1. Introduction
Biological systems are mostly composed of water, and the interactions with water are a central feature of life as we know it [
1,
2,
3,
4,
5]. Solvation influences a wide variety of processes, including protein folding [
6,
7,
8,
9,
10], crystal polymorphism [
11], conformational equilibria [
12,
13,
14,
15] and even basic reaction pathways [
16]. Furthermore, water is one of the main actors in the selectivity of biochemical interactions and has a profound influence on both the kinetics and thermodynamics of protein-protein, protein-nucleic acid and protein-ligand binding [
17]. Any binding event between a ligand and a receptor in aqueous solution is first preceded by the desolvation of water molecules from the binding site and the ligand’s surface. A binding event only occurs if the ligand-receptor interactions can compensate for the loss of ligand-solvent and receptor-solvent interactions and the associated entropy changes [
18,
19,
20]. Given the fundamental importance of the solvent, no biomolecular model is adequate without properly accounting for solvation.
The free energy costs of (de-)solvation are quantified by its solvation free energy, which corresponds to the transfer free energy of the molecule from the gas phase to solution [
21,
22,
23,
24,
25]. In aqueous solution, the solvation free energy is also known as hydration free energy (
). In the molecular mechanics (MM) modeling community,
values have been an essential benchmark quantity for decades [
14,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54]. Furthermore, significant efforts have been invested in the quantum mechanical (QM) community to develop highly accurate implicit solvent methods [
55,
56,
57,
58,
59,
60,
61,
62,
63]. However, when it comes to a hybrid QM/MM approach, where a quantum mechanical solute is embedded in a classical explicit solvent, solvation free energies have received less attention because of the computational cost and complexity of sampling the solvent degrees of freedom.
Gao was a pioneer in determining QM/MM solute–solvent interaction energies for amino acid side chain analogs and nucleotide bases [
64], as well as absolute solvation free energies [
65]. This work was a milestone for QM/MM, and significant efforts have since been invested by many groups all around the world [
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80], making it an indispensable tool in computational chemistry [
81,
82,
83]. It is therefore also not surprising that QM/MM techniques have recently received increasing attention in the context of free energy calculations [
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105]. Focusing on solvation, Stanton, Hartsough and Merz used QM/MM to determine the solvation free energies of ions [
106]. Shoeib et al. studied absolute hydration free energies of ions and small solutes [
107]. Using the quasichemical theory of solutions, Asthagiri, Pratt and Kress calculated the hydration free energy of PBE water [
108], and Weber and Asthagiri provided the hydration free energy of BLYP-D water [
109]. Vapor-liquid equilibria of QM water were studied by McGrath et al. [
110]. Radial distribution functions of QM water have received the attention of multiple groups [
110,
111,
112,
113,
114,
115,
116,
117,
118], as well as the interaction energies of multimers [
119,
120,
121,
122,
123]. Relative solvation free energies were calculated by Reddy, Singh and Erion [
124,
125,
126]. Kamerlin, Haranczyk and Warshel discussed solvation free energies of acetate and methylamine in the context of
calculations [
127]. Shields, Temelso and Archer determined binding free energies of water to small water clusters [
128,
129]. More recently, we have applied QM/MM solvation free energy calculations within the framework of the SAMPL challenges [
79,
130,
131].
One of the most important shortcomings of conventional force fields is the neglect of electronic polarization. During a simulation, the charge distribution of an MM molecule cannot respond to its environment. Since polarizability is known to be important, especially in QM/MM simulations, there is major interest in the use of polarizable force fields such as the CHARMM Drude force field [
132]. Here, we perform simulations with both the CHARMM fixed charge force field and the CHARMM Drude polarizable force field, to discern the benefits and challenges of this new generation of force fields and help lay the groundwork for future development of QM/MM methods with increased predictive capability. It is of particular practical interest to ascertain the degree to which optimization of the QM/MM van der Waals interaction parameters may be needed for different QM methods, and the additional computational efforts of the Drude force field are beneficial. Our recent work [
133] analytically showed that significant additional computational costs can be justified in multi-scale free energy simulations, if the sampling method exhibits a higher phase space overlap with the target QM Hamiltonian. Thus, it can be expected that polarizable force fields and, ultimately, quantum-mechanical methods will play an increasing role in free energy calculations [
134,
135,
136,
137,
138,
139].
