Ag/Pyridine Co-Mediated Oxidative Arylthiocyanation of Activated Alkenes

Abstract
: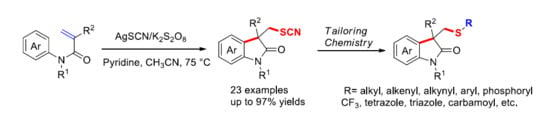
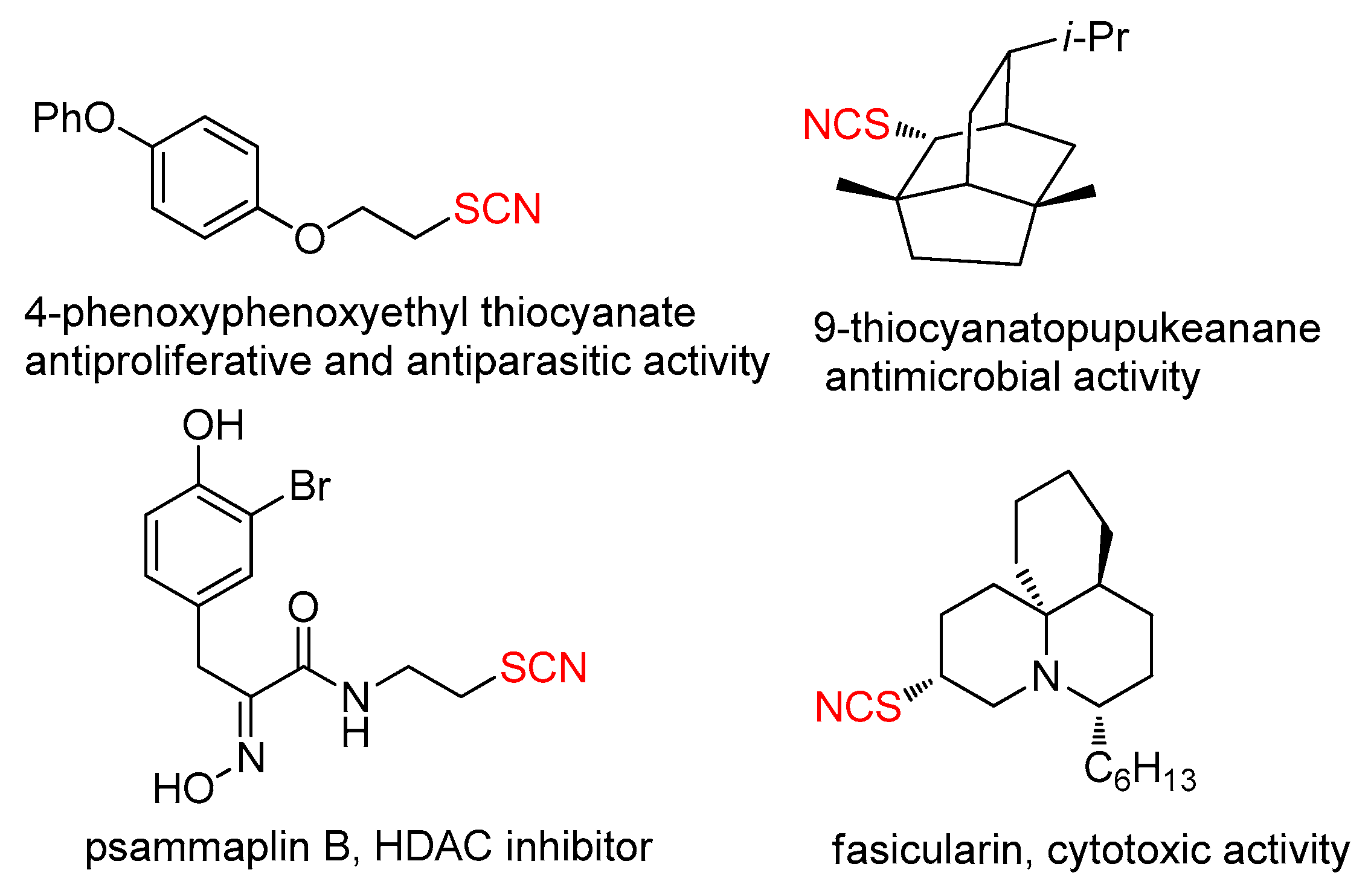
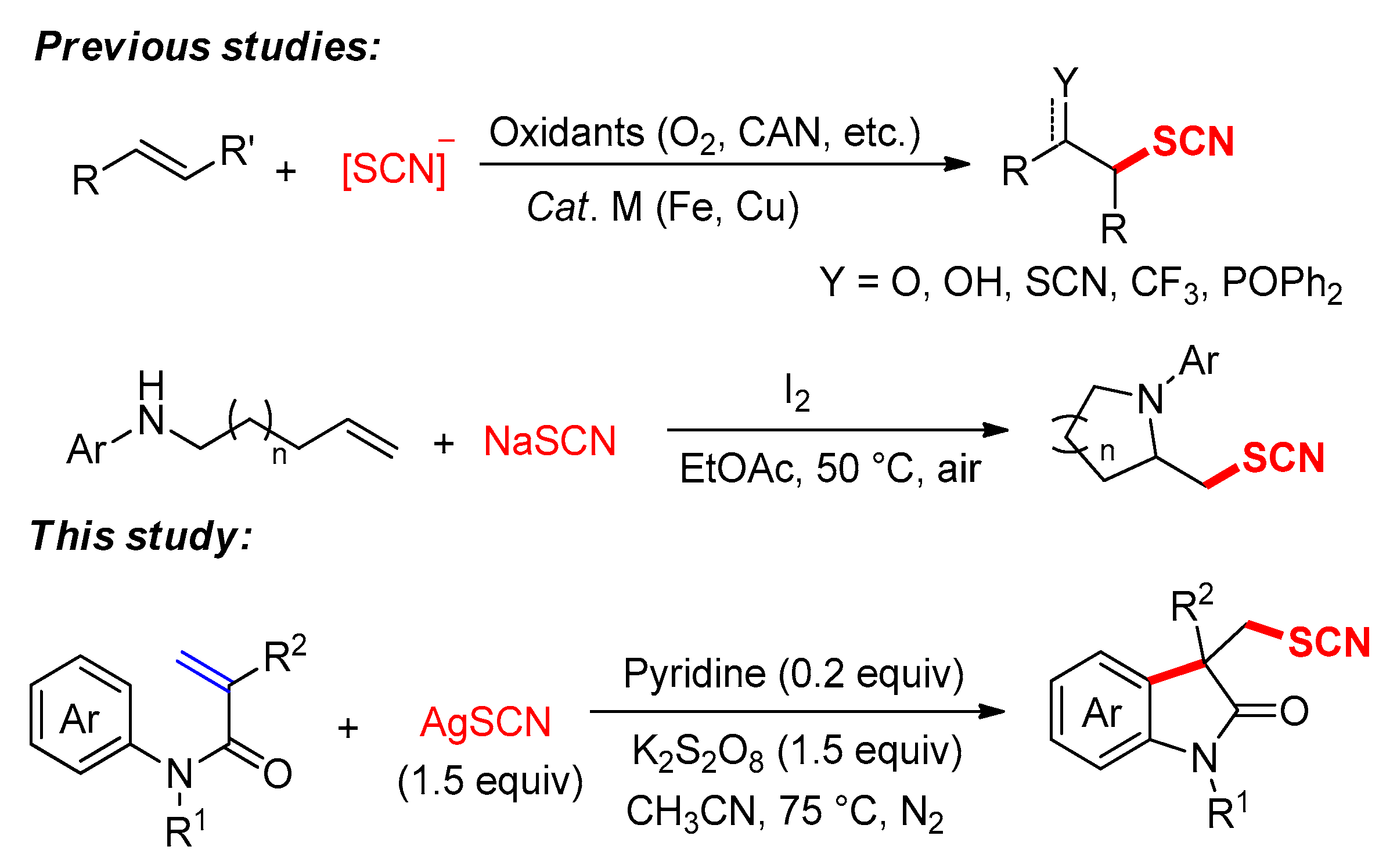
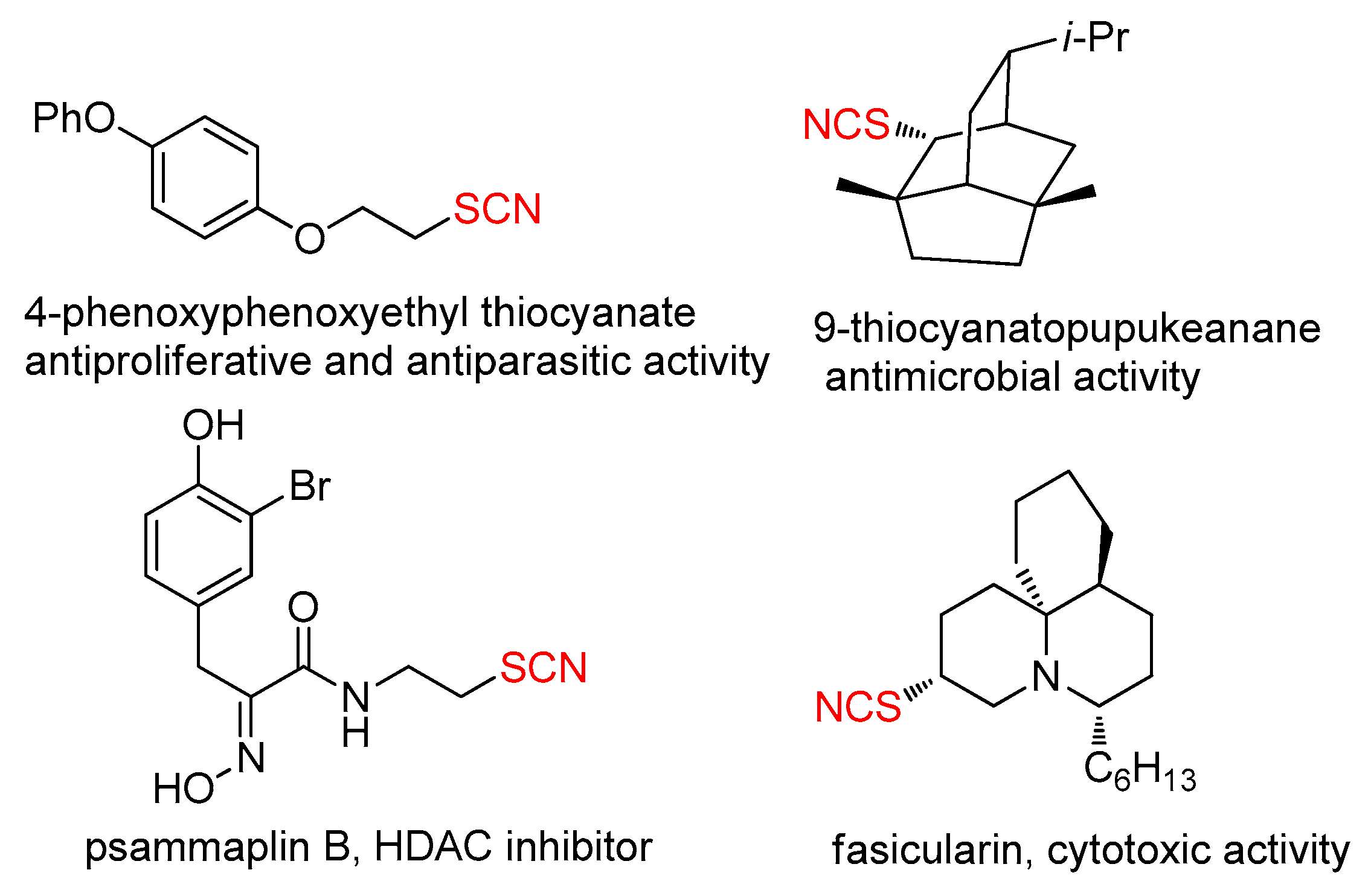
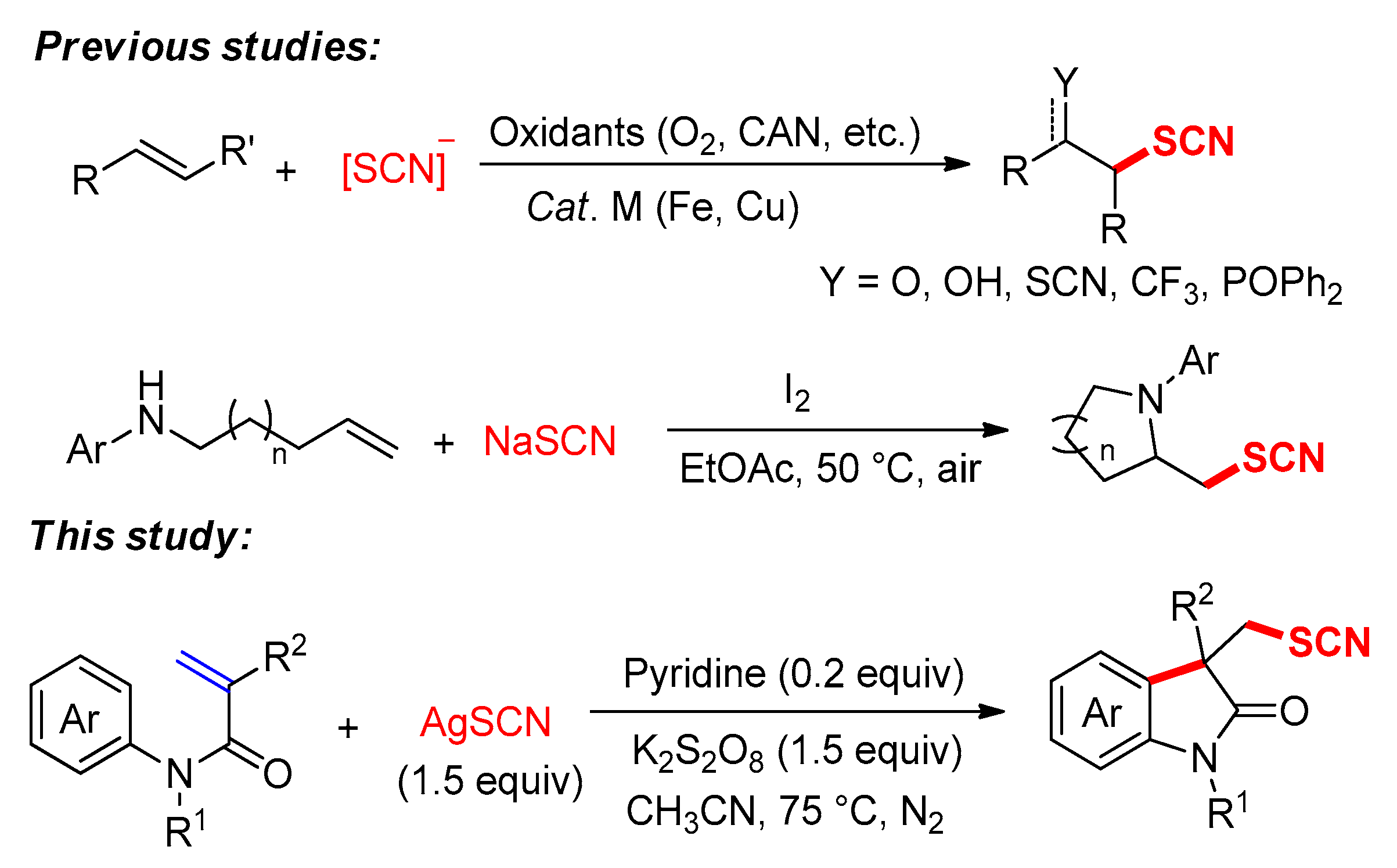
1. Introduction
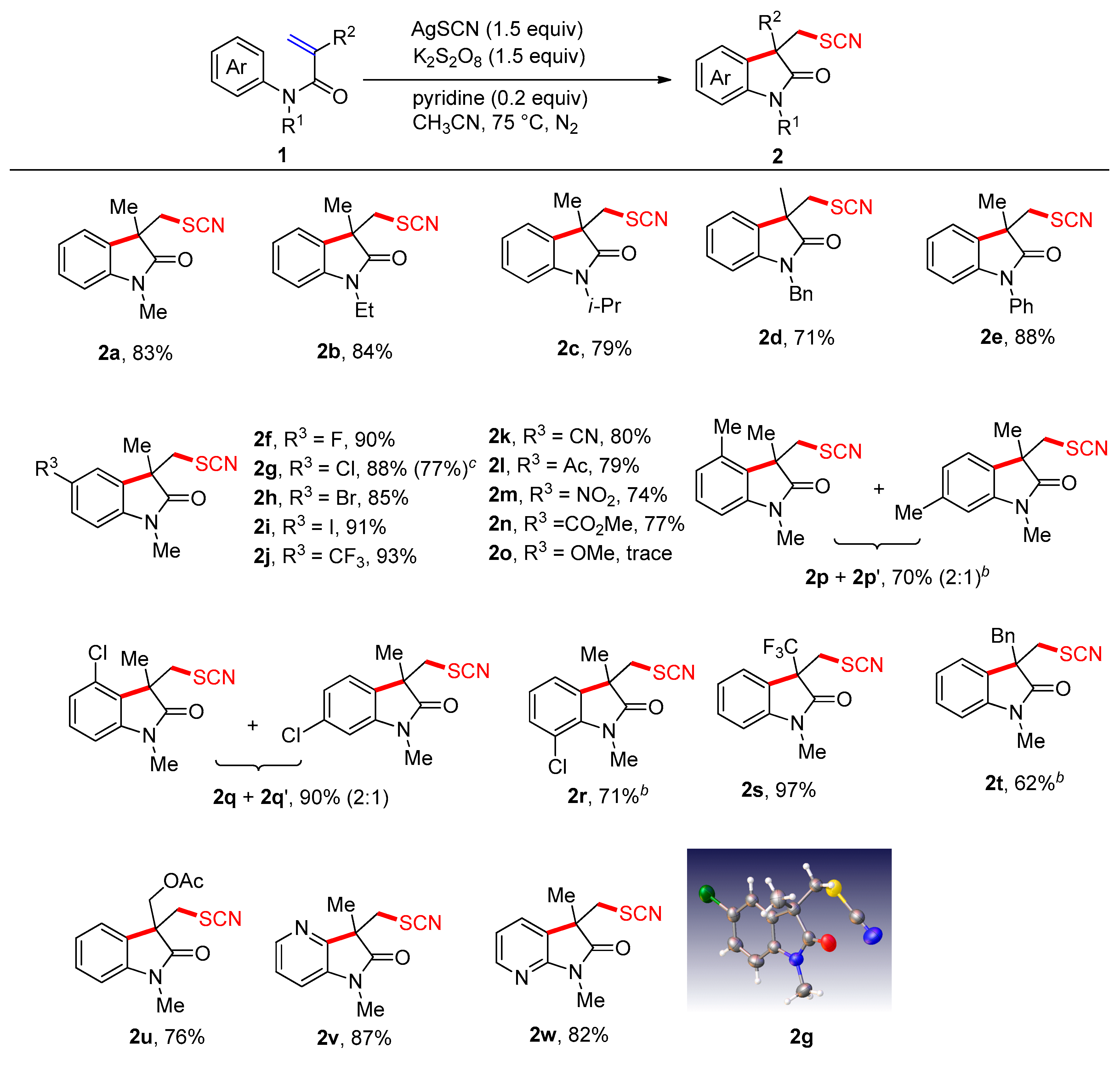
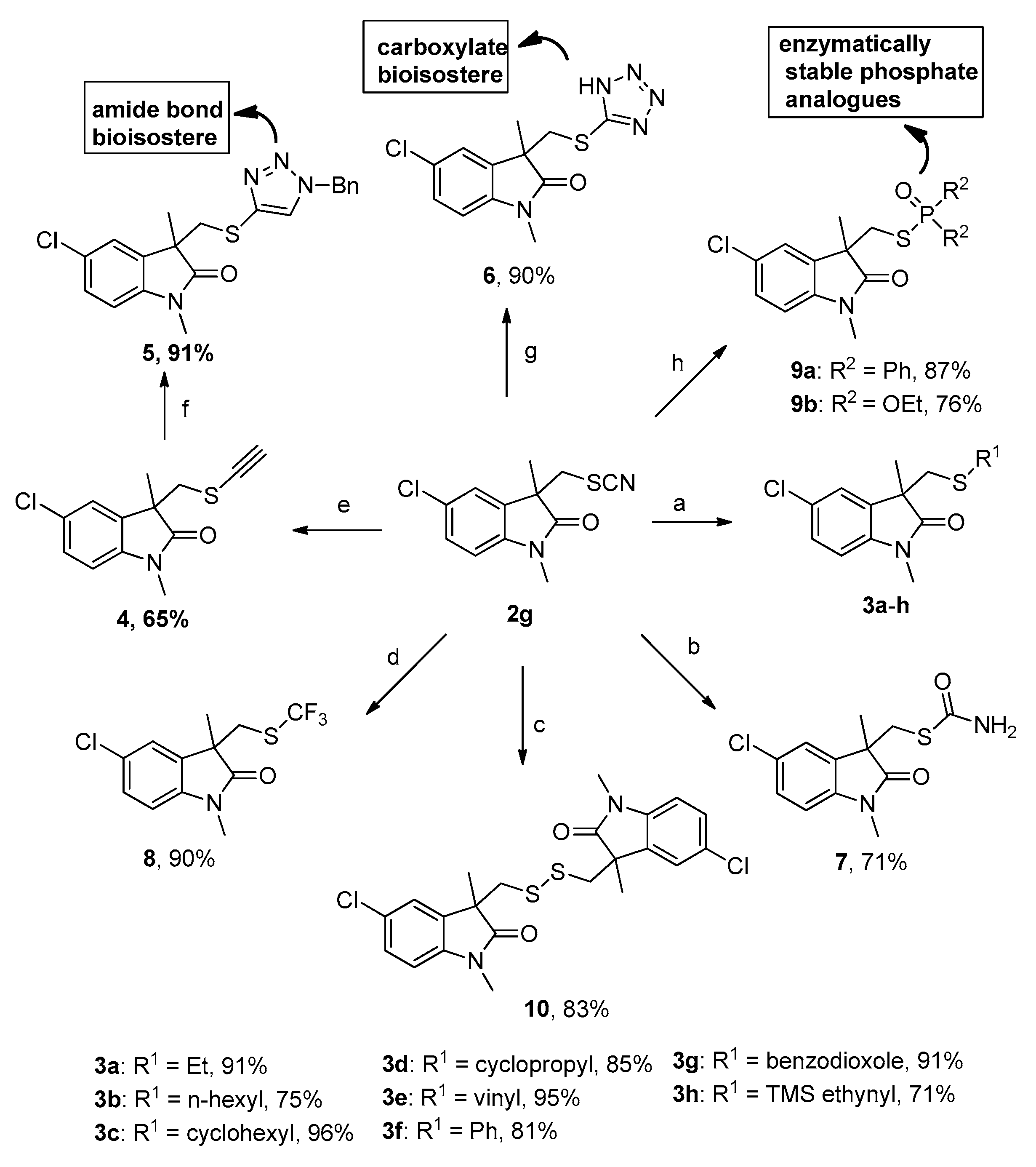
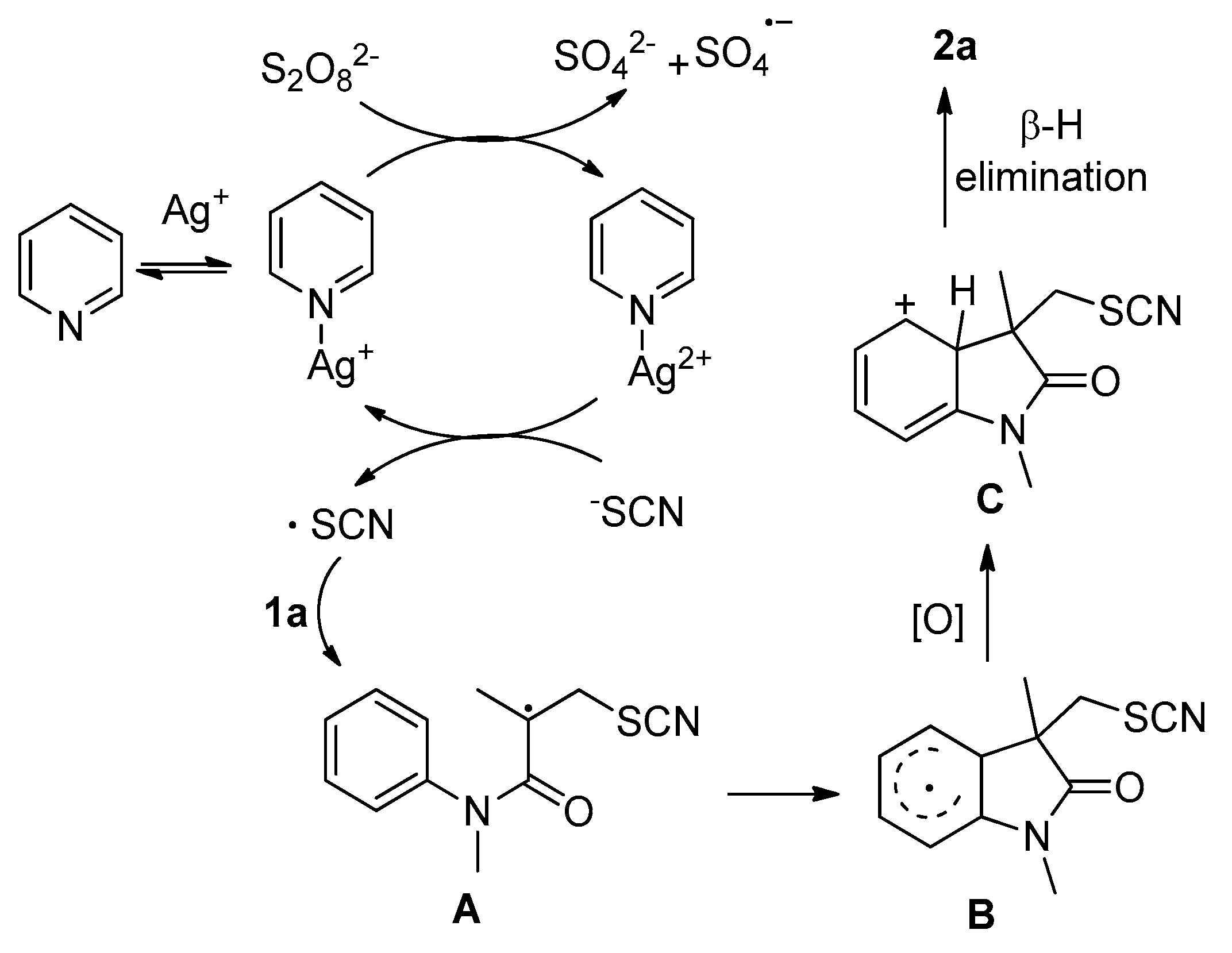
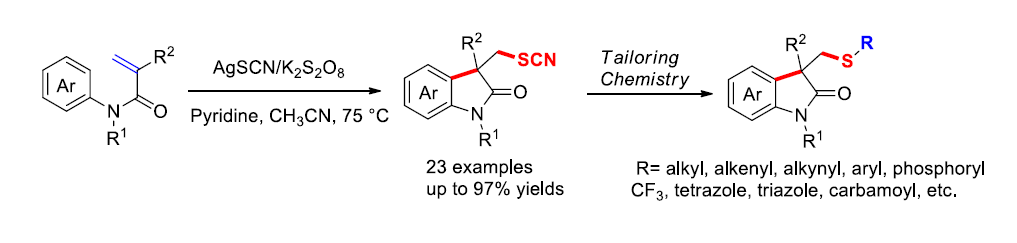
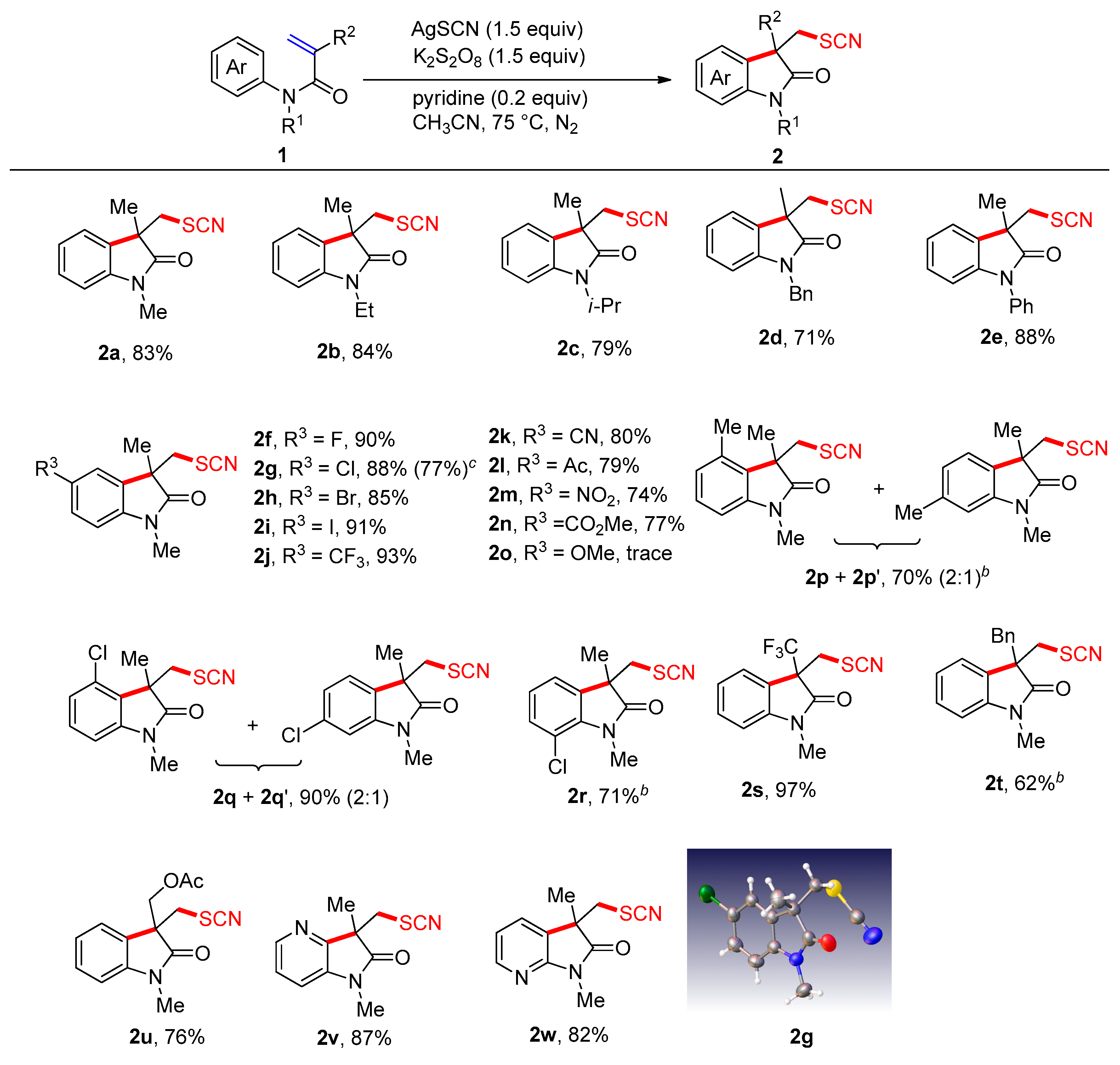
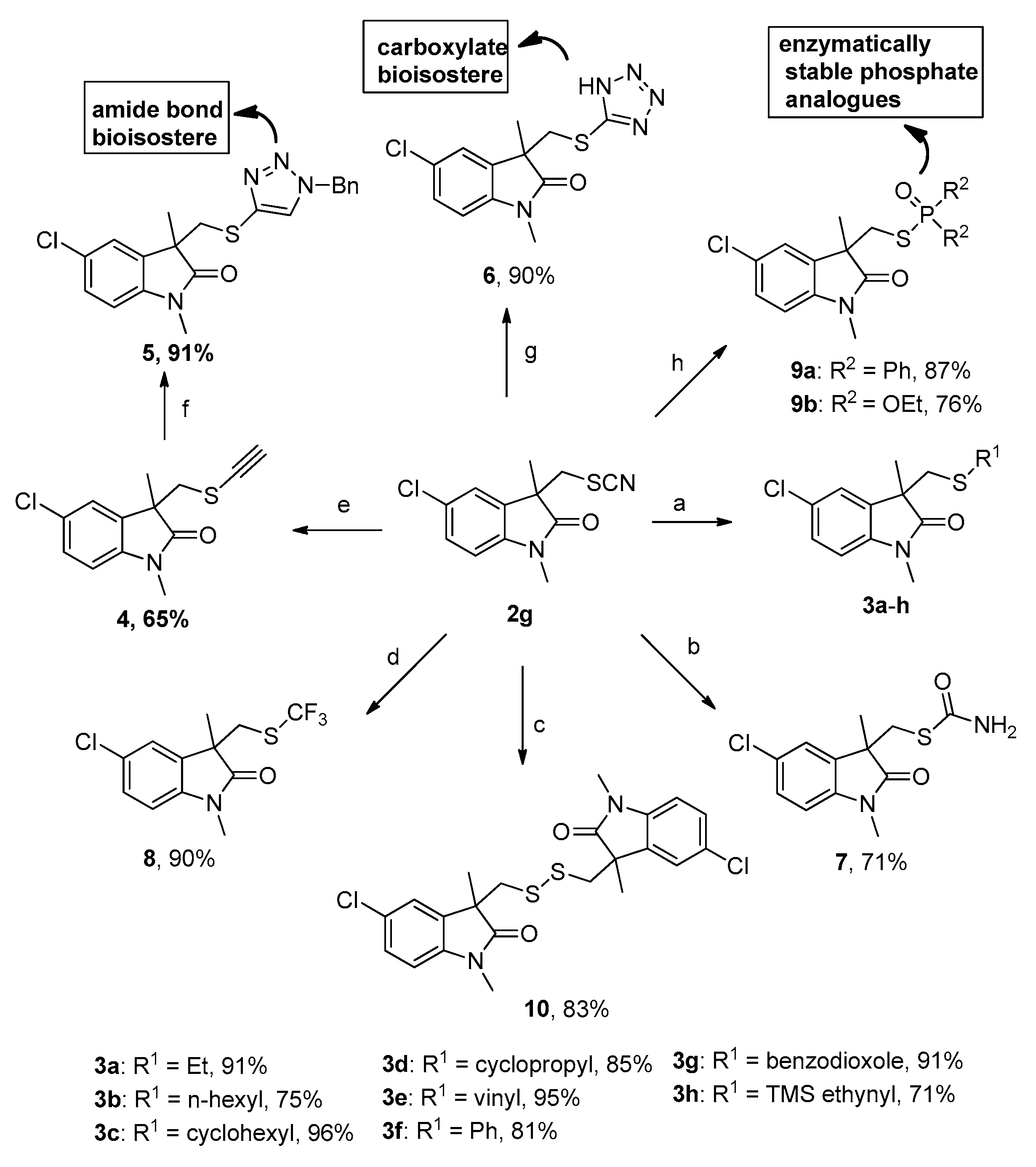
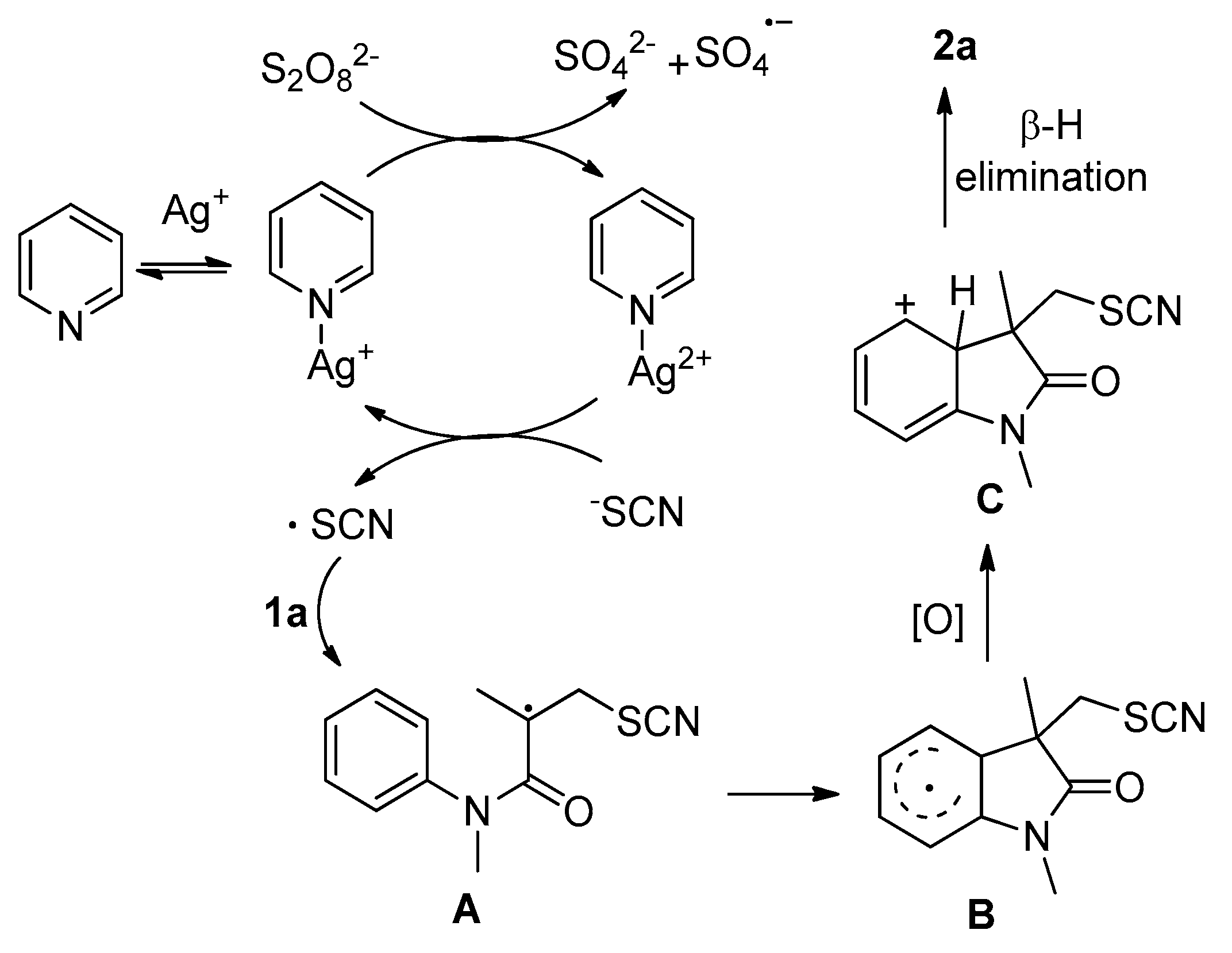
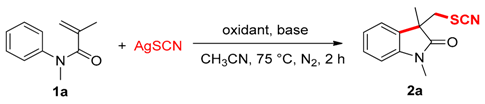
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for Arylthiocyanation of Acrylamides 1
3.3. Procedure for Gram-Scale Preparation of 5-Chloro-1,3-dimethyl-3-(thiocyanatomethyl)indolin-2-one (2g)
3.4. General Procedure for Preparation of Compounds 3a–g
3.5. Procedure for the Preparation of 5-Chloro-1,3-dimethyl-3-((((trimethylsilyl)ethynyl)thio)methyl)indolin-2-one (3h)
3.6. Procedure for the Preparation of 5-Chloro-3-((ethynylthio)methyl)-1,3-dimethylindolin-2-one (4)
3.7. Procedure for the Preparation of Triazole 3-(((1-benzyl-1H-1,2,3-triazol-4-yl)thio)methyl)-5-chloro-1,3-dimethylindolin-2-one (5)
3.8. Procedure for the Preparation of 3-(((1H-tetrazol-5-yl)thio)methyl)-5-chloro-1,3-dimethylindolin-2-one (6)
3.9. Procedure for the Preparation of S-((5-chloro-1,3-dimethyl-2-oxoindolin-3-yl)methyl) carbamothioate (7)
3.10. Procedure for the Preparation of 5-chloro-1,3-dimethyl-3-(((trifluoromethyl)thio)methyl)indolin-2-one (8)
3.11. General Procedure for the Preparation of Compounds 9a–b
