E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
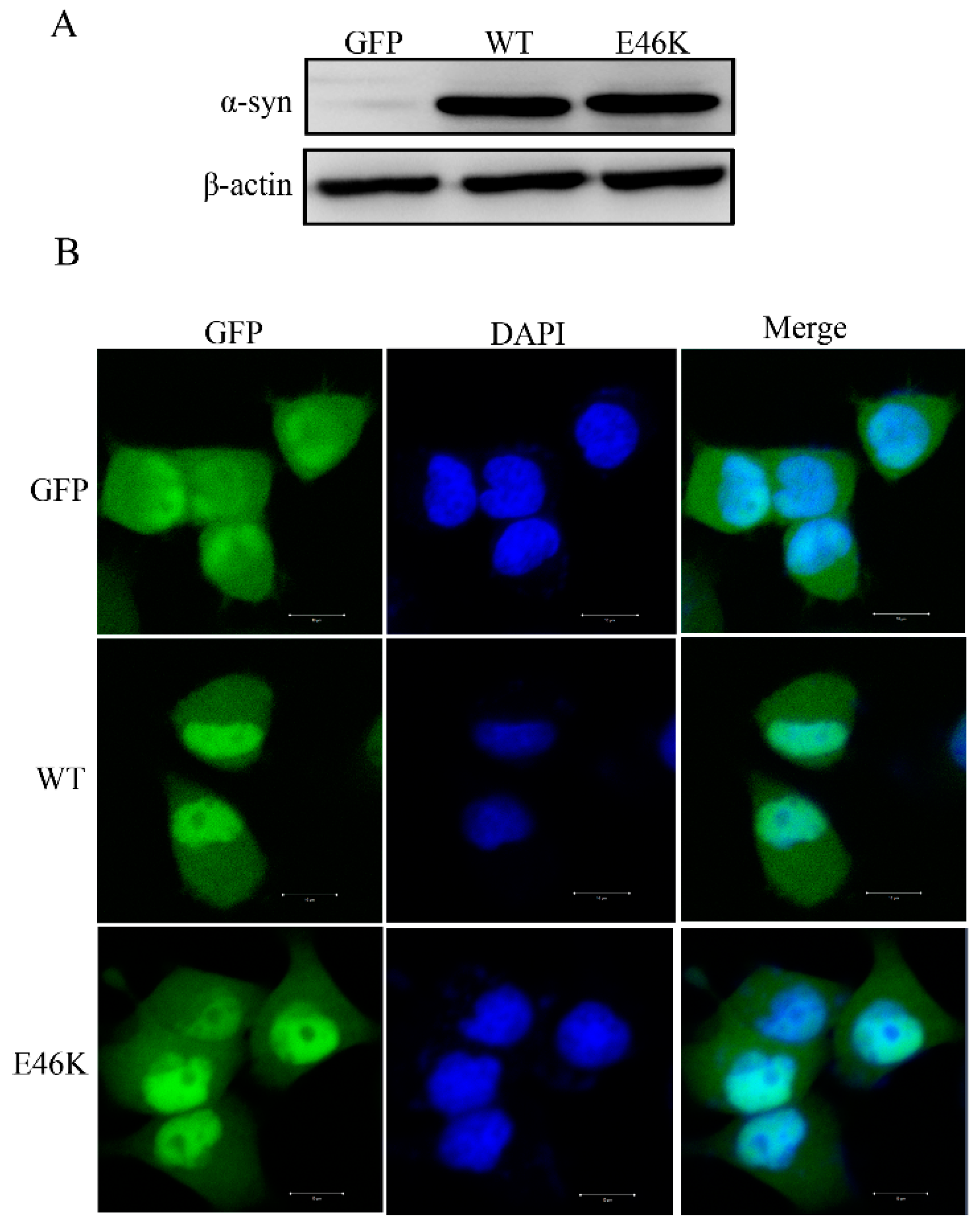
2.1. Establishment of PC12 Cell Stably Overexpressing Human WT and E46K Mutant α-syn
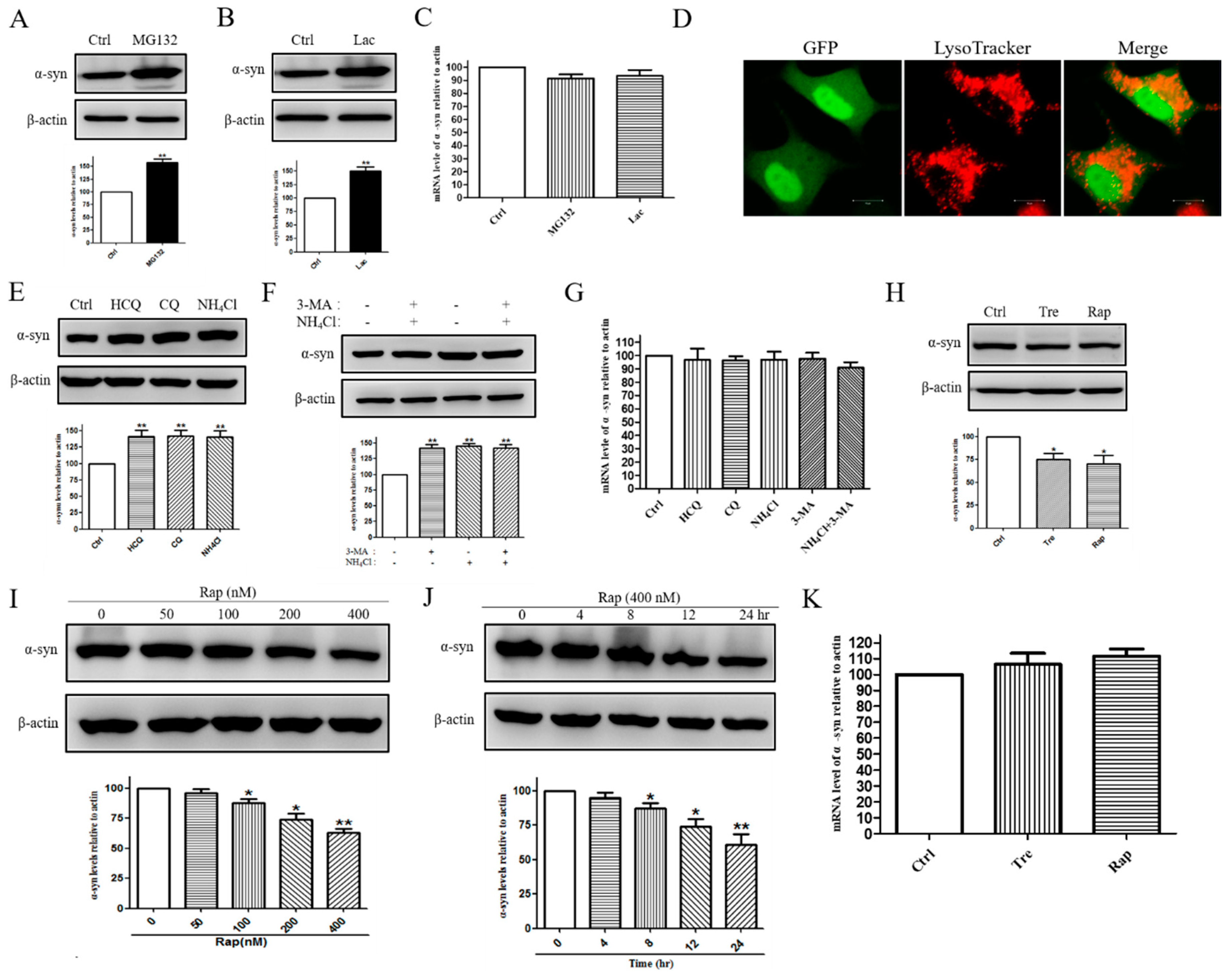
2.2. E46K Mutant α-syn Was Degraded by Proteasome and Macroautophagy Pathway
2.3. WT α-syn Was Degraded by Proteasome and CMA Pathway
2.4. E46K Mutant α-syn Turned over More Slowly Compared with WT α-syn
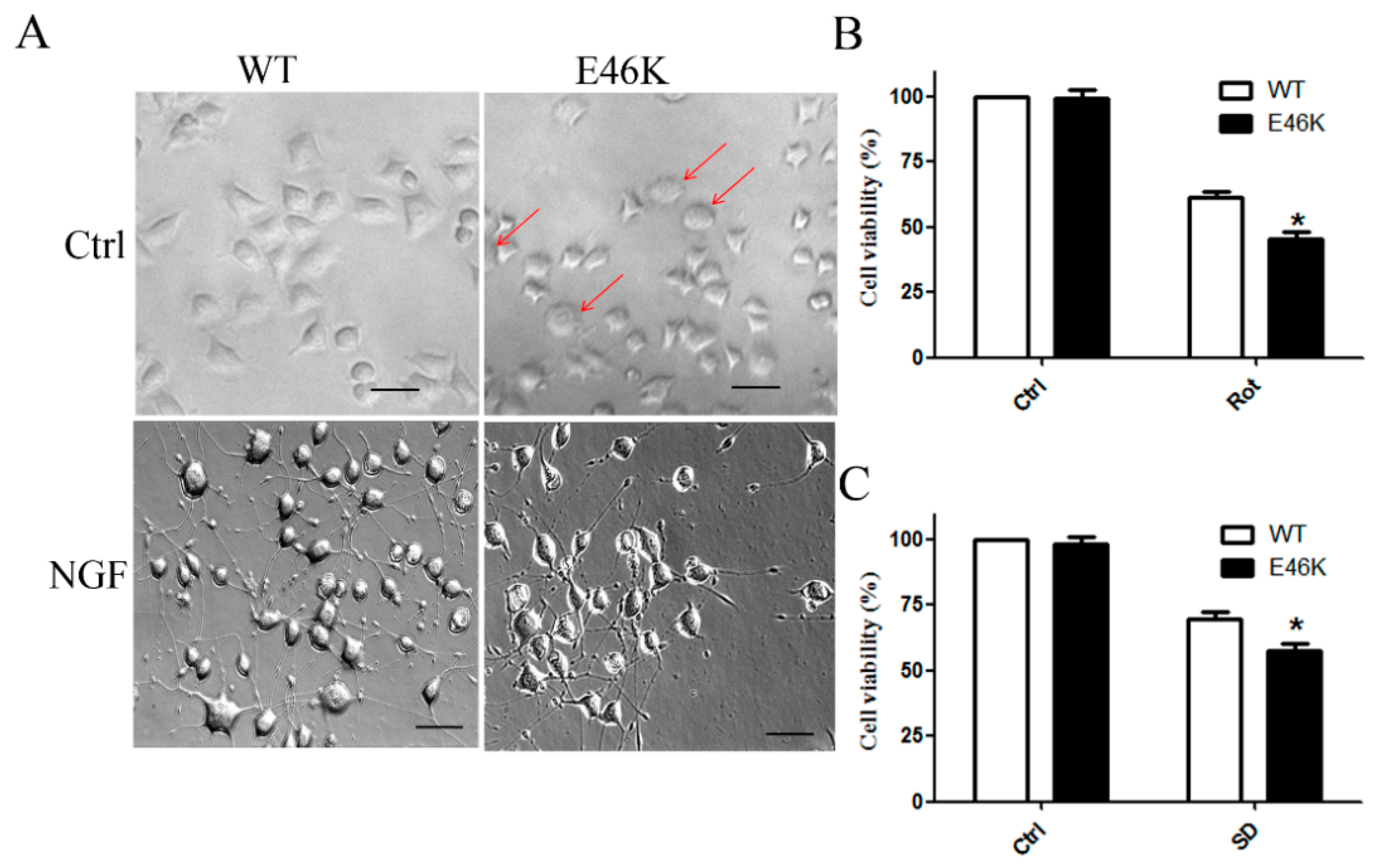
2.5. Cells Overexpressing E46K Mutant α-syn Exhibited Enhanced Vulnerability to Apoptosis Insults
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of PC12 Cells Stably Overexpressing Human WT and E46K Mutant α-Syn
4.3. Drug Treatment
4.4. Western Blot Analysis
4.5. Immunofluorescence Staining
4.6. Staining for Lysosomes
4.7. RT-PCR
4.8. Co-Immunoprecipitation Assay
4.9. Morphological Observation and MTT Assay
4.10. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. α-Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategis. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.; Stefanis, L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Varshavsky, A. The Ubiquitin System, Autophagy, and regulated protein degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. Regulated protein turnover: Snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2017, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. Mammalian Autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Inagaki, F. Mechanisms of autophagy. Ann. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Ktistakis, N.T.; Tooze, S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrelis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Burre, P.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Ozansoy, M.; Başak, A.N. The central theme of Parkinson’s disease: α-synuclein. Mol. Neurobiol. 2013, 47, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Tai, H.C.; Schuman, E.W. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Pickart, C.M. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004, 14, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The autophagy-lysosomal pathway in neurodegeneration: A TFEB perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Geghman, K.D.; Tapias, V.; Sew, T.; Dail, M.K.; Li, C.; Greenamyre, J.T. Expression of E46K-mutant α-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 2013, 240, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Martinze-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanisms insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, X.; Pan, L.; Zhou, P. Trehalose Inhibits A53T Mutant α-Synuclein Overexpression and Neurotoxicity in Transduced PC12 Cells. Molecules 2017, 22, 1293. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jin, J.; Yang, B.; Zhang, W.; Hu, J.; Zhang, Y.; Chen, N. Overexpressed alpha-synuclein regulated the nuclear factor-kappaB signal pathway. Cell. Mol. Neurobiol. 2008, 28, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Yuan, Y.H.; Song, L.K.; Han, N.; Chen, N.H. Over-expression of α-synuclein 98 triggers intracellular oxidative stress and enhances susceptibility to rotenone. Neurosci. Lett. 2011, 491, 148–152. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: All samples are not available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.-q.; Yuan, Y.-h.; Chu, S.-f.; Li, G.-h.; Chen, N.-h. E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules 2018, 23, 2839. https://doi.org/10.3390/molecules23112839
Yan J-q, Yuan Y-h, Chu S-f, Li G-h, Chen N-h. E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules. 2018; 23(11):2839. https://doi.org/10.3390/molecules23112839
Chicago/Turabian StyleYan, Jia-qing, Yu-he Yuan, Shi-feng Chu, Guo-hui Li, and Nai-hong Chen. 2018. "E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway" Molecules 23, no. 11: 2839. https://doi.org/10.3390/molecules23112839
APA StyleYan, J.-q., Yuan, Y.-h., Chu, S.-f., Li, G.-h., & Chen, N.-h. (2018). E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules, 23(11), 2839. https://doi.org/10.3390/molecules23112839