The remainder of this paper is organized as follows: First, we summarize the details of the model systems and simulations. Next, we present the results for the values of twelve simple solutes, using both the fixed charge and the Drude force field. Finally, we compare the performance of MP2, Hartree–Fock, several density functional methods (BLYP, B3LYP, M06-2X) and semi-empirical methods (OM2 and AM1 ) in terms of with QM/MM. This is done for both the fixed charge force field and the Drude force field. We also discuss other aspects that can have an impact on the accuracy of the results and the efficiency of the free energy simulations, including empirically scaling values, using a self-consistent optimization of the Drude particles at each step, or increasing the overlap between the MM force field and the QM target energy function by introducing a tailored MM’ force field. The Appendix includes a comparison of the convergence properties of free energy estimates based on the fixed charge and the Drude force field and also provides the detailed results of all MM free energy sub-steps.
3. Results and Discussion
Before discussing the impact of using QM/MM on the affinity for water, it is illustrative to observe the faithfulness of the solute–water interactions in pure MM. Hydration free energies have been classical benchmark systems for decades. In the CHARMM force field, the compatibility with a particular water model such as TIP3P is a centerpiece of the parameterization strategy, in particular for the charges. Thus, it is expected that the interactions with water are comparable to experiment.
Table 1 lists the hydration free energies for both the CHARMM fixed charge force field (
) and the Drude force field (
). More detailed results, listing the free energy results of the gas phase, electrostatic and van der Waals changes can be found in
Appendix B. Since each simulation was repeated four times, also the corresponding standard deviations of the results are provided. The overall metrics for agreement with experiment are listed in the last three rows. While the fixed charged force field exhibits a root mean squared deviation (RMSD) of
kcal/mol, the Drude force field reaches an RMSD of
kcal/mol. Thus, the Drude force field outperforms the fixed charge force field. Both force fields yield what is considered “chemical accuracy”, but this is most likely a reflection of the simplicity of the test set and the high level of optimization of the parameters. In terms of mean signed deviation, the Drude force field also yields a more favorable result (
kcal/mol compared to
kcal/mol). This indicates some small systematic bias of the fixed charge force field in terms of being overly hydrophobic. The correlation coefficients with the experiment are in both cases excellent (R
of
and
).
The last column of
Table 1 lists the differences between the fixed charge and the Drude force field results. While the deviations for most apolar molecules are not statistically significant, the results for water, acetamide and phenol differ by more than one kcal/mol. Furthermore, several other polar molecules exhibit a change of their
, but all changes improve the agreement with experiment. The only notable exception is cyclohexane, where the deviation from the experimental
increases from ca. half to one kcal/mol. On the other hand, the small differences for methanol and ethanol are a bit surprising.
The increased accuracy of the Drude force field comes at a price though. First, the average CPU times for the aqueous phase simulations increase by at least a factor of two due to the additional Drude and lone pair particles. Second, additional
points were required to achieve approximately the same level of precision as the fixed charge force field. This aspect is more thoroughly discussed in
Appendix A based on the
calculations. The largest differences between the fixed charge and the Drude force field are found for acetamide (
kcal/mol), phenol (
kcal/mol), aniline (
kcal/mol), benzene (
kcal/mol) and water (
kcal/mol).
