Dynamics-Derived Insights into Complex Formation between the CXCL8 Monomer and CXCR1 N-Terminal Domain: An NMR Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
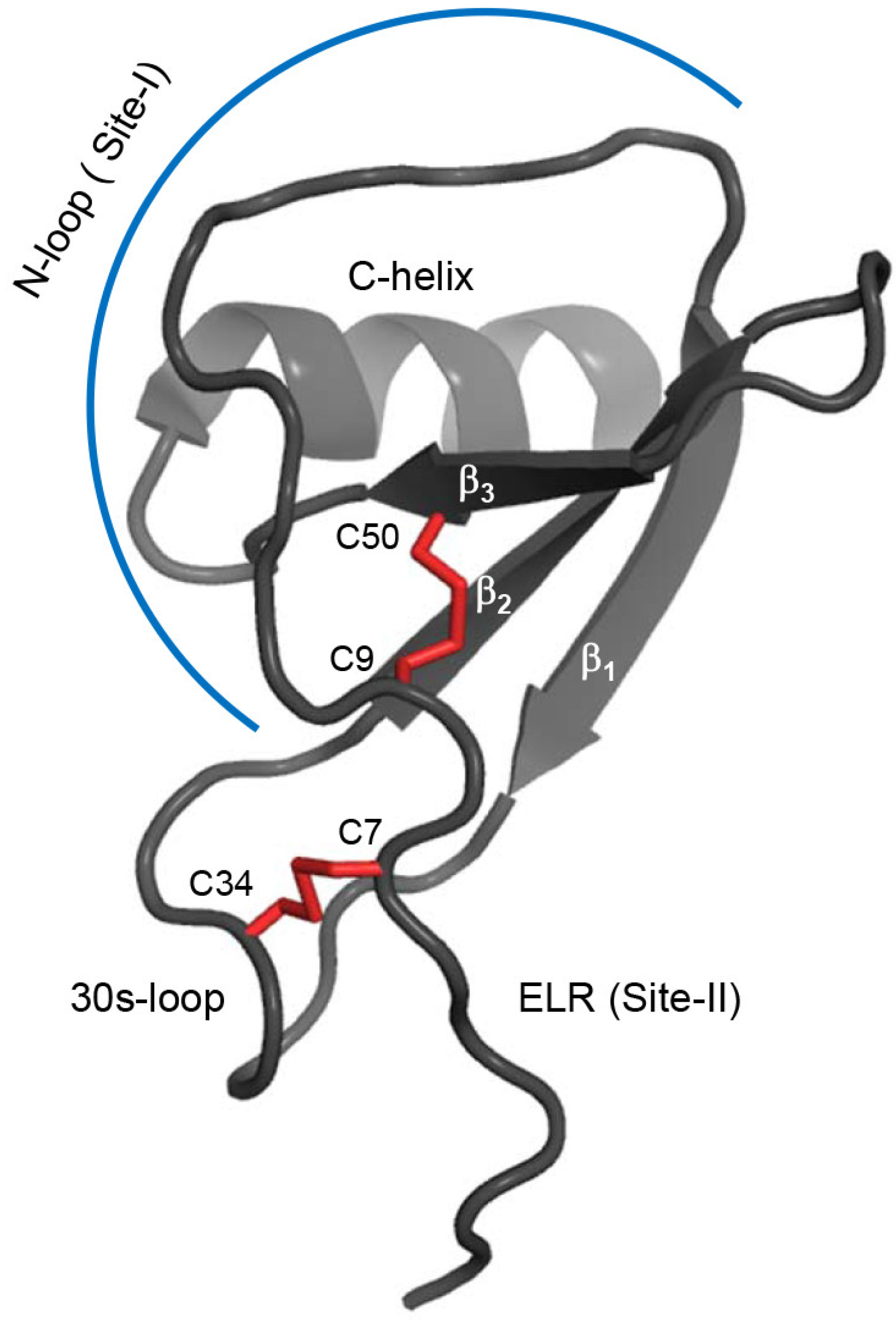
2.1. Design of the CXCL8 Monomer and CXCR1 N-Terminal Domain
2.2. Line Shape Analyses for the 15N-CXCL8–CXCR1 N-Domain Interaction
2.3. Relaxation Measurements and Dynamics of the CXCL8 Monomer
2.4. Relaxation Measurements and Dynamics of the CXCL8 Monomer in the Bound State
2.5. Relaxation Measurements and Dynamics of the CXCR1 N-Domain
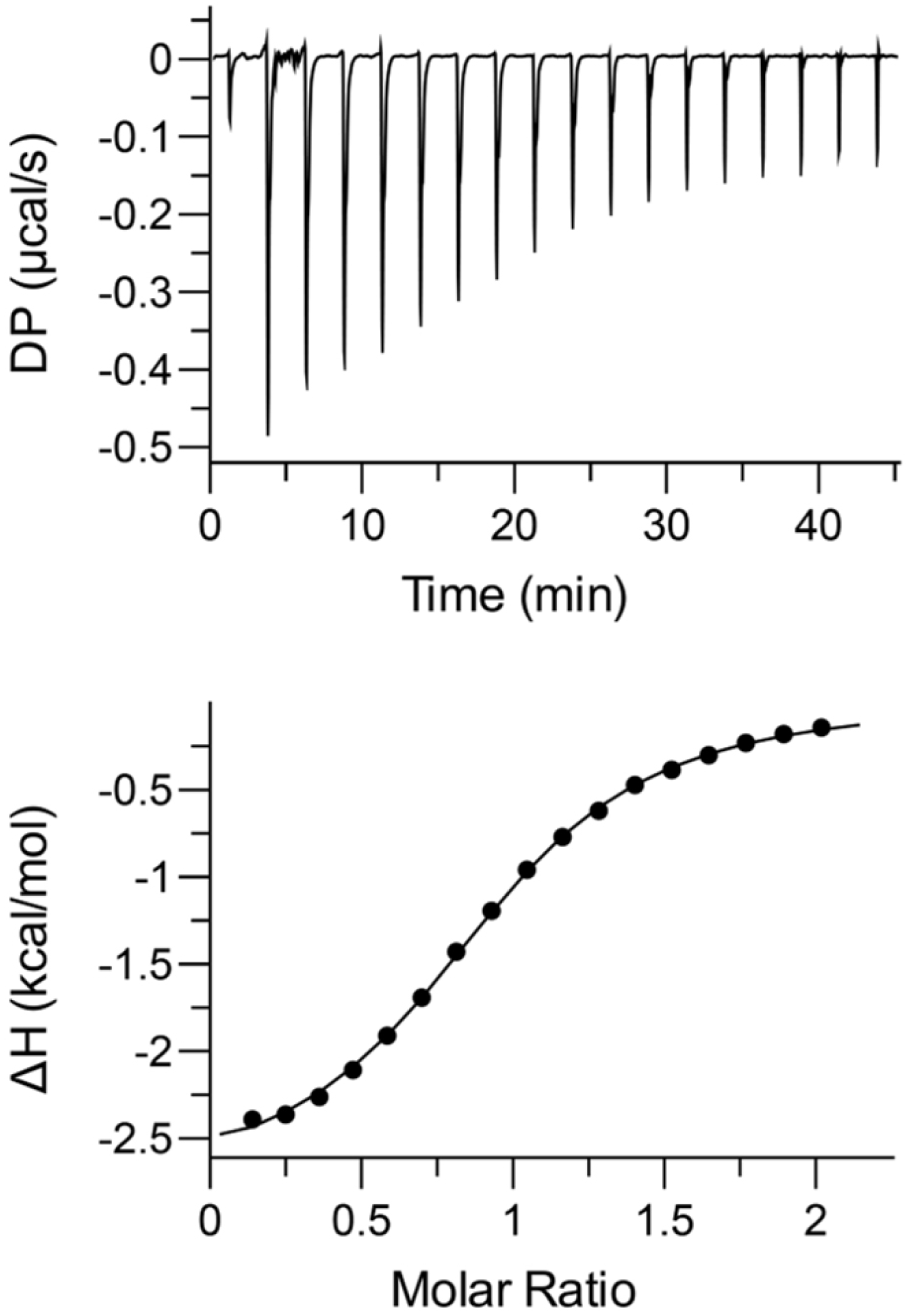
2.6. Thermodynamics of Binding
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Nuclear Magnetic Resonance Experiments
4.3. Nuclear Magnetic Resonance Line Shape Analysis
4.4. Relaxation Measurements
4.5. Isothermal Titration Calorimetry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Galliera, E.; Borroni, E.M.; Corsi, M.M.; Locati, M.; Mantovani, A. Chemokines and chemokine receptors: An overview. Front. Biosci. 2009, 14, 540–551. [Google Scholar] [CrossRef]
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009, 86, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Raghuwanshi, S.K.; Grant, D.J.; Jala, V.R.; Rajarathnam, K.; Richardson, R.M. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 2009, 183, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Das, S.T.; Rajagopalan, L.; Guerrero-Plata, A.; Sai, J.; Richmond, A.; Garofalo, R.P.; Rajarathnam, K. Monomeric and Dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS ONE 2010, 5, e11754. [Google Scholar] [CrossRef] [PubMed]
- Sawant, K.V.; Xu, R.; Cox, R.; Hawkins, H.; Sbrana, E.; Kolli, D.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1-mediated neutrophil trafficking in the lung: Role of CXCR2 activation. J. Innate Immun. 2015, 7, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Dewald, B.; Loetscher, M.; Moser, B.; Baggiolini, M. Structural Requirements for IL-8 Function; Identified by Design of Analogs and CXC Chemokine Hybrids. J. Biol. Chem. 1994, 269, 16075–16081. [Google Scholar] [PubMed]
- Joseph, P.R.; Sarmiento, J.M.; Mishra, A.K.; Das, S.T.; Garofalo, R.P.; Navarro, J.; Rajarathnam, K. Probing the Role of CXC Motif in Chemokine CXCL8 for High Affinity Binding and Activation of CXCR1 and CXCR2 Receptors. J. Biol. Chem. 2010, 285, 29262–29269. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.R.; Sawant, K.; Pedroza, M.; Isley, R.; Garofalo, R.; Richardson, R.; Rajarathnam, K. Identification of a conformational switch in Neutrophil-Activating Chemokines for G protein coupled-receptor activation. Biochem. J. 2013, 456, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Prado, G.N.; Fernando, H.; Clark-Lewis, I.; Navarro, J. Probing receptor binding activity of interleukin-8 dimer using a disulfide trap. Biochemistry 2006, 45, 7882–7888. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Clark-Lewis, I.; Sykes, B.D. 1H NMR Solution Structure of an Active Interleukin-8 Monomer. Biochemistry 1995, 34, 12893–12990. [Google Scholar] [CrossRef]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.T.; Weber, I.T.; St. Charles, R.; Xuan, J.C.; Appella, E.; Yamada, M.; Matsushima, K.; Edwards, B.F.; Clore, G.M.; Gronenborn, A.M. Crystal structure of interleukin 8: Symbiosis of NMR and crystallography. Proc. Natl. Acad. Sci. USA 1991, 88, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Das, B.B.; Casagrande, F.; Tian, Y.; Nothnagel, H.J.; Chu, M.; Kiefer, H.; Maier, K.; De Angelis, A.A.; Marassi, F.M.; et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 2012, 491, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Casagrande, F.; Das, B.B.; Albrecht, L.; Chu, M.; Opella, S.J. Local and global dynamics of the G protein-coupled receptor CXCR1. Biochemistry 2011, 50, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Kovermann, M.; Rogne, P.; Wolf-Watz, M. Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 2016, 49, e6. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011, 20, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.G. Chemical exchange in biomacromolecules: Past, present, and future. J. Magn. Reson. 2014, 241, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, B.L.; Gronenborn, A.M.; Clore, G.M. Analysis of the Backbone Dynamics of Interleukin-8 by 15N Measurements. J. Mol. Biol. 1993, 230, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, A.A.; Holliday, M.J.; Isern, N.G.; Zhang, F.; Camilloni, C.; Huynh, C.; Vendruscolo, M.; Armstrong, G.; Eisenmesser, E.Z. The dynamics of interleukin-8 and its interaction with human CXC receptor I peptide. Protein Sci. 2014, 23, 464–480. [Google Scholar] [CrossRef] [PubMed]
- Berkamp, S.; Park, S.H.; De Angelis, A.A.; Marassi, F.M.; Opella, S.J. Structure of monomeric Interleukin-8 and its interactions with the N-terminal Binding Site-I of CXCR1 by solution NMR spectroscopy. J. Biomol. NMR 2017, 69, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Clubb, R.T.; Omichinski, J.G.; Clore, G.M.; Gronenborn, A.M. Mapping the binding surface of interleukin-8 complexed with an N-terminal fragment of the type 1 human interleukin-8 receptor. FEBS Lett. 1994, 338, 93–97. [Google Scholar] [CrossRef]
