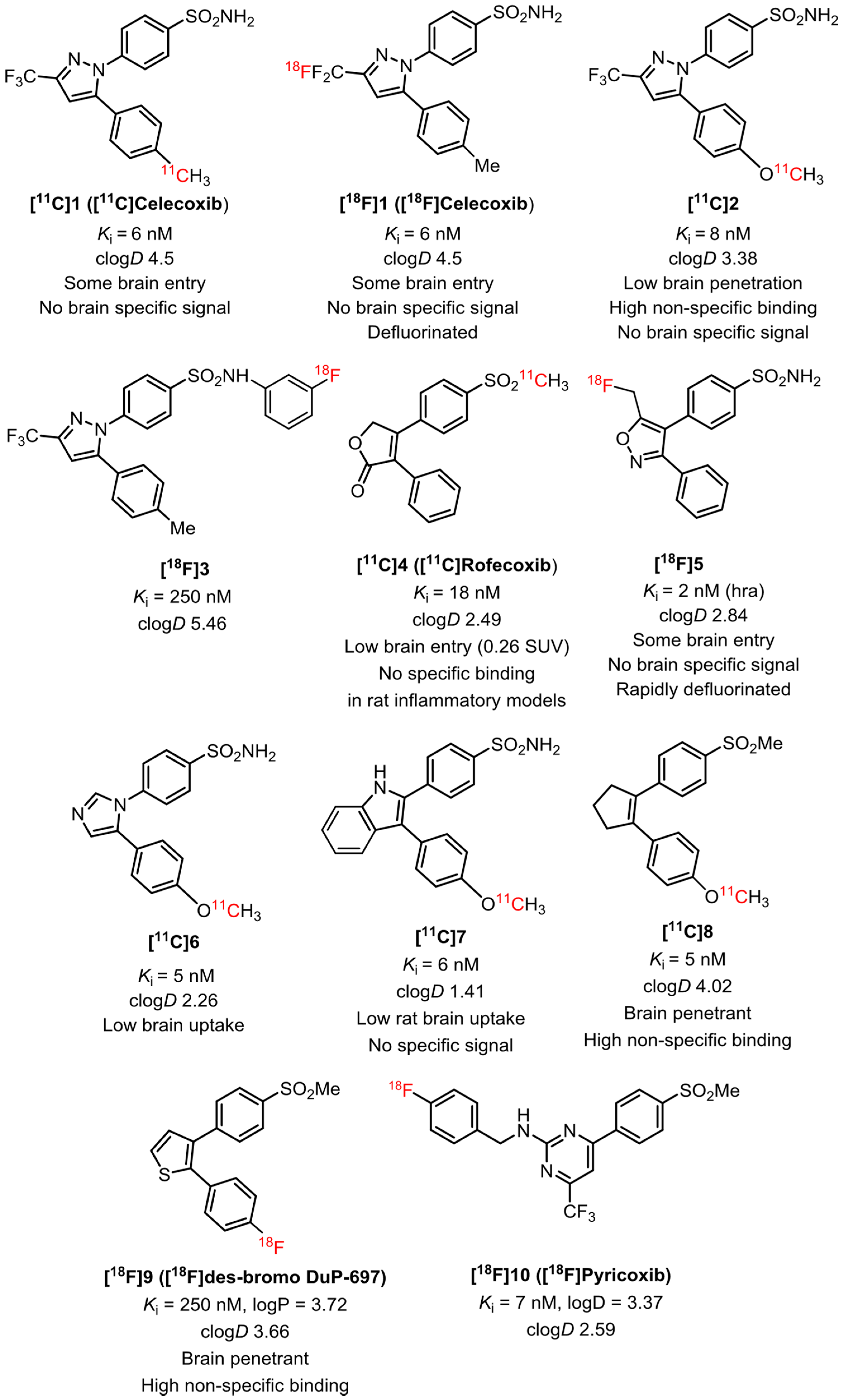
2.1. Target Inhibitor Selection for Radioligand Development
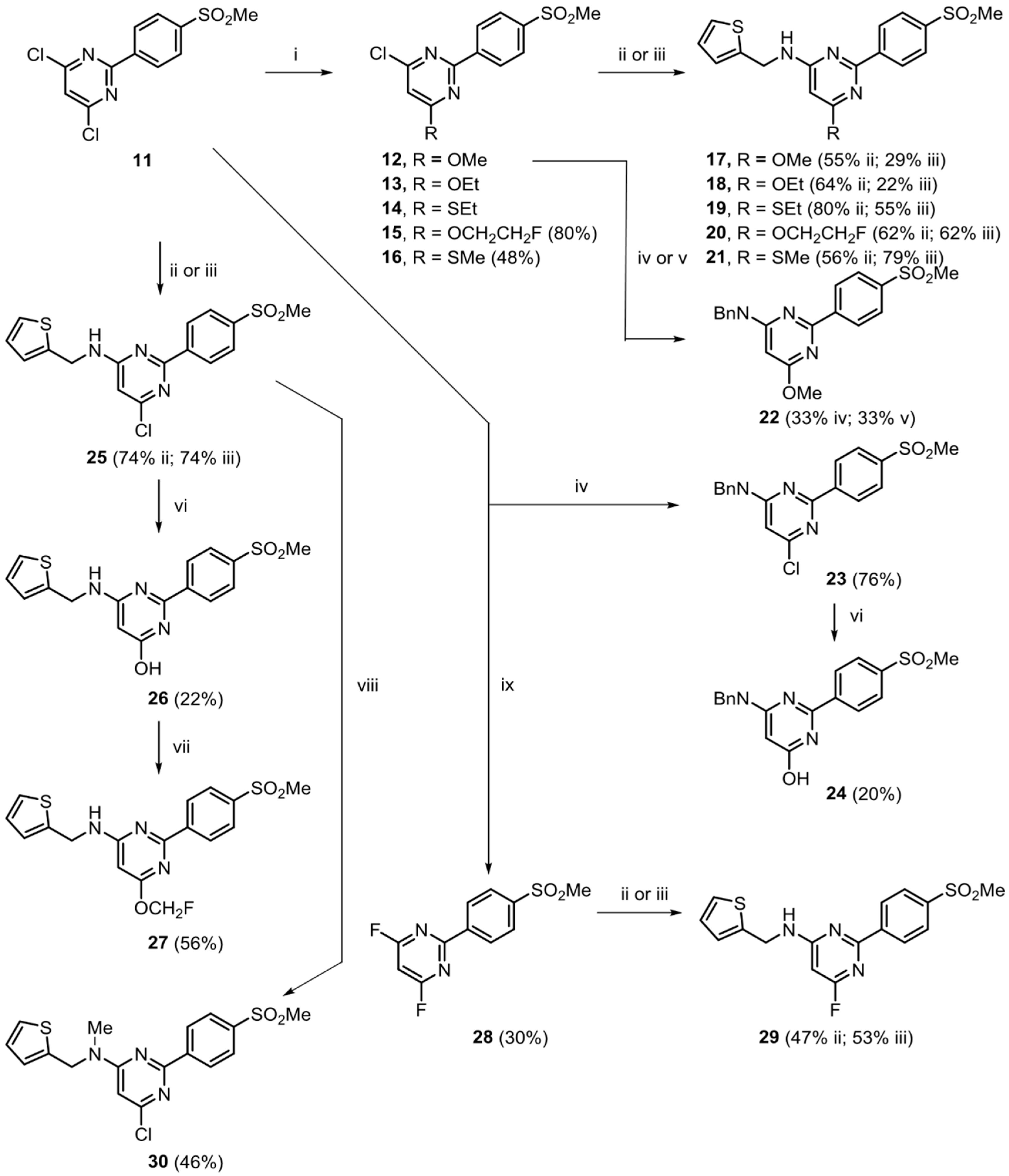
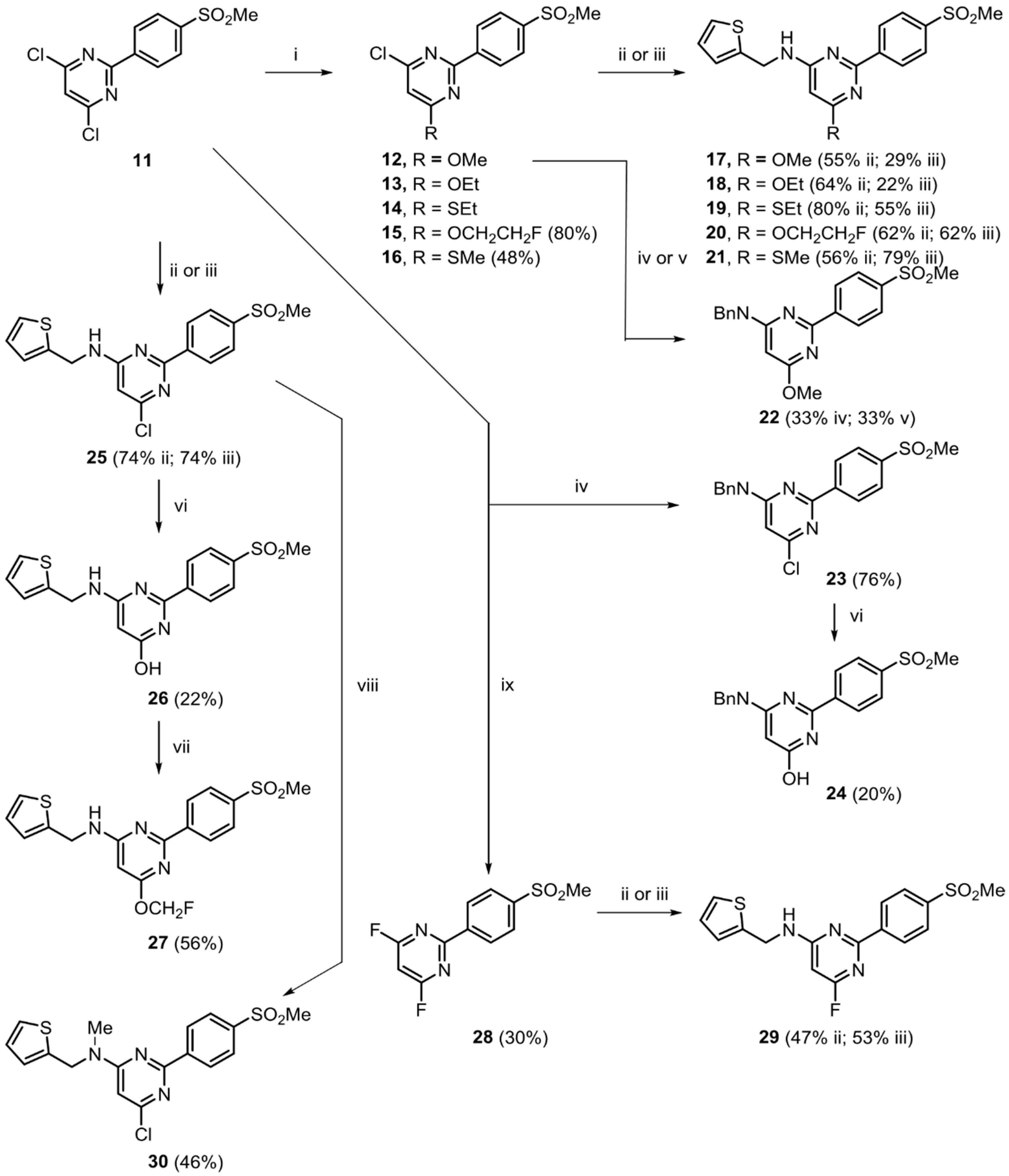
2.2. Chemistry
2.3. COX-1 and COX-2 Inhibition Assays
2.4. Pharmacological Screening
2.5. Selection of Candidates for Radiolabeling
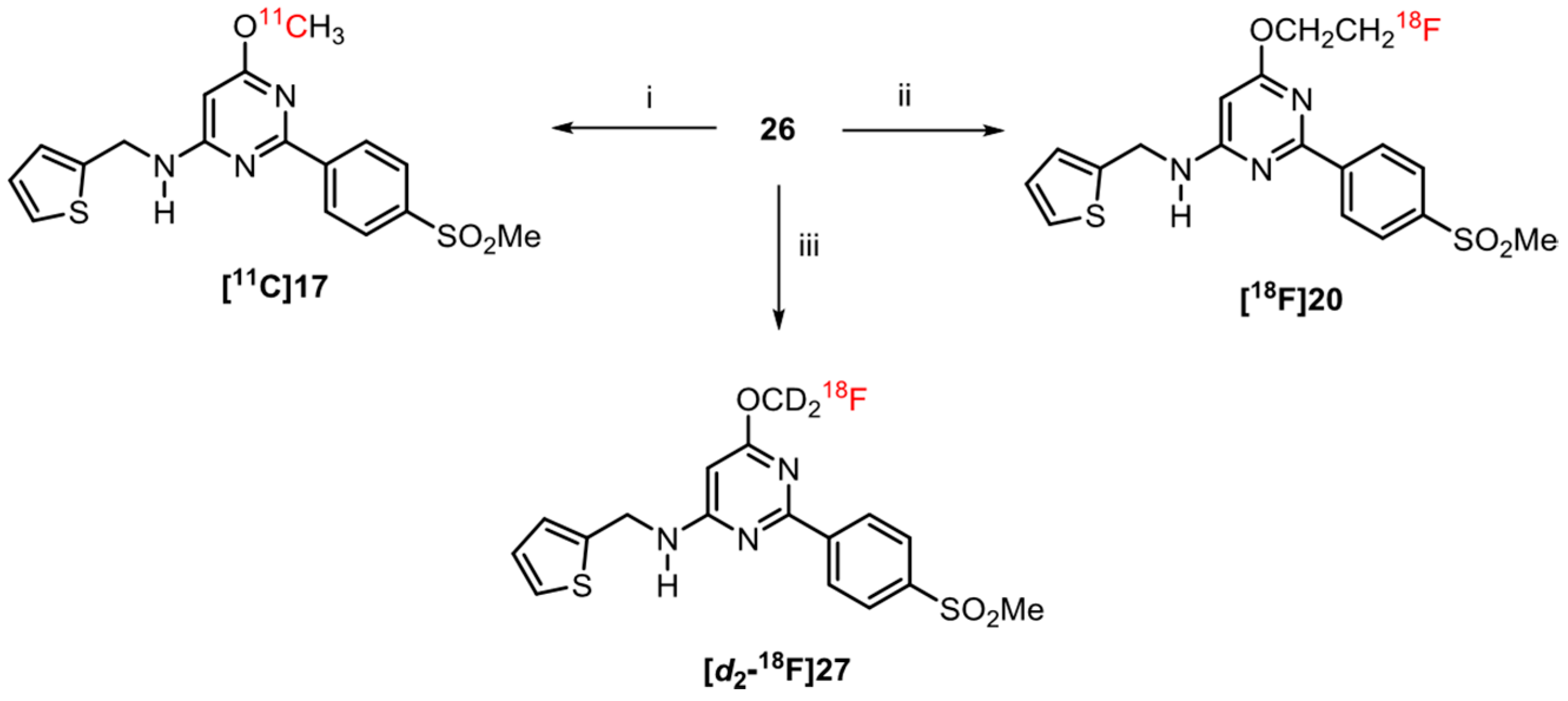
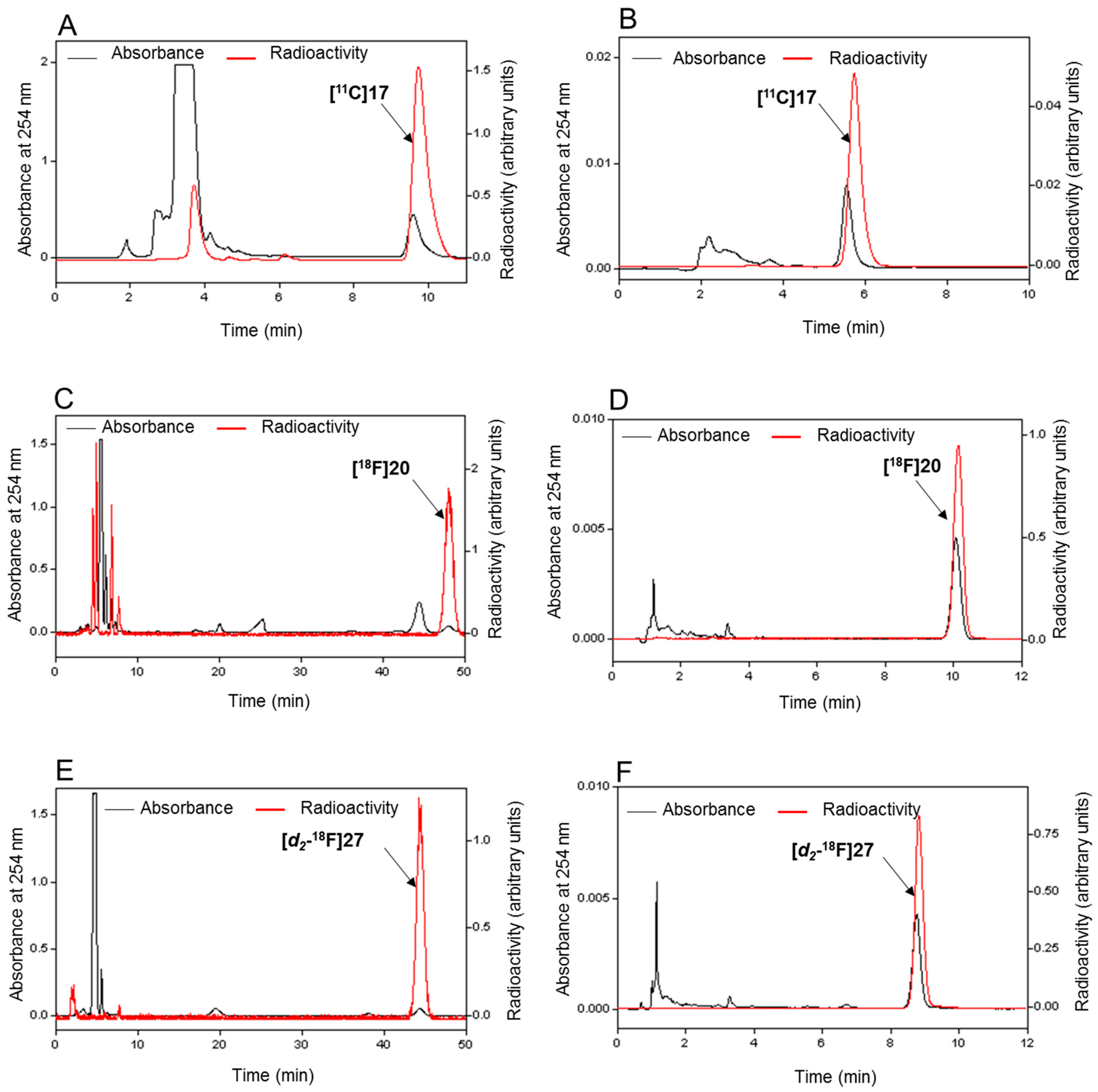
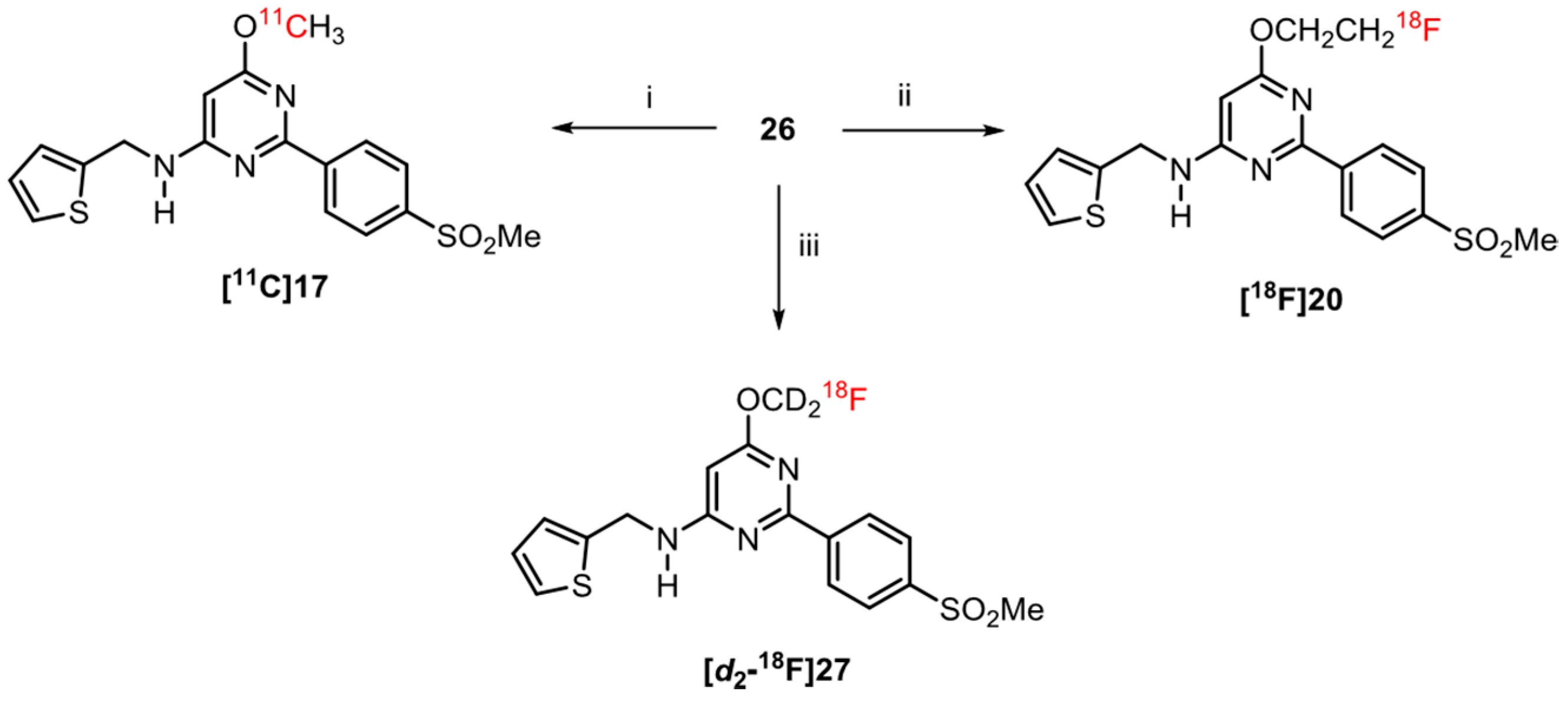
2.6. Radiochemistry
2.7. Calculated and Measured LogD for Prepared Radioligands
3. Experimental
3.1. General
3.2. Synthesis of Target Compounds and Precursors for Radiolabeling
3.2.1. Ether Derivatives of 4,6-Dichloro-2-(4-methylsulfonylphenyl)pyrimidine
3.2.2. Conversion of 4-Halo-2-(4-(methylsulfonyl)phenyl)-pyrimidine Derivatives into 4-Alkylamino Derivatives
3.3. COX-1 and COX-2 Inhibition Assays
3.4. Pharmacological Screening
3.5. Radiochemistry
3.5.1. Radiochemistry Materials and Methods
3.5.2. Production of NCA [11C]Carbon Dioxide
3.5.3. Production of [11C]Iodomethane
3.5.4. Radiosynthesis of [11C]17
3.5.5. Production of NCA [18F]Fluoride Ion
3.5.6. Radiosynthesis of [18F]20
3.5.7. Radiosynthesis of [d2-18F]27
3.6. LogD Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; Horrocks, L.A.; Farooqui, A.A. Cyclooxygenases, lipoxygenase, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Rev. 2006, 52, 201–243. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Kim, B.S.; Im, H.-I. Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int. Neurourol. J. 2016, 20 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Eberstål, S.; Sandén, E.; Fritzell, S.; Darabi, A.; Visse, E.; Siesjö, P. Intratumor COX-2 inhibition enhances GM-CSF immunotherapy against established mouse GL261 brain tumor. Int. J. Cancer 2014, 134, 2748–2753. [Google Scholar] [CrossRef] [PubMed]