The
values obtained from different QM/MM methods based on trajectories with the CHARMM fixed charge force field are presented in
Table 2. The columns are ordered based on the RMSD of the corresponding method from experimental hydration free energies, starting with the lowest RMSD on the left. The last six rows again represent the root mean squared deviation from experiment, the mean signed deviation and the Pearson correlation coefficient. RMSD, MSD and R
are given twice: once for the complete dataset (unmarked) and once for all molecules except ethanol and acetamide (marked with asterisks). The two molecules were omitted because of the high standard deviations of more than one kcal/mol in some calculations (ethanol in the case of the fixed charge force field and acetamide because of problems encountered with the Drude force field). This allows a direct comparison of the converged parts of the two datasets.
Overall, the QM/MM results with electrostatic embedding and CHARMM TIP3P water in the MM region are slightly disappointing. The RMSD vary between and kcal/mol, which is worse than the pure MM result of kcal/mol. This finding can partly be explained by the high level of optimization of the MM force field. Furthermore, the QM methods were not adapted to cancel some of the shortcomings of the TIP3P water model.
Before discussing the results in more detail, we want to validate our protocol for obtaining QM/MM hydration free energies based on the existing literature. The
values for water are in good agreement with relative free energy results between MM and QM based on QM/MM sampling with Monte Carlo simulations by Shaw, Woods and Mulholland [
181]. Table 1 of [
181] lists a free energy difference between QM water and CHARMM TIP3P water of
kcal/mol for MP2, while we obtain a difference of
kcal/mol. The discrepancies for BLYP (
versus our
kcal/mol) and HF (
versus
kcal/mol) are higher, but this can be explained by the use of different basis sets (Shaw et al. used aug-cc-pVDZ, while we employed 6-31G(d)). Furthermore, the BLYP and M06-2X
values exhibit an average deviation of
and
kcal/mol from the results published in Table 4 of [
141]. The small discrepancies can be explained by the use of rigid gas-phase geometries for the solutes in [
141] and by the high uncertainty of the ethanol result here. For B3LYP, the
values for ethane (
kcal/mol) and methanol (
kcal/mol) are in excellent agreement with previously published hydration free energy differences (
kcal/mol here compared to
kcal/mol in Table 1 of [
80] and
kcal/mol in Figure 7 of [
177]). The relatively good agreement with previously published results, in conjunction with the simplicity of the solutes and the high number of QM/MM potential energy evaluations, supports our findings.
In terms of the compatibility of different QM methods with CHARMM TIP3P water based on the RMSD from experiment, the OM2 method seems to be the best (RMSD =
kcal/mol), followed by BLYP (
kcal/mol), B3LYP (
kcal/mol), M06-2X (
kcal/mol), MP2 (
kcal/mol), AM1 (
kcal/mol) and HF (
kcal/mol). This finding agrees with the ranking by Shaw et al. based on the free energy difference between QM and MM water (BLYP < MP2 < HF) [
181]. To some degree, it is surprising that the semi-empirical method OM2 and the pure functional BLYP clearly outperform more advanced QM methods. As discussed in Section IV E and Table S14 of [
141], the QM/MM electrostatics become more attractive as the amount of Hartree–Fock exchange increases from BLYP to B3LYP to M06-2X to HF/MP2. With fixed QM/MM van der Waals interactions, the hydration free energies become more negative. The MSD are
kcal/mol for BLYP,
kcal/mol for B3LYP,
kcal/mol for M06-2X and
kcal/mol for Hartree–Fock. Thus, the QM/MM results are significantly too hydrophilic. Although the CHARMM charges are based on Hartree–Fock calculations [
145], the results imply that Hartree–Fock itself is not particularly suited for QM/MM simulations, due to the large systematic bias in favor of solute–solvent interactions. However, since the QM/MM
values are highly correlated with the experimental data, it is possible to address this shortcoming by scaling the interactions. This is illustrated in
Appendix C.
