
Nuciferine Inhibits Proinflammatory Cytokines via the PPARs in LPS-Induced RAW264.7 Cells


 and
and
Abstract
1. Introduction
2. Results
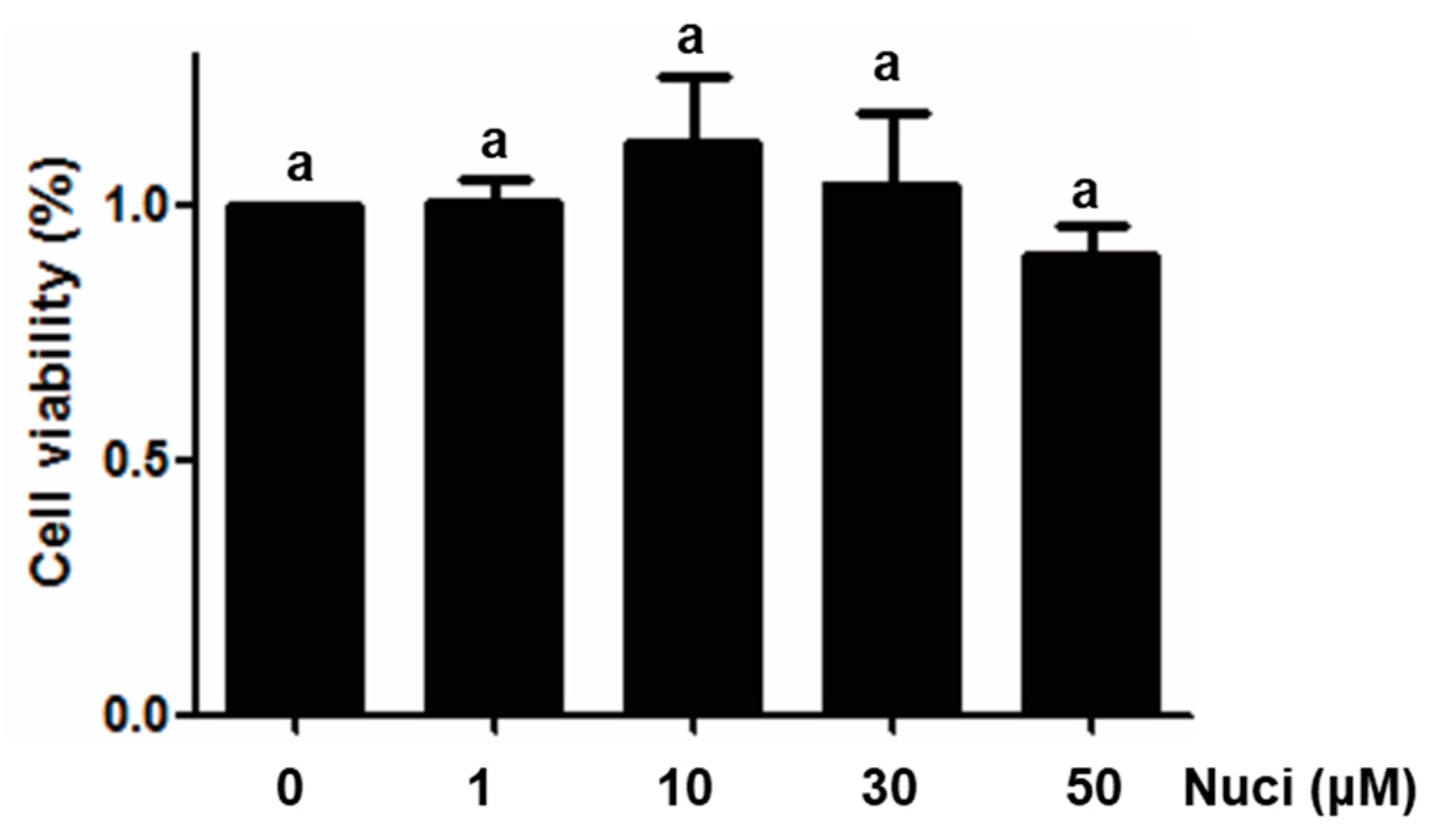
2.1. Cytotoxicity of Nuciferine on RAW264.7 Cells
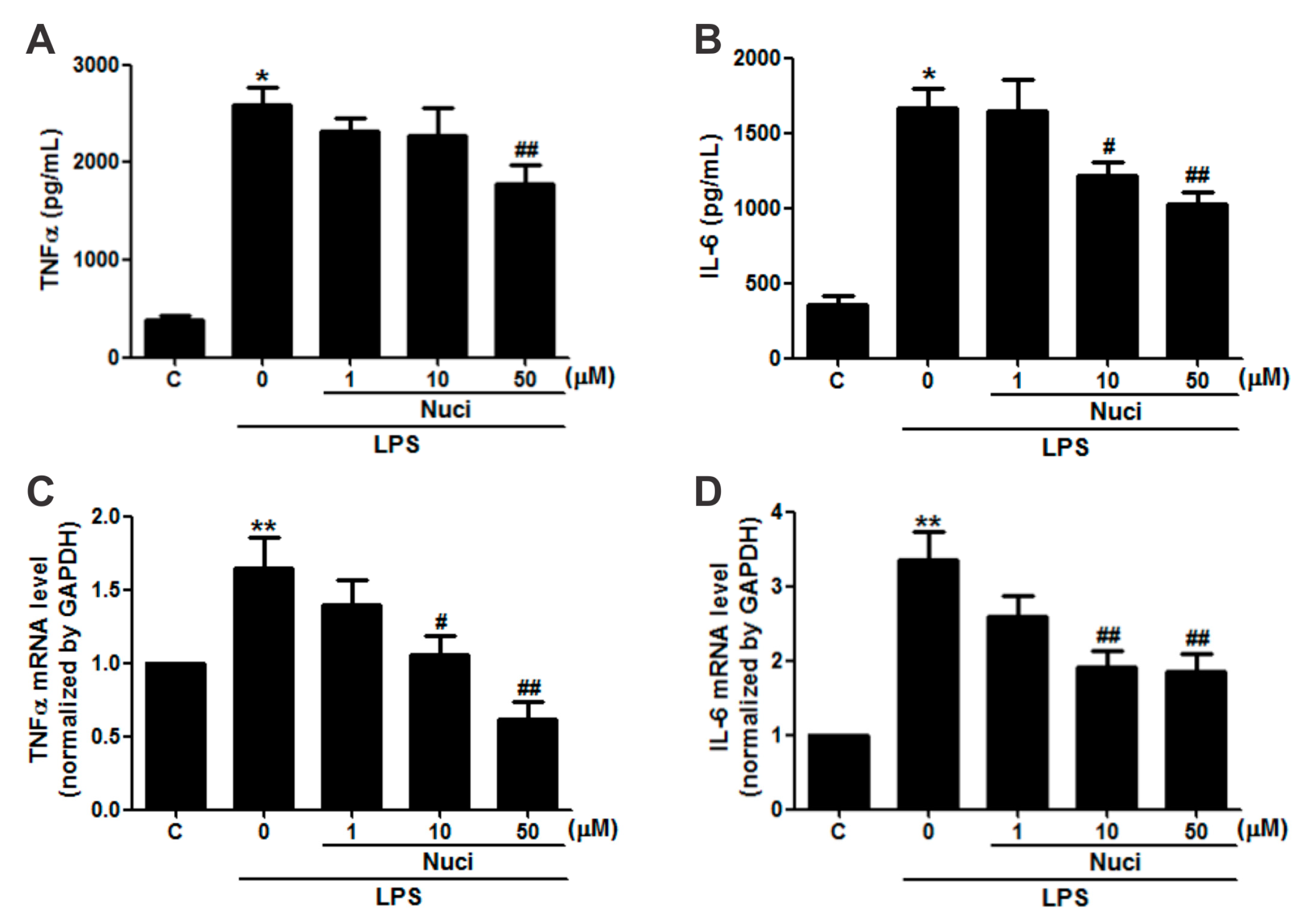
2.2. Nuciferine Inhibited IL-6 and TNFα Production in LPS-Induced RAW264.7 Cells
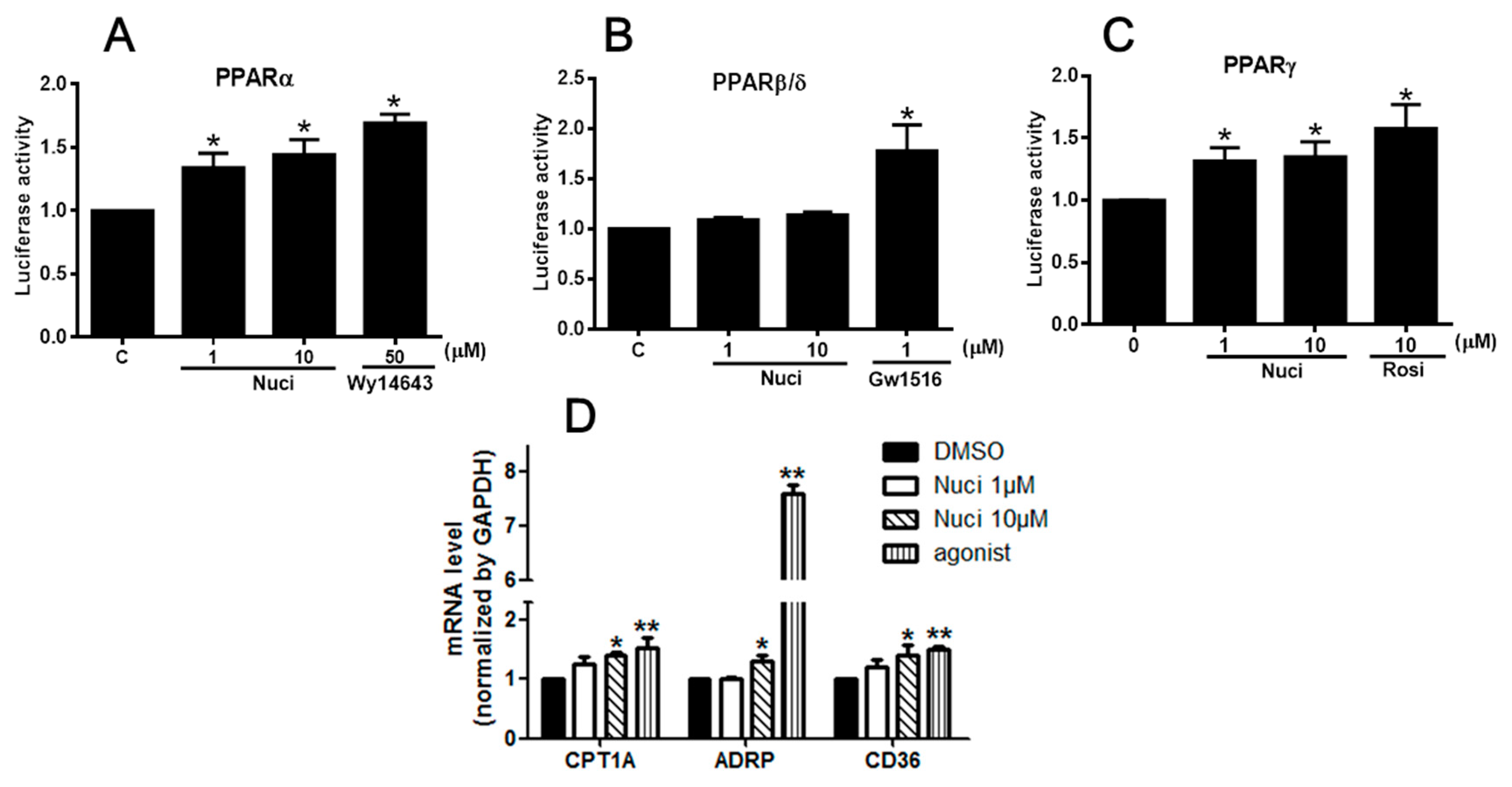
2.3. Nuciferine Increased the PPARs Activity
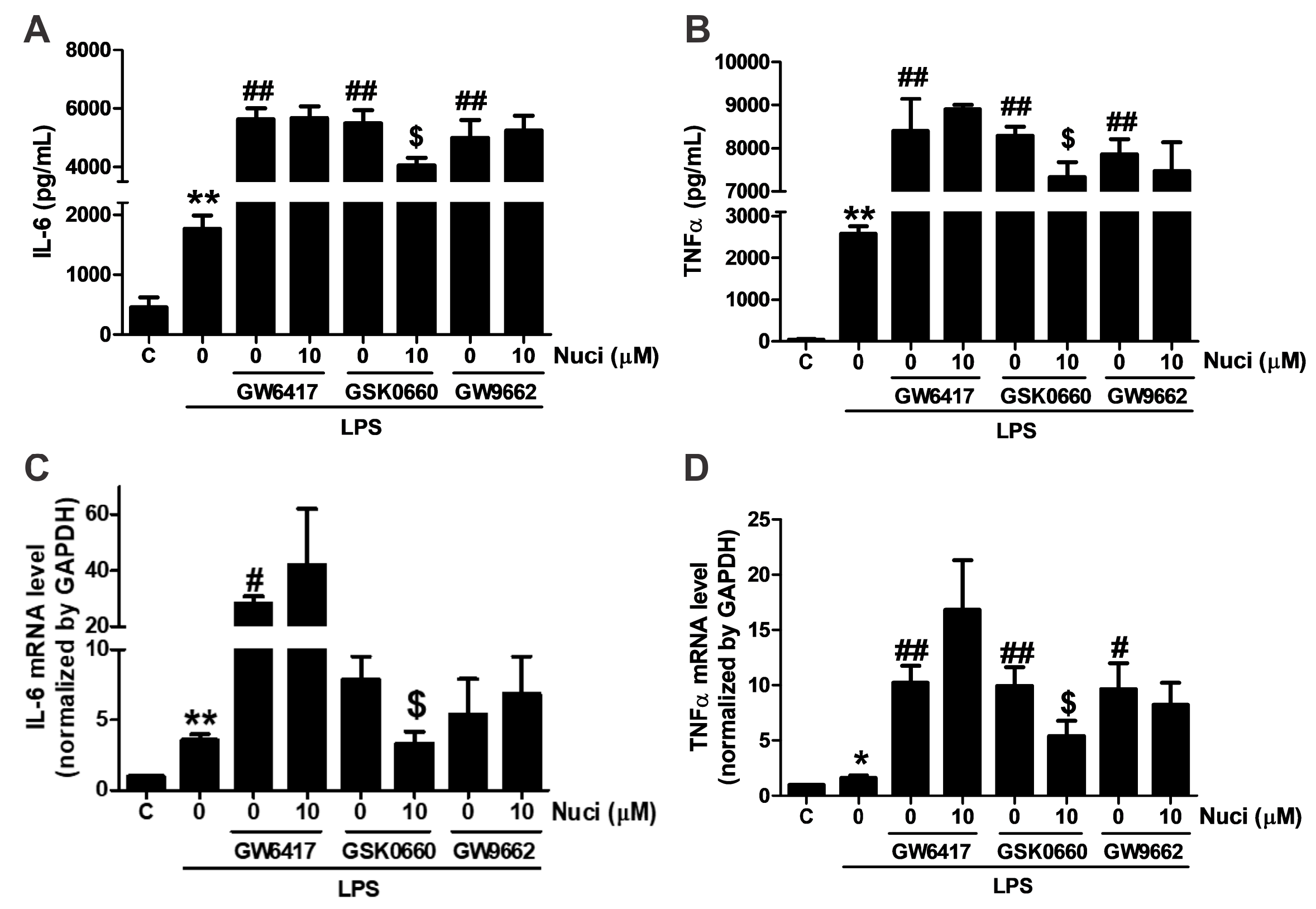
2.4. Antagonists of PPARα and PPARγ Abolished the Anti-Inflammatory Effects of Nuciferine
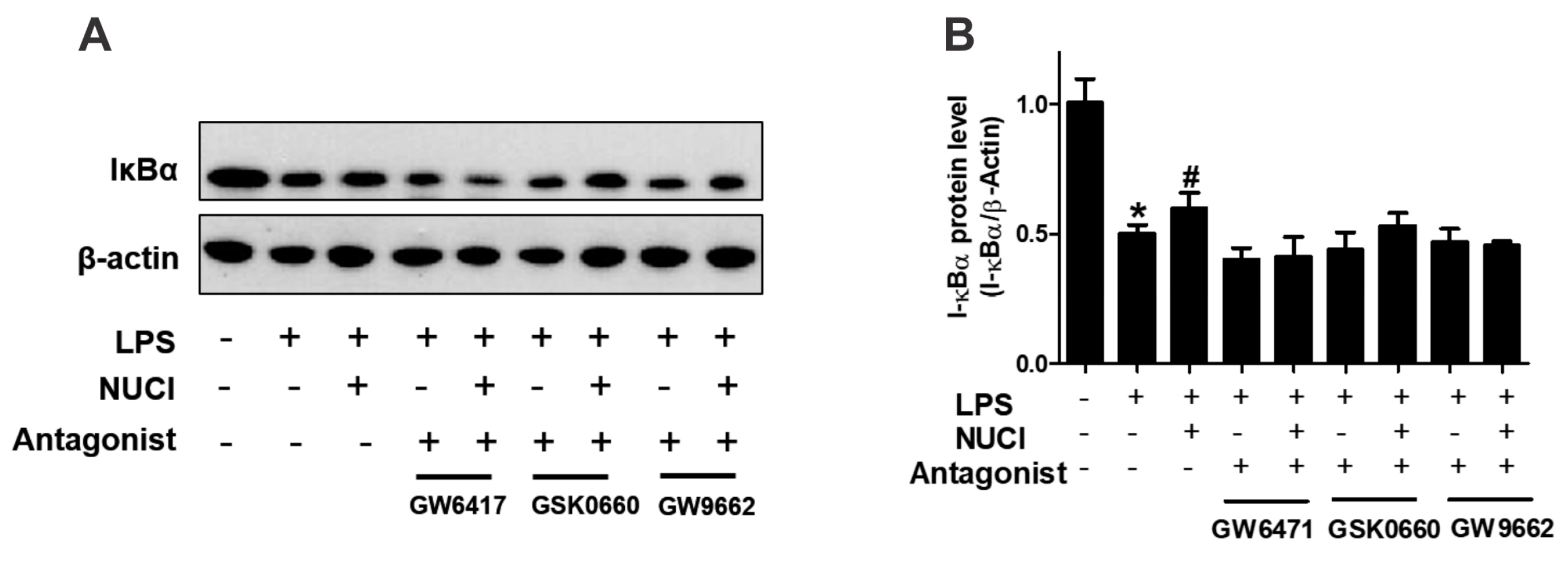
2.5. Nuciferine Decreased LPS Induced IκB-α Degradation through PPARs Activition
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cytotoxicity
4.4. IL-6 and TNFα Levels Determination
4.5. PPARs Luciferase Reporter Assay
4.6. RNA Isolation and Analysis
4.7. Immunoblotting
4.8. Data Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boteanu, R.M.; Suica, V.I.; Uyy, E.; Ivan, L.; Dima, S.O.; Popescu, I.; Simionescu, M.; Antohe, F. Alarmins in chronic noncommunicable diseases: Atherosclerosis, diabetes and cancer. J. Proteomics 2017, 153, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Pendino, K.J. Macrophages and inflammatory mediators in tissue injury. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, W.; Shi, M.; Yang, Z.; Zhang, X.; Gong, P. Docosahexaenoic acid attenuates LPS-stimulated inflammatory response by regulating the PPARgamma/NF-κB pathways in primary bovine mammary epithelial cells. Res. Vet. Sci. 2017, 112, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Flores-Bastías, O.; Karahanian, E. Neuroinflammation produced by heavy alcohol intake is due to loops of interactions between Toll-like 4 and TNF receptors, peroxisome proliferator-activated receptors and the central melanocortin system: A novel hypothesis and new therapeutic avenues. Neuropharmacology 2018, 128, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Gautam, L.N.; Adhikari, D.; Karki, R. A Comprehensive Review on Chemical Profiling of Nelumbo Nucifera: Potential for Drug Development. Phytother. Res. 2017, 31, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, J.; Zhang, D.; Du, X.; Han, L.; Lv, C.; Li, Y.; Wang, R.; Wang, B.; Huang, Y. Nuciferine and paeoniflorin can be quality markers of Tangzhiqing tablet, a Chinese traditional patent medicine, based on the qualitative, quantitative and dose-exposure-response analysis. Phytomedicine 2018, 44, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, G.; He, Y.; Xu, B.; Mi, X.; Wang, H.; Wang, Z. Pronuciferine and nuciferine inhibit lipogenesis in 3T3-L1 adipocytes by activating the AMPK signaling pathway. Life Sci. 2015, 136, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.; Ta, T.N.; Pham, T.H.; Nguyen, Q.T.; Pham, H.D.; Mishra, S.; Nyomba, B.L. Nuciferine stimulates insulin secretion from beta cells-an in vitro comparison with glibenclamide. J. Ethnopharmacol. 