- Zweers, M.C.; de Boer, T.N.; van Roon, J.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Mastbergen, S.C. Celecoxib: Considerations regarding its potential disease-modifying properties in osteoarthritis. Arthritis Res. Ther. 2011, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.; O’Banion, M.K. Cyclooxygenases in the central nervous system: Implications for treatment of neurological disorders. Curr. Pharm. Des. 2000, 6, 1755–1776. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; Veerhuis, R.; Janssen, I.; van Elk, E.-J.; Rozemuller, A.J.M.; Eikelenboom, P. The role of cyclooxygenase-1 and -2 activity in prostaglandin E2 secretion by culture human adult microglia: Implications for Alzheimer’s disease. Brain Res. 2002, 951, 218–226. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Garavito, M.R. The cyclooxygenase-2 structure: New drugs for an old target? Nat. Struct. Biol. 1996, 3, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Bakhle, Y.S. Structure of COX-1 and COX-2 enzyme and their interaction with inhibitors. Drugs Today 1999, 35, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Plount Price, M.L.; Jorgensen, W. Analysis of binding inhibitory potencies of celecoxib analogues with COX-1 and COX-2 from combined docking and Monte Carlo simulation and insight into the COX-2/COX-1 selectivity. J. Am. Chem. Soc. 2000, 122, 9455–9466. [Google Scholar] [CrossRef]
- Habeb, A.G.; Rao, P.N.R.; Knaus, E.E.W. Design and syntheses of diarylisoxazoles: Novel inhibitors of cyclooxygenase-2 (COX-2) with analgesic antiinflammatory activity. Drug Dev. Res. 2000, 51, 273–286. [Google Scholar] [CrossRef]
- Penning, T.; Talley, J.J.; Bertenshaw, S.; Carter, J.; Collins, P.; Docter, S.; Graneto, M.; Lee, L.; Malecha, J.; Miyashiro, J.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Desmond, R.; Dolling, U.; Marcune, B.; Tillyer, R.; Tschaen, D. Process for making phenylheterocycles useful as COX-2 inhibitors. World Patent WO96/08482, 21 March 1996. [Google Scholar]