The
values obtained from different QM/MM methods based on trajectories with the CHARMM Drude force field are presented in
Table 3. The columns are again ordered based on the RMSD of the corresponding method from experimental
values, starting with the lowest RMSD on the left. Except for the two semi-empirical methods, the rank order of the QM methods based on RMSD is actually inverted, with Hartree–Fock (RMSD =
kcal/mol) followed by MP2 (
kcal/mol), M06-2X (
kcal/mol), B3LYP (
kcal/mol) and BLYP (
kcal/mol). However, the RMSD is not a reliable measure here, since the acetamide results are far from converged, with standard deviations between
and
kcal/mol. As more thoroughly discussed in a recent paper, high standard deviations in multi-scale free energy simulations can be an indicator that the MM energy minimum is very far away from the QM energy minimum [
133]. Indeed, when comparing the optimal C–C bond length of acetamide of MM (
Å for the types CD201A and CD33C) with, e.g., the bond length of an energy optimized structure with OM2 (
Å), there is a clear discrepancy of
Å, which leads to substantial energy differences. Given that the equilibrium bond lengths of C–C bonds are typically between
and
Å in the CHARMM force field, this is a clear indication for a typo in the Drude parameter file for acetamide. A further investigation is in progress.
Ignoring the flawed acetamide results and focusing on the metrics marked with an asterisk, the overall results of most methods (except BLYP and AM1) are surprisingly similar, with RMSD * between
and
kcal/mol and MSD * between mere
and
kcal/mol. While the RMSD * are a little bit higher than the best results for the CHARMM TIP3P water model (RMSD * between
and
kcal/mol), the consistency between most methods and the low systematic errors can be regarded as a sign of better compatibility with QM/MM methods. Given that the development of polarizable Drude force fields is still in its early stages, one can still expect some improvements in the future. The AM1 semi-empirical method is among the most inaccurate methods in the test set, with RMSD of
kcal/mol for the fixed charge model and
kcal/mol for the Drude model. In the light of such results, it is somewhat surprising that the popular AM1-BCC method to determine MM charges [
182,
183], which builds upon AM1, is as effective as it is when it comes to hydration free energies [
44].
Another aspect that can influence the accuracy of the Drude oscillator model is the use of the extended Lagrangian formalism [
184], in which Drude particles have a mass and kinetic energy. This implies that the particles do not necessarily reside at the energy minimum at each step. Also in our QM/MM energy evaluations, the Drude particles in the MM region were not relaxed in response to the QM wave function. To evaluate the effect of relaxing the Drude particles, five steps of conjugate gradient energy minimization were performed with QM/MM after an MM SCF optimization of the Drude particles. The resulting hydration free energies for Hartree–Fock with the extended Lagrangian approach (HF-EL) and based on the self-consistent optimization of the Drude particles (HF-SCOD) are shown in
Table 4. While the overall agreement with experiment in terms of the RMSD does not change significantly with the use of self-consistent Drude particles (RMSD of
and
kcal/mol), the solvent affinity increases in all cases (as it should). For the Hartree–Fock calculations, this leads to a lower systematic error in terms of MSD of a mere
kcal/mol (instead of
kcal/mol). The average change of
kcal/mol is lower than the average standard deviation of ca.
kcal/mol, so most differences here are not statistically significant.