3.12. Procedure for the Preparation of 3,3′-(disulfanediylbis(methylene))bis(5-chloro-1,3-dimethylindolin-2-one) (10)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Elhalem, E.; Bailey, B.N.; Docampo, R.; Ujváry, I.; Szajnman, S.H.; Rodriguez, J.B. Design, Synthesis, and Biological Evaluation of Aryloxyethyl Thiocyanate Derivatives against Trypanosoma cruzi. J. Med. Chem. 2002, 45, 3984. [Google Scholar] [CrossRef] [PubMed]
- Yasman, Y.; Edrada, R.A.; Wray, V.; Proksch, P. New 9-Thiocyanatopupukeanane Sesquiterpenes from the Nudibranch Phyllidia varicosa and Its Sponge-Prey Axinyssa aculeate. J. Nat. Prod. 2003, 66, 1512. [Google Scholar] [CrossRef] [PubMed]
- Piña, I.C.; Gautschi, J.T.; Wang, G.-Y.-S.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the Sponge Pseudoceratina purpurea: Inhibition of Both Histone Deacetylase and DNA Methyltransferase. J. Org. Chem. 2003, 68, 3866. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Abe, H.; Aoyagi, S.; Kibayashi, C.; Gates, K.S. DNA damage by fasicularin. J. Am. Chem. Soc. 2005, 127, 15004. [Google Scholar] [CrossRef] [PubMed]
- Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Recent advances in the chemistry of organic thiocyanates. Chem. Soc. Rev. 2016, 45, 494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liebeskind, L.S. Palladium-catalyzed, copper (I)-mediated coupling of boronic acids and benzylthiocyanate. A cyanide-free cyanation of boronic acids. Org. Lett. 2006, 8, 4331. [Google Scholar] [CrossRef] [PubMed]
- Demko, Z.P.; Sharpless, K.B. An intramolecular [2+3] cycloaddition route to fused 5-heterosubstituted tetrazoles. Org. Lett. 2001, 3, 4091. [Google Scholar] [CrossRef] [PubMed]
- D’hooghe, M.; Waterinckx, A.; De Kimpe, N. A novel entry toward 2-imino-1, 3-thiazolidines and 2-imino-1, 3-thiazolines by ring transformation of 2-(thiocyanomethyl) aziridines. J. Org. Chem. 2005, 70, 227. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ganesh, M.; Seidel, D. Catalytic enantioselective synthesis of α, β-diamino acid derivatives. J. Am. Chem. Soc. 2009, 131, 11648. [Google Scholar] [CrossRef] [PubMed]