2012, 142, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Liu, Y.L.; Yang, Y.; Zhang, D.M.; Kong, L.D. Nuciferine restores potassium oxonate-induced hyperuricemia and kidney inflammation in mice. Eur. J. Pharmacol. 2015, 747, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Zhao, X.J.; Chen, T.Y.; Liu, Y.L.; Jiao, R.Q.; Zhang, J.H.; Ma, C.; Liu, J.H.; Pan, Y.; Kong, L.D. Nuciferine Alleviates Renal Injury by Inhibiting Inflammatory Responses in Fructose-Fed Rats. J. Agric. Food Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Munetsuna, E.; Yamada, H.; Ando, Y.; Yamazaki, M.; Taromaru, N.; Nagura, A.; Ishikawa, H.; Suzuki, K.; Teradaira, R.; et al. High fructose consumption induces DNA methylation at PPARalpha and CPT1A promoter regions in the rat liver. Biochem. Biophys. Res. Commun. 2015, 468, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kanno, Y.; Li, W.; Wakatabi, H.; Sasaki, T.; Koike, K.; Nemoto, K.; Li, H. Picrasidine N Is a Subtype-Selective PPARβ/δ Agonist. J. Nat. Prod. 2016, 79, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Zhao, S.; Yu, B.; Wang, X.; Matyal, R.; Li, Y.; Jiang, Z. High-density lipoprotein increases the uptake of oxidized low density lipoprotein via PPARgamma/CD36 pathway in inflammatory adipocytes. Int. J. Biol. Sci. 2015, 11, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Luo, Y.; Wang, D.; Li, J.; Wu, X.; Mei, W. Sodium salicylate modulates inflammatory responses through AMP-activated protein kinase activation in LPS-stimulated THP-1 cells. J. Cell. Biochem. 