- Barter, E.F.; Stone, M.J. Synergistic interactions between chemokine receptor elements in recognition of interleukin-8 by soluble receptor mimics. Biochemistry 2012, 51, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.R.; Rajarathnam, K. Solution NMR characterization of WT CXCL8 monomer and dimer binding to CXCR1 N-terminal domain. Protein Sci. 2015, 24, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Fernando, H.; Chin, C.; Rösgen, J.; Rajarathnam, K. Dimer Dissociation is Essential for Interleukin-8 (IL-8) Binding to CXCR1 Receptor. J. Biol. Chem. 2004, 279, 36175–36178. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, L.; Rajarathnam, K. Ligand Selectivity and Affinity of Chemokine Receptor CXCR1: Role of N-terminal Domain. J. Biol. Chem. 2004, 279, 30000–30008. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, L.; Rösgen, J.; Bolen, W.D.; Rajarathnam, K. Novel use of an osmolyte to dissect multiple thermodynamic linkages in a chemokine ligand-receptor system. Biochemistry 2005, 44, 12932–12939. [Google Scholar] [CrossRef] [PubMed]
- Fernando, H.; Nagle, G.; Rajarathnam, K. Thermodynamic Characterization of Interleukin-8 Binding to CXCR1 Receptor N-domain. FEBS J. 2007, 274, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.; Joseph, P.R.; Rajarathnam, K. Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain: Role of coupled interactions and dynamics. Biochemistry 2009, 48, 8795–8805. [Google Scholar] [CrossRef] [PubMed]
- Skelton, N.J.; Quan, C.; Reilly, D.; Lowman, H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 1999, 7, 157–168. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef] [PubMed]
- Loria, J.P.; Rance, M.; Palmer, A.G. A relaxation compensated CPMG sequence for characterizing chemical exchange. J. Am. Chem. Soc. 1999, 121, 2331–2332. [Google Scholar] [CrossRef]
- Wang, C.; Grey, M.J.; Palmer, A.G. CPMG sequences with enhanced sensitivity to chemical exchange. J. Biomol. NMR 2001, 21, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Markin, C.J.; Spyracopoulos, L. Increased precision for analysis of protein-ligand dissociation constants determined from chemical shift titrations. J. Biomol. NMR 2012, 53, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Markin, C.J.; Spyracopoulos, L. Accuracy and precision of protein-ligand interaction kinetics determined from chemical shift titrations. J. Biomol. NMR 2012, 54, 355–376. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Sykes, B.D.; Kay, C.M.; Dewald, B.; Geiser, T.; Baggiolini, M.; Clark-Lewis, I. Neutrophil activation by monomeric interleukin-8. Science 1994, 264, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Lowman, H.B.; Fairbrother, W.J.; Slagle, P.H.; Kabakoff, R.; Liu, J.; Shire, S.; Hebert, C.A. Monomeric variants of IL-8: Effects of side chain substitutions and solution conditions upon dimer formation. Protein Sci. 1997, 6, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Hayes, G.L.; Darbha, N.S.; Meyer, E.; LiWang, P.J. Investigation of CC and CXC chemokine quaternary state mutants. Biochem. Biophys. Res. Commun. 2005, 338, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Kay, C.M.; Clark-Lewis, I.; Sykes, B.D. Characterization of quaternary structure of interleukin-8 and functional implications. Methods Enzymol. 1997, 287, 89–105. [Google Scholar] [PubMed]
- Joseph, P.R.; Poluri, K.M.; Rajagopalan, L.; Gangavarupu, P.; Raghuwanshi, S.K.; Richardson, R.; Garofalo, R.P.; Rajarathnam, K. Proline Mutagenesis as a Strategy for Design of Monomeric Proteins. Biophys. J. 2013, 105, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Schumacher, C.; Baggiolini, M.; Moser, B. Structure-Activity Relationship of Interleukin-8 Determined Using Chemically Synthesized Analogs. J. Biol. Chem. 1991, 266, 23128–23134. [Google Scholar] [PubMed]
- Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 2. Analysis of Experimental Results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Clore, G.M.; Szabo, A.; Bax, A.; Kay, L.E.; Driscoll, P.C.; Gronenborn, A.M. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990, 112, 4989–4991. [Google Scholar] [CrossRef]
- Mandel, A.M.; Akke, M.; Palmer, A.G. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 1995, 246, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.; Loria, J.P. FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. J. Biomol. NMR 2003, 26, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Gagne, S.M.; Tsuda, S.; Spyracopoulos, L.; Kay, L.E.; Sykes, B.D. Backbone and methyl dynamics of the regulatory domain of troponin C: Anisotropic rotational diffusion and contribution of conformational entropy to calcium affinity. J. Mol. Biol. 1998, 278, 667–686. [Google Scholar] [CrossRef] [PubMed]
- Tjandra, N.; Wingfield, P.; Stahl, S.; Bax, A. Anisotropic rotational diffusion of perdeuterated HIV protease from 15N NMR relaxation measurements at two magnetic fields. J. Biomol. NMR 1996, 8, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Tjandra, N.; Feller, S.E.; Pastor, R.W.; Bax, A. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 1995, 117, 12562–12566. [Google Scholar] [CrossRef]