Because the convergence of some of the QM/MM
results was not satisfactory, we also explored the possibility to improve this situation by employing a tailored force field (denoted as MM’). By adopting bonded terms that match more closely the bond lengths and angles encountered in the target QM method, the phase space overlap is supposed to be increased, which also improves the convergence of the free energy calculation between MM and QM [
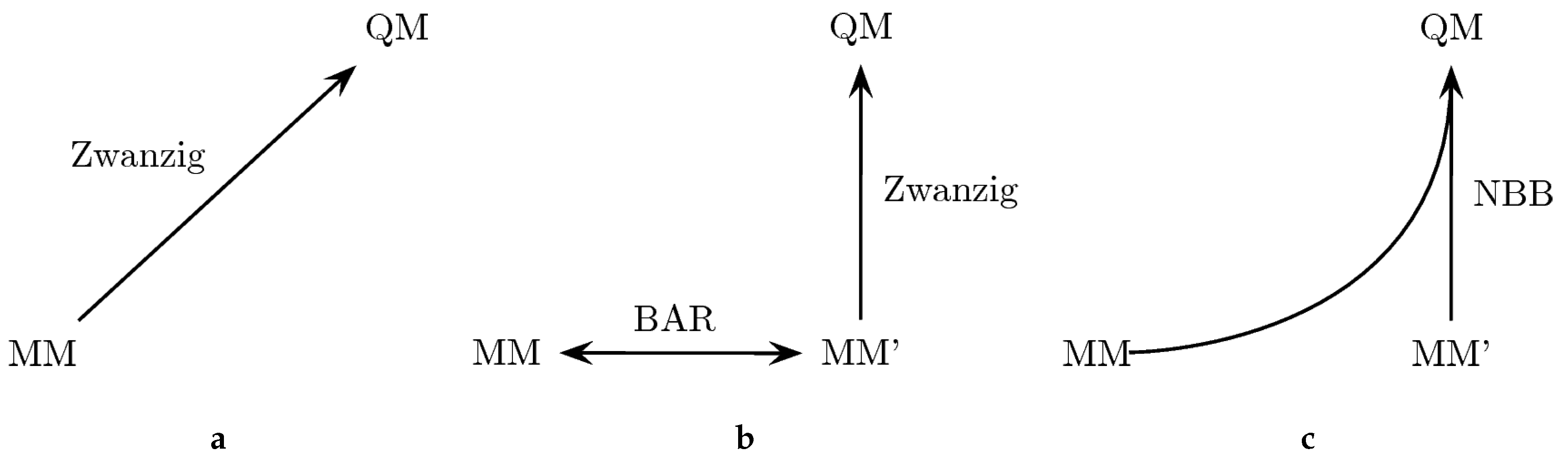
133]. The approach is outlined in
Figure 2. In particular, we explored three different ways to perform the “bookend” corrections: (a) using the Zwanzig equation [
178] to directly calculate the free energy difference between the MM force field and the QM Hamiltonian; (b) generating an MM’ tailored force field with optimized parameters to increase the phase space overlap with the QM Hamiltonian; the free energy difference between the original force field and the tailored force field can be calculated with Bennett’s acceptance ratio method (BAR) [
161], while the free energy difference between the modified MM’ force field and the QM state is calculated with the Zwanzig equation; (c) combining all the potential energy data from MM, MM’ and QM with the Non-Boltzmann–Bennett equation [
79,
80,
180].
A comparison of the results of the three theoretically equivalent approaches is given in
Table 5. The third column (MM→QM) reflects the
values from direct free energy calculations between MM and QM energy surfaces using the Zwanzig equation. In principle, the results here should correspond to those in the third column of
Table 2 (OM2). However, since different trajectories and setups were employed, one can expect some small discrepancies. The overall RMSD (
compared to
kcal/mol) and MSD (
versus
kcal/mol) are similar compared to
Table 2, which serves as another verification of the approach. The third column of
Table 5 shows the results obtained using the tailored MM’ force field to calculate the free energy difference to the QM state. The
values of Columns 2 and 3 should also match within the corresponding uncertainties, since the end points are the same. Indeed, except for aniline, the differences between the two columns are below 0.2–0.3 kcal/mol, which also corresponds to the average standard deviation of the results (shown in the last line). Importantly, the average standard deviation is a little bit lower for the MM→MM’→QM transformation, due to the increased phase space overlap between the MM’ and the QM state. The last column shows the result of an NBB calculation that combines the potential energy data of the two transformations in an optimal way. The fact that the NBB results are almost identical to the MM→MM’→QM transformation further indicates that there is more phase space overlap between the MM’ and QM state, thus dominating the NBB calculation. However, the overall improvement is rather small, which signifies that the original bonded parameters were already well optimized.