2018, 119, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xie, G.; Xu, Y.; Ma, L.; Tong, C.; Fan, D.; Du, F.; Yu, H. PEP-1-MsrA ameliorates inflammation and reduces atherosclerosis in apolipoprotein E deficient mice. J. Transl. Med. 2015, 13, 316. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Du, B.; Xu, B. Anti-inflammatory effects of phytochemicals from fruits, vegetables, and food legumes: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, Y.; Guo, S.; Yang, J.; Jiang, K.; Zhao, G.; Qiu, C.; Deng, G. Nuciferine Ameliorates Inflammatory Responses by Inhibiting the TLR4-Mediated Pathway in Lipopolysaccharide-Induced Acute Lung Injury. Front. Pharmacol. 2017, 8, 939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Zhang, J.G.; Wu, X.; Liu, Y.; Gu, S.Y.; Zhu, G.H.; Wang, Y.Z.; Liu, G.L.; Li, X.Y. Nuciferine downregulates Per-Arnt-Sim kinase expression during its alleviation of lipogenesis and inflammation on oleic acid-induced hepatic steatosis in HepG2 cells. Front. Pharmacol. 2015, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yi, D.D.; Guo, J.L.; Xiang, Z.X.; Deng, L.F.; He, L. Nuciferine, extracted from Nelumbo nucifera Gaertn, inhibits tumor-promoting effect of nicotine involving Wnt/β-catenin signaling in non-small cell lung cancer. J. Ethnopharmacol. 