- Iranpoor, N.; Firouzabadi, H.; Shaterian, H.R. Efficient conversion of thiols to thiocyanates by in situ generated Ph3P(SCN)2. Tetrahedron Lett. 2002, 43, 3439. [Google Scholar] [CrossRef]
- Ju, Y.; Kumar, D.; Varma, R.S. Revisiting nucleophilic substitution reactions: microwave-assisted synthesis of azides, thiocyanates, and sulfones in an aqueous medium. J. Org. Chem. 2006, 71, 6697. [Google Scholar] [CrossRef] [PubMed]
- Iranpoor, N.; Firouzabadi, H.; Nowrouzi, N. Preparation of thiocyanates and isothiocyanates from alcohols, thiols, trimethylsilyl-, and tetrahydropyranyl ethers using triphenylphosphine/2,3-dichloro-5,6-dicyanobenzoquinone (DDQ)/n-Bu4NSCN system. Tetrahedron 2006, 62, 5498. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Jung, S.H.; Chae, J. A facile and green protocol for nucleophilic substitution reactions of sulfonate esters by recyclable ionic liquids [bmim][X]. Synlett 2012, 23, 2692. [Google Scholar] [CrossRef]
- Falck, J.R.; Gao, S.; Prasad, R.N.; Koduru, S.R. Electrophilic α-thiocyanation of chiral and achiral N-acyl imides. A convenient route to 5-substituted and 5, 5-disubstituted 2, 4-thiazolidinediones. Bioorg. Med. Chem. Lett. 2008, 18, 1768. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.-S.; Wu, D.; Karmaker, P.G.; Yin, H.-Q.; Chen, F.-X. Enantioselective Organocatalyzed Direct α-Thiocyanation of Cyclic β-Ketoesters by N-Thiocyanatophthalimide. Org. Lett. 2018, 20, 1600. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Qiu, J.-S.; Karmaker, P.G.; Yin, H.-Q.; Chen, F.-X. N-Thiocyanatosaccharin: A “Sweet” Electrophilic Thiocyanation Reagent and the Synthetic Applications. J. Org. Chem. 2018, 83, 1576. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Wang, S.-F.; Jiang, Q.