4. Conclusions
In this work, we computed hydration free energies for twelve simple solutes to determine an effective choice of QM method to use in combination with explicit solvent. Here, we focused on the fixed charge CHARMM TIP3P and the polarizable SWM4 water model in the CHARMM force field. As a reference, we first provided hydration free energies based on pure MM simulations. Both the fixed charge (RMSD = kcal/mol) and the Drude force field simulations (RMSD = kcal/mol) exhibit excellent agreement with the experimental data and are well converged with respect to conformational sampling.
For QM/MM hydration free energy calculations based on the CHARMM CGenFF fixed charge force field, the best results were obtained with the OM2 semi-empirical method (RMSD =
kcal/mol) and the BLYP method (RMSD =
kcal/mol). The other methods (B3LYP, M06-2X, MP2, AM1 and Hartree–Fock) yielded RMSD between
and
kcal/mol. This ranking of QM methods agrees with the previous observation that the systematic error of hydration free energies of QM/MM methods with CHARMM TIP3P water increases systematically with the amount of Hartree–Fock exchange [
141]. Therefore, we recommend using either OM2 or BLYP for QM/MM simulations in aqueous solution with CHARMM TIP3P water. This QM/MM protocol was also successfully applied to the calculation of distribution coefficients in SAMPL5 [
130], which reflects the change from a hydrophilic to a hydrophobic environment.
As for the QM/MM hydration free energy calculations based on the CHARMM Drude force field, the best results were obtained with the OM2 semi-empirical method (RMSD = kcal/mol). However, the ranking of the other methods is nearly reversed, with Hartree–Fock (RMSD = kcal/mol) outperforming MP2, M06-2X, B3LYP, BLYP and AM1. The MP2, M06-2X and Hartree–Fock methods perform slightly better with the Drude force field in terms of RMSD, and their systematic error is significantly lower. Thus, if a potential bias from the solute–solvent interactions is a concern, it might be advisable to employ the Drude force field for QM/MM simulations with those methods. However, the performance of QM/MM with the Drude force field is only marginally better. Furthermore, the Drude accuracy between the extended-Lagrangian (EL) and self-consistent optimization implementations is statistically indistinguishable, but can slightly affect the systematic bias.
Overall, the OM2 semi-empirical method shows the best performance for both datasets with RMSD of and kcal/mol, while the AM1 semi-empirical method exhibits the worst performance with RMSD of and kcal/mol. The PM3 semi-empirical method was omitted in the manuscript because of its RMSD of and kcal/mol, further demonstrating the high variability in the quality of semi-empirical methods. However, both the accuracy and robustness of the OM2 hydration free energy results are very encouraging, especially since the OM2 parametrization did not include solvation free energies. This makes the method suitable for improving the quality of MM free energy predictions via post-processing, as OM2 can be applied to thousands of snapshots within mere minutes on a commodity laptop.
Our results also corroborate the conclusions of a recent study by Ganguly, Boulanger and Thiel [
185]. The effect of MM polarization via Drude particles on QM/MM hydration free energies is only moderate compared to the well-developed CHARMM fixed charge force field. Fixed charge force fields are well tested, faster and more robust than the recently developed polarizable force fields. Therefore, they will most likely continue to play a significant role in computational chemistry. While polarization is a highly relevant physical effect, Drude force fields still neglect other important factors such as charge penetration, coupling of polarization with many-body exchange, dispersion and charge transfer [
186,
187,
188]. In addition, the impact of Drude point charges in proximity to the QM region is still unclear at this point.
The force field parameters (e.g., the van der Waals parameters) will likely have to be adapted according to the target QM function. Thus, some form of tailored MM’ force field will be beneficial for future applications of QM/MM in multi-scale free energy simulations. The need for improvement is highlighted by the systematic errors of QM/MM in the kcal/mol range, as well as the clear superiority of the MM results compared to QM/MM. Our results show that spending computer power to add all the right physics to the QM region in a QM/MM simulation will be in vain if the MM description of the solvent environment is not compatible with the QM description of the solute.


 ,
, 

{kind=link}
{kind=link}
{kind=link}