2015, 165, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, J.; Chu, H.; Zhang, X.; Wang, Z.; Wang, H. Purification and characterization of aporphine alkaloids from leaves of Nelumbo nucifera Gaertn and their effects on glucose consumption in 3T3-L1 adipocytes. Int. J. Mol. Sci. 2014, 15, 3481–3494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Deng, J.; Liu, D.; Tuo, X.; Xiao, L.; Lai, B.; Yao, Q.; Liu, J.; Yang, H.; Wang, N. Nuciferine ameliorates hepatic steatosis in high-fat diet/streptozocin-induced diabetic mice through a PPARalpha/PPARgamma coactivator-1alpha pathway. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, T.; Wortmann, A.; Schumann, T.; Finkernagel, F.; Lieber, S.; Roth, K.; Toth, P.M.; Diederich, W.E.; Nist, A.; Stiewe, T.; et al. The transcriptional PPARβ/δ network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res. 2015, 43, 5033–5051. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Rachez, C.; Freedman, L.P. Discrete roles for peroxisome proliferator-activated receptor gamma and retinoid X receptor in recruiting nuclear receptor coactivators. Mol. Cell. Biol. 2000, 20, 8008–8017. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Paumelle, R.; Caron, S.; Hennuyer, N.; Mansouri, R.; Gervois, P.; Staels, B.; Haegeman, G.; De Bosscher, K. PPARα blocks glucocorticoid receptor α-mediated transactivation but cooperates with the activated glucocorticoid receptor α for transrepression on NF-κB. Proc. Natl. Acad. Sci. USA 2009, 106, 7397–7402. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.T.; Nishiyama, K.; Matsuo, Y.; Kuwamura, M.; Morioka, A.; Nakajima, H.; Takeuchi, T. PPARalpha contributes to colonic protection in mice with DSS-induced colitis. Int. Immunopharmacol. 