-W.; Cheng, C.-G.; Xiao, X.-H.; Zhu, G.-G. Palladium-Catalyzed Site-Selective sp3 C–H Bond Thiocyanation of 2-Aminofurans. J. Org. Chem. 2018, 83, 716. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Yadav, L.D.S. Visible-light-induced direct α-C(sp3)–H thiocyanation of tertiary amines. Tetrahedron Lett. 2015, 56, 6696. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Reddy, U.V.S.; Chary, D.N. Iron (III) chloride as mild and efficient reagent for the α-thiocyanation of ketones: An expedient synthesis of α-oxo thiocyanates. Synthesis 2008, 8, 1283–1287. [Google Scholar] [CrossRef]
- Liu, S.; Guo, Z.; Wang, Y.; Wang, T.; Wu, L.-Q. Trichloroisocyanuric Acid as a Novel and Versatile Reagent for the Rapid α-Thiocyanation of Ketones. Bull. Korean Chem. Soc. 2011, 32, 3760. [Google Scholar] [CrossRef]
- Wu, D.; Yang, X.; Wu, L.-Q. SelectfluorTM: A novel and efficient reagent for the rapid α-thiocyanation of ketones. J. Chem. Sci. 2012, 124, 901. [Google Scholar] [CrossRef]
- Wu, L.; Yang, X.-J. Efficient α-thiocyanation of ketones using pyridinium hydrobromide Perbromide. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 748–753. [Google Scholar]
- Jiao, J.; Nguyen, L.X.; Patterson, D.R.; Flowers, R.A., II. An efficient and general approach to β-functionalized ketones. Org. Lett. 2007, 9, 1323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, J.T.; Yu, Y.; Guan, Z.; He, Y.H. Photocatalytic anion oxidation achieves direct aerobic difunctionalization of alkenes leading to β-thiocyanato alcohols. Tetrahedron 2018, 74, 3038. [Google Scholar] [CrossRef]
- Liu, K.; Li, D.P.; Zhou, S.F.; Pan, X.Q.; Shoberu, A.; Zou, J.P. Molecular oxygen induced free radical oxythiocyanation of styrenes leading to α-oxothiocyanates. Tetrahedron 2015, 71, 4031. [Google Scholar] [CrossRef]