- Talley, J.J.; Brown, D.L.; Carter, J.S.; Graneto, M.J.; Koboldt, C.M.; Masferrer, J.L.; Perkins, W.E.; Rogers, R.S.; Shaffer, A.F.; Zhang, Y.Y.; et al. 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, Valdecoxib: A potent and selective inhibitor of COX-2. J. Med. Chem. 2000, 43, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Prabhakara, J.; Majo, V.J.; Simpson, N.R.; Van Heertum, R.L.; Mann, J.J.; Kumar, J.S.D. Synthesis of [11C]celecoxib: A potential PET probe for imaging COX-2 expression. J. Label. Comp. Radiopharm. 2005, 48, 887–895. [Google Scholar] [CrossRef]
- Ji, B.; Kumata, K.; Onoe, H.; Kaneko, H.; Zhang, M.R.; Seki, C.; Ono, M.; Shukuri, M.; Tokunaga, M.; Minamihisamatsu, T.; et al. Assessment of radioligands for PET imaging of cyclooxygenase-2 in an ischemic neuronal injury model. Brain Res. 2013, 1533, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; Van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of [18F]-4-[5-(4-methylphenyl)-3(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorg. Med. Chem. Lett. 2007, 15, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, Y.; Kawamura, K.; Wang, W.-F.; Ishiwata, K.; Yamamota, F.; Kuwano, T.; Ono, M.; Maeda, M. Radiosynthesis and in vivo evaluation of 11C-labeled 1,5-diarylpyrazole derivatives for mapping cyclooxygenases. Ann. Nucl. Med. 2005, 19, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Tietz, O.; Bhardwaj, A.; Marshall, A.; Way, J.; Wuest, M.; Wuest, F. Design, synthesis, and evaluation of an 18F-labeled radiotracer based on celecoxib–NBD for positron emission tomography (PET) imaging of cyclooxygenase-2 (COX-2). ChemMedChem 2015, 10, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.F.J.; Doorduin, J.; Dierckx, R.A.; van Waarde, A. Evaluation of [11C]rofecoxib as PET tracer for cyclooxygenase 2 overexpression in rat models of inflammation. Nucl. Med. Biol. 2008, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Comley, R.A.; Passchier, J.; Willemsen, A.; Wall, A.; Bergstrom, M.; Langstrom, B.; Pruim, J.; Wishart, M.; Rabiner, E.; Gunn, R.N.; et al. Uptake and regional distribution of [11C]rofecoxib in human brain. NeuroImage 2010, 52, S135–S136. [Google Scholar] [CrossRef]