2010, 10, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Barish, G.D.; Atkins, A.R.; Downes, M.; Olson, P.; Chong, L.W.; Nelson, M.; Zou, Y.; Hwang, H.; Kang, H.; Curtiss, L.; et al. PPARδ regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4271–4276. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xiao, L.; Wang, N. Peroxisome proliferator-activated receptor α ligands and modulators from dietary compounds: Types, screening methods and functions. J. Diabetes 2017, 9, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rodas, M.C.; Valenzuela, R.; Echeverria, F.; Rincon-Cervera, M.A.; Espinosa, A.; Illesca, P.; Munoz, P.; Corbari, A.; Romero, N.; Gonzalez-Manan, D.; et al. Supplementation with Docosahexaenoic Acid and Extra Virgin Olive Oil Prevents Liver Steatosis Induced by a High-Fat Diet in Mice through PPAR-alpha and Nrf2 Upregulation with Concomitant SREBP-1c and NF-kB Downregulation. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Silva-Veiga, F.M.; Rachid, T.L.; de Oliveira, L.; Graus-Nunes, F.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. GW0742 (PPAR-β agonist) attenuates hepatic endoplasmic reticulum stress by improving hepatic energy metabolism in high-fat diet fed mice. Mol. Cell. Endocrinol. 2018, 474, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, P.; Kulurkar, P.; Singh, D.; Kumar, D.; Patial, V. Iridoid glycosides fraction from Picrorhiza kurroa attenuates cyclophosphamide-induced renal toxicity and peripheral neuropathy via PPAR-gamma mediated inhibition of inflammation and apoptosis. Phytomedicine 2017, 36, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Imanifooladi, A.A.; Yazdani, S.; Nourani, M.R. The role of nuclear factor-κB in inflammatory lung disease. Inflamm. Allergy-Drug Targets 2010, 9, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; Gervois, P.; Fruchart, J.C.; Staels, B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J. Biol. Chem. 2000, 275, 36703–36707. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Donato, M.T.; Gomez-Lechon, M.J. General Cytotoxicity Assessment by Means of the MTT Assay. Methods Mol. Biol. 2015, 1250, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.T.; Shakibakho, S.; So, E.Y.; Mui, A.L.F. Transfecting RAW264.7 Cells with a Luciferase Reporter Gene. J. Visualized Exp. 2015, 100. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xiao, L.; Yuan, Y.; Luo, X.; Jiang, M.; Ni, J.; Wang, N. Procyanidin B2 inhibits NLRP3 inflammasome activation in human vascular endothelial cells. Biochem. Pharmacol. 2014, 92, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nuciferine (μM) | LPS (ng/mL) | TNFα | IL-6 | ||
|---|---|---|---|---|---|
| Protein (pg/mL) | mRNA | Protein (pg/mL) | mRNA | ||
| 0 | 0 | 380.51 ± 51.27 | 0.99 ± 0.01 | 352.01 ± 60.02 | 1.01 ± 0.01 |
| 0 | 500 | 2584.46 ± 179.60 * | 1.64 ± 0.21 ** | 1663.71 ± 137.20 * | 3.34 ± 0.39 ** |