- Yang, H.; Duan, X.-H.; Zhao, J.-F.; Guo, L.N. Transition-Metal-Free Tandem Radical Thiocyanooxygenation of Olefinic Amides: A New Route to SCN-Containing Heterocycles. Org. Lett. 2015, 17, 1998. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-N.; Gu, Y.-R.; Yang, H.; Hu, J. Transition-metal free thiocyanooxygenation of functionalized alkenes: facile routes to SCN-containing dihydrofurans and lactones. Org. Biomol. Chem. 2016, 14, 3098. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Reddy, B.V.S.; Gupta, M.K. Ferric (III) chloride-promoted efficient thiocyanation of arylalkenes: A facile synthesis of dithiocyanates. Synthesis 2004, 12, 1983. [Google Scholar] [CrossRef]
- Nair, V.; Nair, L.G. A very efficient cerium (IV) ammonium nitrate (CAN) mediated thiocyanation of aralkenes: Formation of dithiocyanates. Tetrahedron Lett. 1998, 39, 4585. [Google Scholar] [CrossRef]
- Tao, Z.K.; Li, C.K.; Zhang, P.Z.; Shoberu, A.; Zou, J.P.; Zhang, W.T. Phosphinoyl Radical-Initiated 1, 2-Bifunctional Thiocyanodiphenylphosphinoylation of Alkenes. J. Org. Chem. 2018, 83, 2418. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.L.; Wang, F.; Chen, P.H.; Liu, G.S. Copper-Catalyzed Intermolecular Trifluoromethylthiocyanation of Alkenes: Convenient Access to CF3-Containing Alkyl Thiocyanates. Org. Lett. 2015, 17, 2438. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Hussain, M.I.; Zhang, X.; Shi, J.; Hu, W.; Xiong, Y. Aerobic intramolecular aminothiocyanation of unactivated alkenes promoted by in situ generated iodine thiocyanate. Tetrahedron 2018, 74, 2669. [Google Scholar] [CrossRef]
- Karmaker, P.G.; Qiu, J.; Wu, D.; Yin, H.; Chen, F.X. Free Radical Cyclization of N-Arylacrylamides: Mild and Facile Synthesis of 3-Thiocyanato Oxindoles. Synlett 2018, 29, 954. [Google Scholar]