- Toyokuni, T.; Kumar, J.S.D.; Walsh, J.C.; Shapiro, A.; Talley, J.J.; Phelps, M.E.; Herschman, H.R.; Barrio, J.R.; Satymurthy, N. Synthesis of 4-(5-[18F]fluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide, a new [18F]fluorinated analogue of valdecoxib, as a potential radiotracer for imaging cyclooxygenase-2 with positron emission tomography. Bioorg. Med. Chem. Lett. 2005, 15, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fujisaka, Y.; Kawamura, K.; Ishiwata, K.; Qinggeletu, F.Y.; Mukai, T.; Maeda, M. Radiosynthesis and evaluation of 11C-labeled diaryl-substituted imidazole and indole derivatives for mapping cyclooxygenase-2. Biol. Pharm. Bull. 2006, 29, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Kniess, T.; Bergmann, R.; Pietszsch, J. Synthesis and evaluation in vitro and in vivo of a 11C-labeled cyclooxygenase-2 (COX-2) inhibitor. Bioorg. Med. Chem. 2008, 16, 7662–7670. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.F.J.; van Waarde, A.; Buursma, A.R.; Vaalburg, W. Synthesis and in vivo evaluation of 18F-desbromo-DuP-697 as a PET tracer for cyclooxygenase-2 expression. J. Nucl. Med. 2003, 44, 1700–1706. [Google Scholar] [PubMed]
- Leblanc, Y.; Guathier, J.; Ethier, D.; Guay, J.; Mancinin, J.; Reindeau Tagari, P.; Vickers, P.; Wong, E.; Prasit, P. Synthesis and biological evaluation of 2,3-diarylthiophenes as selective COX-2 and COX-1 inhibitors. Bioorg. Med. Chem. Lett. 1995, 5, 2123–2128. [Google Scholar] [CrossRef]