| 1 | 500 | 2315.98 ± 146.64 | 1.39 ± 0.17 | 1643.94 ± 209.69 | 2.59 ± 0.28 |
| 10 | 500 | 2139.87 ± 275.53 | 1.05 ± 0.13 # | 1216.91 ± 88.18 # | 1.92 ± 0.20 ## |
| 50 | 500 | 1772.82 ± 203.58 ## | 0.61 ± 0.11 ## | 1028.78 ± 74.61 ## | 1.85 ± 0.21 ## |
| Nuci (μM) | PPARα | PPARβ/δ | PPARγ |
|---|---|---|---|
| 0 | 1.03 ± 0.04 | 0.99 ± 0.01 | 1.02 ± 0.01 |
| 1 | 1.34 ± 0.11 * | 1.03 ± 0.19 | 1.31 ± 0.09 * |
| 10 | 1.44 ± 0.11 * | 1.13 ± 0.27 | 1.35 ± 0.12 * |
| agonist | 1.69 ± 0.06 * | 1.17 ± 0.22 * | 1.57 ± 0.13 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Deng, J.; Liu, D.; Tuo, X.; Yu, Y.; Yang, H.; Wang, N. Nuciferine Inhibits Proinflammatory Cytokines via the PPARs in LPS-Induced RAW264.7 Cells. Molecules 2018, 23, 2723. https://doi.org/10.3390/molecules23102723
Zhang C, Deng J, Liu D, Tuo X, Yu Y, Yang H, Wang N. Nuciferine Inhibits Proinflammatory Cytokines via the PPARs in LPS-Induced RAW264.7 Cells. Molecules. 2018; 23(10):2723. https://doi.org/10.3390/molecules23102723
Chicago/Turabian StyleZhang, Chao, Jianjun Deng, Dan Liu, Xingxia Tuo, Yan Yu, Haixia Yang, and Nanping Wang. 2018. "Nuciferine Inhibits Proinflammatory Cytokines via the PPARs in LPS-Induced RAW264.7 Cells" Molecules 23, no. 10: 2723. https://doi.org/10.3390/molecules23102723
APA StyleZhang, C., Deng, J., Liu, D., Tuo, X., Yu, Y., Yang, H., & Wang, N. (2018). Nuciferine Inhibits Proinflammatory Cytokines via the PPARs in LPS-Induced RAW264.7 Cells. Molecules, 23(10), 2723. https://doi.org/10.3390/molecules23102723