- Schurr, J.M.; Babcock, H.P.; Fujimoto, B.S. A Test of the Model-Free Formulas. Effects of Anisotropic Rotational Diffusion and Dimerization. J. Magn. Reson. Ser. B 1994, 105, 211–224. [Google Scholar] [CrossRef]
- Spyracopoulos, L.; Lewis, M.J.; Saltibus, L.F. Main chain and side chain dynamics of the ubiquitin conjugating enzyme variant human Mms2 in the free and ubiquitin-bound States. Biochemistry 2005, 44, 8770–8781. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Dewald, B.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Disulfide Bridges in Interleukin-8 Probed Using Non-Natural Disulfide Analogs: Dissociation of Roles in Structure and Function. Biochemistry 1999, 38, 7653–7658. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Spyracopoulos, L.; Lavigne, P.; Kim, K.S.; Clark-lewis, I.; Sykes, B.D. Backbone dynamics of the human CC chemokine eotaxin: Fast motions, slow motions, and implications for receptor binding. Protein Sci. 1999, 8, 2041–2054. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.L.; Stone, M.J. Backbone dynamics of the CC-chemokine eotaxin-2 and comparison among the eotaxin group chemokines. Proteins 2003, 50, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Mayer, K.L.; Stone, M.J. Backbone dynamics of the human CC-chemokine eotaxin. J. Biomol. NMR 1999, 15, 115–124. [Google Scholar] [CrossRef] [PubMed]
- LiWang, A.C.; Cao, J.J.; Zheng, H.; Lu, Z.; Peiper, S.C.; LiWang, P.J. Dynamics Study on the Anti-Human Immunodeficiency Virus Chemokine Viral Macrophage-Inflammatory Protein-II (VMIP-II) Reveals a Fully Monomeric Protein. Biochemistry 1999, 38, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Li, Y.; Rohrer, T.; Gentz, R. Solution Structure and Dynamics of Myeloid Progenitor Inhibitor Factor-1 (MPIF-1): A Novel Monomeric CC Chemokine. J. Biol. Chem. 2001, 276, 4909–4916. [Google Scholar] [CrossRef] [PubMed]
- Perozzo, R.; Folkers, G.; Scapozza, L. Thermodynamics of protein-ligand interactions: History, presence, and future aspects. J. Recept. Signal Transduct. Res. 2004, 24, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.; Sawant, K.V.; Sarmiento, J.; Navarro, J.; Rajarathnam, K. Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 2013, 288, 12244–12252. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Gustavsson, M.; Zheng, Y.; Stephens, B.S.; Handel, T.M. What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annu. Rev. Biophys. 2017, 46, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.R.; Kalodimos, C.G. Protein activity regulation by conformational entropy. Nature 2012, 488, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikova, O.K.; Sykes, B.D. Backbone dynamics of SDF-1α determined by NMR: Interpretation in the presence of monomer-dimer equilibrium. Protein Sci. 2006, 15, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Mayer, K.L.; Mayer, M.R.; Stone, M.J. NMR solution structure and backbone dynamics of the CC chemokine eotaxin-3. Biochemistry 2001, 40, 7820–7831. [Google Scholar] [CrossRef] [PubMed]