- Tietz, O.; Wuest, M.; Marshall, A.; Gubrecht, D.; Hamman, I.; Wang, M.; Bergman, C.; Way, J.D.; Wuest, F. PET imaging of cyclooxygenase-2 (COX-2) in a preclinical colorectal cancer model. EJNMMI Res. 2016, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Kniess, T.; Pietzsch, J. Radiolabeled COX-2 inhibitors for non-invasive visualization of COX-2 expression and activity—A critical update. Molecules 2013, 18, 6311–6355. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET Radiotracers: Crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.F. Imaging of cyclooxygenase (COX-2) expression; potential uses in diagnosis and drug evaluation. Curr. Pharm. Des. 2006, 12, 3847–3856. [Google Scholar] [CrossRef] [PubMed]
- Orjales, A.; Mosquera, R.; Lopez, B.; Olivera, R.; Labeaga, L.; Nuῆez, M.T. Novel 2-(4methylsulfonylphenyl)pyrimidine derivates as highly potent and specific COX-2 inhibitors. Bioorg. Med. Chem. 2008, 16, 2183–2199. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C. Preparation of an o-substituted benzamidine by the Pinner method. A literature clarification. Org. Prep. Proced. Int. 1990, 22, 643–645. [Google Scholar] [CrossRef]
- Pews, G.R.; Puckett, W.E. 1,1,1-Trichloro-3-[5-(2,4,6-trifluoropyrimidyl)]-3,4-epoxybutane. J. Fluorine Chem. 1989, 42, 179–186. [Google Scholar] [CrossRef]
- Cheung, C.W.; Buchwald, S.L. Palladium-catalyzed hydroxylation of aryl and heteroaryl halides enabled by the use of a palladacycle precatalyst. J. Org. Chem. 2014, 79, 5351–5358. [Google Scholar] [CrossRef] [PubMed]
- Huff, R.G.; Bayram, E.; Tan, H.; Knutson, S.T.; Knaggs, M.H.; Richon, A.B.; Santiago, P., II; Fetrow, J.S. Chemical and structural diversity in cyclooxygenase protein active sites. Chem. Diversity 2005, 2, 1533–1552. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.; Ulin, J.; Dahlström, K.; Jensen, M. Synthesis of [11C]iodomethane by iodination of [11C]methane. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Savolainen, H.; Cantore, M.; Colabufo, N.A.; Elsinga, P.H.; Windhorst, A.D.; Luurtsema, G. Synthesis and preclinical evaluation of three novel fluorine-18 labeled radiopharmaceuticals for P-glycoprotein PET imaging at the blood–brain barrier. Mol. Pharmacol. 