- Fafarman, A.T.; Webb, L.J.; Chuang, J.I.; Boxer, S.G. Site-specific conversion of cysteine thiols into thiocyanate creates an IR probe for electric fields in proteins. J. Am. Chem. Soc. 2006, 128, 13356. [Google Scholar] [CrossRef] [PubMed]
- Slocum, J.D.; Webb, L.J. Nitrile probes of electric field agree with independently measured fields in green fluorescent protein even in the presence of hydrogen bonding. J. Am. Chem. Soc. 2016, 138, 6561. [Google Scholar] [CrossRef] [PubMed]
- Sigala, P.A.; Fafarman, A.T.; Bogard, P.E.; Boxer, S.G.; Herschlag, D. Do ligand binding and solvent exclusion alter the electrostatic character within the oxyanion hole of an enzymatic active site? J. Am. Chem. Soc. 2007, 129, 12104. [Google Scholar] [CrossRef] [PubMed]
- Malik, G.; Swyka, R.A.; Tiwari, V.K.; Fei, X.; Applegate, G.A.; Berkowitz, D.B. A thiocyanopalladation/carbocyclization transformation identified through enzymatic screening: Stereocontrolled tandem C–SCN and C–C bond formation. Chem. Sci. 2017, 8, 8050. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-F.; Tan, D.-H.; Chen, Y.; Lv, W.-X.; Liu, X.-G.; Li, Q.; Wang, H. Direct radical trifluoromethylthiolation and thiocyanation of aryl alkynoate esters: mild and facile synthesis of 3-trifluoromethylthiolated and 3-thiocyanated coumarins. Org. Chem. Front. 2015, 2, 1511. [Google Scholar] [CrossRef]
- Mandal, S.; Bera, T.; Dubey, G.; Saha, J.; Laha, J.K. Uses of K2S2O8 in Metal-Catalyzed and Metal-Free Oxidative Transformations. ACS Catal. 2018, 8, 5085. [Google Scholar] [CrossRef]
- Zhang, M.-Z.; Ji, P.-Y.; Liu, Y.-F.; Xu, J.-W.; Guo, C.-C. Disulfides as Sulfonylating Precursors for the Synthesis of Sulfone-Containing Oxindoles. Adv. Synth. Catal. 2016, 358, 2976. [Google Scholar] [CrossRef]
- Bruehlman, R.J.; Verhoek, F.H. The basic strengths of amines as measured by the stabilities of their complexes with silver ions. J. Am. Chem. Soc. 1948, 70, 1401. [Google Scholar] [CrossRef]