- Mizoue, L.S.; Bazan, J.F.; Johnson, E.C.; Handel, T.M. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry 1999, 38, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- David, K.S.; Oliveira, E.R.A.; Horta, B.A.C.; Valente, A.P.; de Paula, V.S. Insights into CC Chemokine Ligand 2/Chemokine Receptor 2 Molecular Recognition: A Step Forward toward Antichemotactic Agents. Biochemistry 2017, 56, 3197–3210. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.J.; Louis, J.M.; Gronenborn, A.M. A protein contortionist: Core mutations of GB1 that induce dimerization and domain swapping. J. Mol. Biol. 2003, 333, 141–152. [Google Scholar] [CrossRef]
- Spyracopoulos, L. A suite of Mathematica notebooks for the analysis of protein main chain 15N NMR relaxation data. J. Biomol. NMR 2006, 36, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.K.; Rance, M.; Chazin, W.J.; Palmer, A.G., 3rd. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J. Biomol. NMR 1997, 9, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Bieri, M.; Gooley, P.R. Automated NMR relaxation dispersion data analysis using NESSY. BMC Bioinform. 2011, 12, 421. [Google Scholar] [CrossRef] [PubMed]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, P.R.B.; Spyracopoulos, L.; Rajarathnam, K. Dynamics-Derived Insights into Complex Formation between the CXCL8 Monomer and CXCR1 N-Terminal Domain: An NMR Study. Molecules 2018, 23, 2825. https://doi.org/10.3390/molecules23112825
Joseph PRB, Spyracopoulos L, Rajarathnam K. Dynamics-Derived Insights into Complex Formation between the CXCL8 Monomer and CXCR1 N-Terminal Domain: An NMR Study. Molecules. 2018; 23(11):2825. https://doi.org/10.3390/molecules23112825
Chicago/Turabian StyleJoseph, Prem Raj B., Leo Spyracopoulos, and Krishna Rajarathnam. 2018. "Dynamics-Derived Insights into Complex Formation between the CXCL8 Monomer and CXCR1 N-Terminal Domain: An NMR Study" Molecules 23, no. 11: 2825. https://doi.org/10.3390/molecules23112825
APA StyleJoseph, P. R. B., Spyracopoulos, L., & Rajarathnam, K. (2018). Dynamics-Derived Insights into Complex Formation between the CXCL8 Monomer and CXCR1 N-Terminal Domain: An NMR Study. Molecules, 23(11), 2825. https://doi.org/10.3390/molecules23112825