2015, 12, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Chin, F.T.; Morse, C.L.; Shetty, H.U.; Pike, V.W. Automated radiosynthesis of [18F]SPA-RQ for imaging human brain NK1 receptors with PET. J. Label. Comp. Radiopharm. 2006, 49, 17–31. [Google Scholar] [CrossRef]
- Kuchar, M.; Mamat, C. Methods to increase the metabolic stability of 18F-radiotracers. Molecules 2015, 20, 16186–16220. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, S.S.; Anderson, K.B.; Jenko, K.J.; Luckenbaugh, D.A.; Innis, R.B.; Pike, V.W. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human. J. Pharm. Sci. 2012, 101, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Assay Kits. Available online: https://www.caymanchem.com/app/template/Products.vm/active/kits (accessed on 11 January 2017).
- Singh, P.; Shrestra, S.; Cortes-Salva, M.-Y.; Jenko, K.J.; Zoghbi, S.S.; Morse, C.L.; Innis, R.B.; Pike, V.W. 3-Substituted 1,5-diaryl-1H-1,2,4-triazoles as prospective PET radioligands for imaging brain COX-1 in monkey. Part 1: Synthesis and pharmacology. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.-M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry—Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [PubMed]
- Christman, D.R.; Finn, R.D.; Karlstrøm, K.; Wolf, A.P. The production of ultra high specific activity 11Clabeled hydrogen cyanide, carbon dioxide, carbon monoxide and methane via the 14N(p,α)11C reaction. Int. J. Appl. Radiat. Isot. 1975, 26, 435–442. [Google Scholar] [CrossRef]
- Briard, E.; Zoghbi, S.S.; Imaizumi, M.; Gourley, J.P.; Shetty, H.U.; Hong, J.; Cropley, V.; Fujita, M.; Innis, R.B.; Pike, V.W. Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J. Med. Chem. 2008, 51, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.; Shrestha, S.; Eldridge, M.; Cortes, M.; Yu, Z.