- Vosburgh, W.C.; Cogswell, S.A. Complex Ions. VIII. Pyridine—Silver Ions. J. Am. Chem. Soc. 1943, 65, 2412. [Google Scholar] [CrossRef]
- Alexiev, A.; Bontchev, P.R. A new catalytic reaction for determination of silver. Mikrochim. Acta 1970, 58, 13. [Google Scholar] [CrossRef]
- Bonchev, P.R.; Aleksiev, A.A. Use of Marcus’s theory for selecting activators of homogeneous catalytic reactions. Theor. Exp. Chem. 1975, 9, 144. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Salehi, P.; Sardarian, A.R.; Seddighi, M. Oxidation of Benzylic Hydrocarbons to Carbonyl Compounds by Tetrapyridinesilver(II) Peroxydisulfate Ag(Py)4S2O8 Under Non-Aqueous and Aprotic Condition. Synthetic Commun. 1991, 21, 1121. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Salehi, P.; Mohammadpour-Baltork, I. Tetrakis (pyridine) silver (II) Peroxodisulfate, (Ag (py) 4) S2O8, a Reagent for the Oxidative Transformations. Bull. Chem. Soc. Jpn. 1992, 65, 2878. [Google Scholar] [CrossRef]
- Nikolaev, A.; Legault, C.Y.; Zhang, M.; Orellana, A. The Acid-Free Cyclopropanol-Minisci Reaction Reveals the Catalytic Role of Silver–Pyridine Complexes. Org. Lett. 2018, 20, 796. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Flowers, R.A. Uncovering the mechanism of the Ag (I)/persulfate-catalyzed cross-coupling reaction of arylboronic acids and heteroarenes. J. Am. Chem. Soc. 2013, 135, 4672. [Google Scholar] [CrossRef] [PubMed]
- Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H. Difunctionalization of Acrylamides through C–H Oxidative Radical Coupling: New Approaches to Oxindoles. Synthesis 2015, 47, 1195. [Google Scholar] [CrossRef]
- Chen, J.-R.; Yu, X.-Y.; Xiao, W.-J. Tandem radical cyclization of N-arylacrylamides: an emerging platform for the construction of 3, 3-disubstituted oxindoles. Synthesis 2015, 47, 604. [Google Scholar] [CrossRef]