-X.; Lehmann, M.; Kim, M.-J.; Singh, P.; Fredericks, M.; Tye, G.; et al. Novel PET radioligands show that COX-2, but not COX-1, is induced by neuroinflammation in rhesus macaque. Biol. Psychiatry 2018, 83 (Suppl. 1), S160. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are all available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
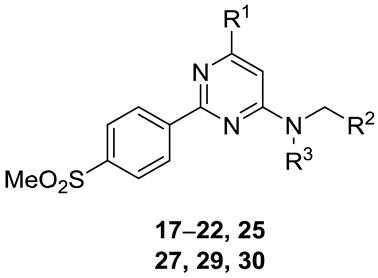
| # | R1 | R2 | R3 | clogD | tPSA (Å2) | HBD | Hetero-Atoms | M.Wt. (Da) | HWB a IC50 (nM) | Selectivity vs. COX-1 b |
| 17 | OMe | 2-Thi c | H | 2.80 | 80.1 | 1 | 8 | 375 | 0.4 | >22,004 |
| 18 | OEt | 2-Thi | H | 2.88 | 80.1 | 1 | 8 | 389 | 0.3 | >33,333 |
| 19 | SEt | 2-Thi | H | 3.47 | 70.9 | 1 | 8 | 405 | 1.2 | >8403 |
| 20 | O(CH2)2F | 2-Thi | H | 3.09 | 80.1 | 1 | 9 | 407 | ||
| 21 | SMe | 2-Thi | H | 3.34 | 70.9 | 1 | 8 | 391 | ||
| 22 | OMe | Ph | H | 2.71 | 80.1 | 1 | 7 | 369 | 5.1 | >19,646 |
| 25 | Cl | 2-Thi | H | 2.74 | 70.9 | 1 | 8 | 379 | 1.2 | >81,300 |
| 27 | OCH2F | 2-Thi | H | 2.72 | 80.2 | 1 | 9 | 393 | ||
| 29 | F | 2-Thi | H | 2.91 | 70.9 | 1 | 8 | 363 | ||
| 30 | Cl | 2-Thi | Me | 3.47 | 62.1 | 0 | 8 | 394 | ||
| Optimal parameter space | 2–3 | <90 | low | <9 | <500 | low nM | >100 | |||
 | |||||||
|---|---|---|---|---|---|---|---|
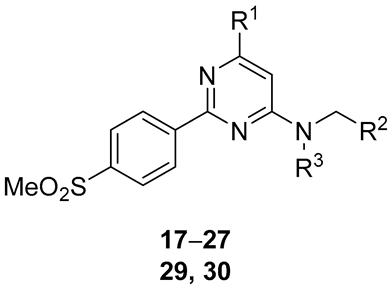
| # | R1 | R2 | R3 | IC50 (nM) a | |||
| COX-1 | COX-2 | ||||||
| Monkey WB | Human WB | Monkey WB | Human WB | ||||
| 17 | OMe | 2-Thi b | H | >1000 | >1000 >8800 c | 2.58 ± 0.23 | 2.89 ± 0.15 0.4 c |
| 18 | OEt | 2-Thi | H | >10,000 d >105 c | 3 d 0.3 c | ||
| 19 | SEt | 2-Thi | H | >1000 | >1000 >10,000 c | 52.3 ± 0.01 | 3.52 ± 0.43 1.2 c |
| 20 | O(CH2)2F | 2-Thi | H | 1.28 e | 1.9 e | ||
| 21 | SMe | 2-Thi | H | >1000 | >1000 | 4.83 ± 2.31 | 2.65 ± 0.45 |
| 22 | OMe | Ph | H | >1000 | >1000 >105 c | 14.6 ± 10.1 | 7.35 ± 1.35 5.1 c |
| 23 | Cl | Ph | H | >105 c | 10.9c | ||
| 24 | OH | Ph | H | >1000 | >1000 >105 c | > 1000 d | >1000 d 49.3 c |