- Mu, X.; Wu, T.; Wang, H.Y.; Guo, Y.L.; Liu, G. Palladium-catalyzed oxidative aryltrifluoromethylation of activated alkenes at room temperature. J. Am. Chem. Soc. 2012, 134, 878. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–10 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Oxidant | Base (equiv.) | Yield b (%) |
|---|---|---|---|
| 1 | DTBP | none | 0 |
| 2 | Oxone | none | 0 |
| 3 | PhI(OAc) | none | 0 |
| 4 | selectfluor | none | trace |
| 5 | CAN | none | 15 |
| 6 | K2S2O8 | none | 0 |
| 7 | K2S2O8 | NaHCO3 (1) | 0 |
| 8 | K2S2O8 | Cs2CO3 (1) | trace |
| 9 | K2S2O8 | HMPA (1) | 0 |
| 10 | K2S2O8 | Et3N (1) | 0 |
| 11 | K2S2O8 | DBU (1) | 55 |
| 12 | K2S2O8 | pyridine (1) | 83 |
| 13 c | K2S2O8 | Pyridine (0.2) | 85 |
| 14 c | K2S2O8 | Pyridine (0.1) | 64 |
| 15 | none | Pyridine (0.2) | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, D.-L.; Du, J.-X.; Chu, W.-M.; Ma, C.-Y.; Tao, J.-Y.; Feng, W.-H. Ag/Pyridine Co-Mediated Oxidative Arylthiocyanation of Activated Alkenes. Molecules 2018, 23, 2727. https://doi.org/10.3390/molecules23102727
Kong D-L, Du J-X, Chu W-M, Ma C-Y, Tao J-Y, Feng W-H. Ag/Pyridine Co-Mediated Oxidative Arylthiocyanation of Activated Alkenes. Molecules. 2018; 23(10):2727. https://doi.org/10.3390/molecules23102727
Chicago/Turabian StyleKong, De-Long, Jian-Xun Du, Wei-Ming Chu, Chun-Ying Ma, Jia-Yi Tao, and Wen-Hua Feng. 2018. "Ag/Pyridine Co-Mediated Oxidative Arylthiocyanation of Activated Alkenes" Molecules 23, no. 10: 2727. https://doi.org/10.3390/molecules23102727
APA StyleKong, D.-L., Du, J.-X., Chu, W.-M., Ma, C.-Y., Tao, J.-Y., & Feng, W.-H. (2018). Ag/Pyridine Co-Mediated Oxidative Arylthiocyanation of Activated Alkenes. Molecules, 23(10), 2727. https://doi.org/10.3390/molecules23102727