| 25 | Cl | 2-Thi | H | >10,000 d >105 c | 4 d 1.2 c | ||
| 26 | OH | 2-Thi | H | >105 c | 10 d | 79.4 c | |
| 27 | OCH2F | 2-Thi | H | >1000 | >1000 | 2.26 ± 0.45 | 2.26 ± 0.63 |
| 29 | F | 2-Thi | H | 2.5 d | |||
| 30 | Cl | 2-Thi | Me | > 108 d | >1000 | ||
| Radioligand | cLogD | LogD (n = 6) |
|---|---|---|
| [11C]17 | 2.80 | 3.74 ± 0.02 |
| [18F]20 | 3.09 | 2.94 ± 0.32 |
| [d2-18F]27 | 2.72 | 3.70 ± 0.12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Salva, M.Y.; Shrestha, S.; Singh, P.; Morse, C.L.; Jenko, K.J.; Montero Santamaria, J.A.; Zoghbi, S.S.; Innis, R.B.; Pike, V.W. 2-(4-Methylsulfonylphenyl)pyrimidines as Prospective Radioligands for Imaging Cyclooxygenase-2 with PET—Synthesis, Triage, and Radiolabeling. Molecules 2018, 23, 2850. https://doi.org/10.3390/molecules23112850
Cortes-Salva MY, Shrestha S, Singh P, Morse CL, Jenko KJ, Montero Santamaria JA, Zoghbi SS, Innis RB, Pike VW. 2-(4-Methylsulfonylphenyl)pyrimidines as Prospective Radioligands for Imaging Cyclooxygenase-2 with PET—Synthesis, Triage, and Radiolabeling. Molecules. 2018; 23(11):2850. https://doi.org/10.3390/molecules23112850
Chicago/Turabian StyleCortes-Salva, Michelle Y., Stal Shrestha, Prachi Singh, Cheryl L. Morse, Kimberly J. Jenko, Jose A. Montero Santamaria, Sami S. Zoghbi, Robert B. Innis, and Victor W. Pike. 2018. "2-(4-Methylsulfonylphenyl)pyrimidines as Prospective Radioligands for Imaging Cyclooxygenase-2 with PET—Synthesis, Triage, and Radiolabeling" Molecules 23, no. 11: 2850. https://doi.org/10.3390/molecules23112850
APA StyleCortes-Salva, M. Y., Shrestha, S., Singh, P., Morse, C. L., Jenko, K. J., Montero Santamaria, J. A., Zoghbi, S. S., Innis, R. B., & Pike, V. W. (2018). 2-(4-Methylsulfonylphenyl)pyrimidines as Prospective Radioligands for Imaging Cyclooxygenase-2 with PET—Synthesis, Triage, and Radiolabeling. Molecules, 23(11), 2850. https://doi.org/10.3390/molecules23112850