Synthesis and Application of Scaffolds of Chitosan-Graphene Oxide by the Freeze-Drying Method for Tissue Regeneration

 ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. CS Extraction from the Mycelium of A. niger
2.2. Deacetylation Degree and Characterization of CS by 1H-NMR
2.3. Characterization of CS by FTIR Spectroscopy
2.4. Molecular Weight Determination of CS
2.5. Synthesis of the GO
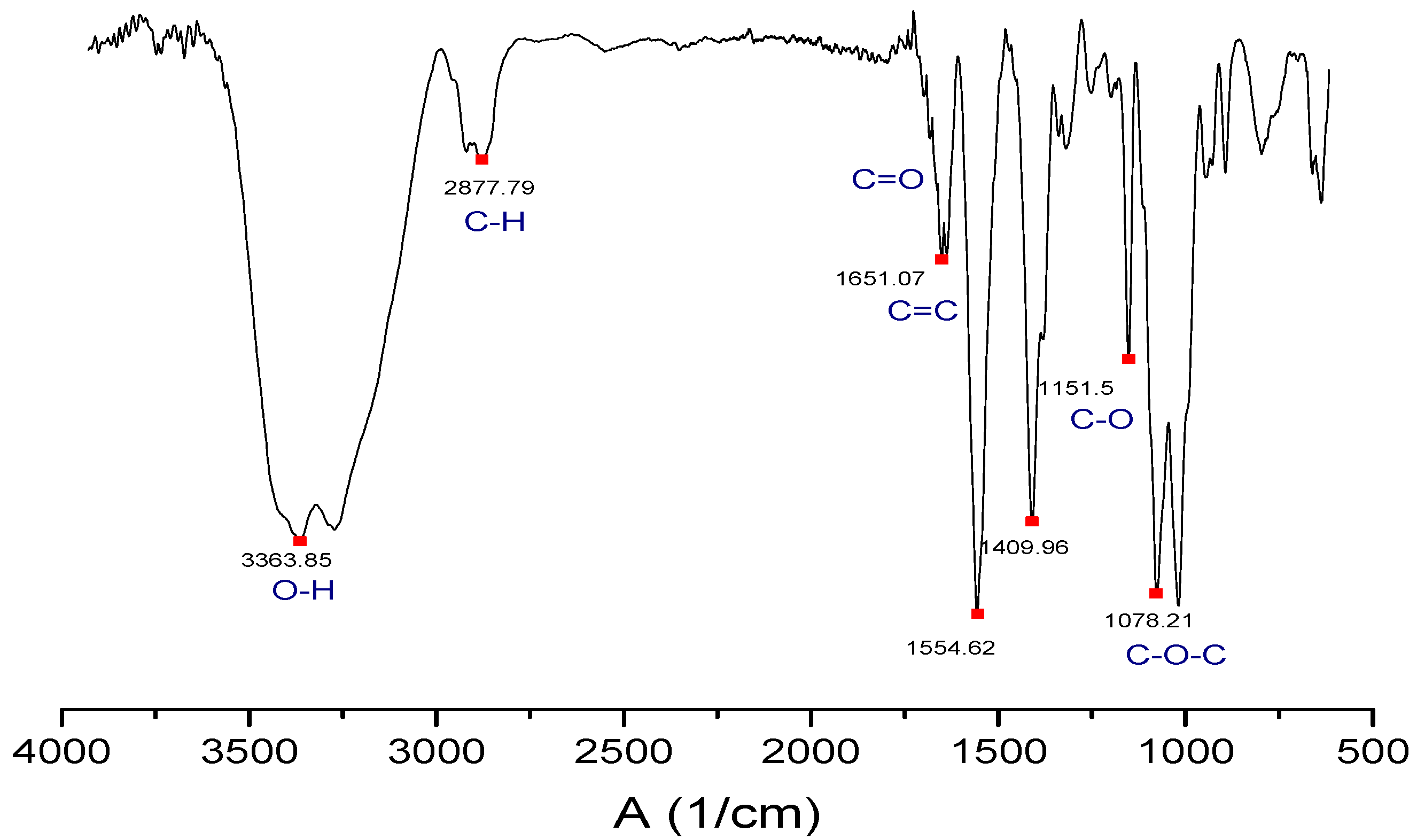
2.6. Characterization of the GO by FTIR Spectroscopy
2.7. Characterization of the GO by Raman Spectroscopy
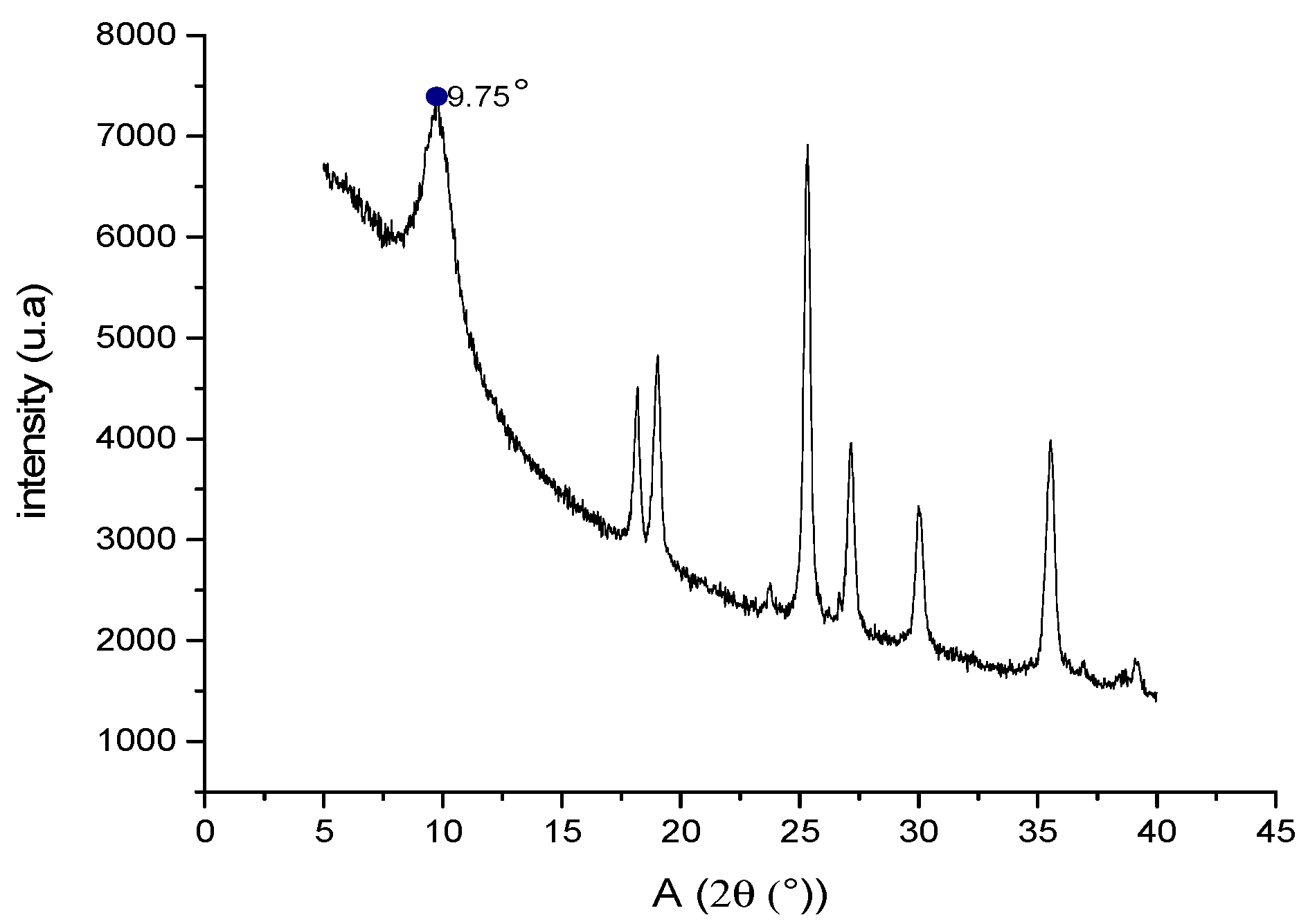
2.8. Characterization of GO by X-ray Diffraction (XRD)
2.9. Atomic Force Microscopy (AFM) Studies of GO
2.10. Particle Size Study of GO by Dynamic Light Scattering
2.11. Preparation of CS/GO Scaffolds by Freeze-drying Method
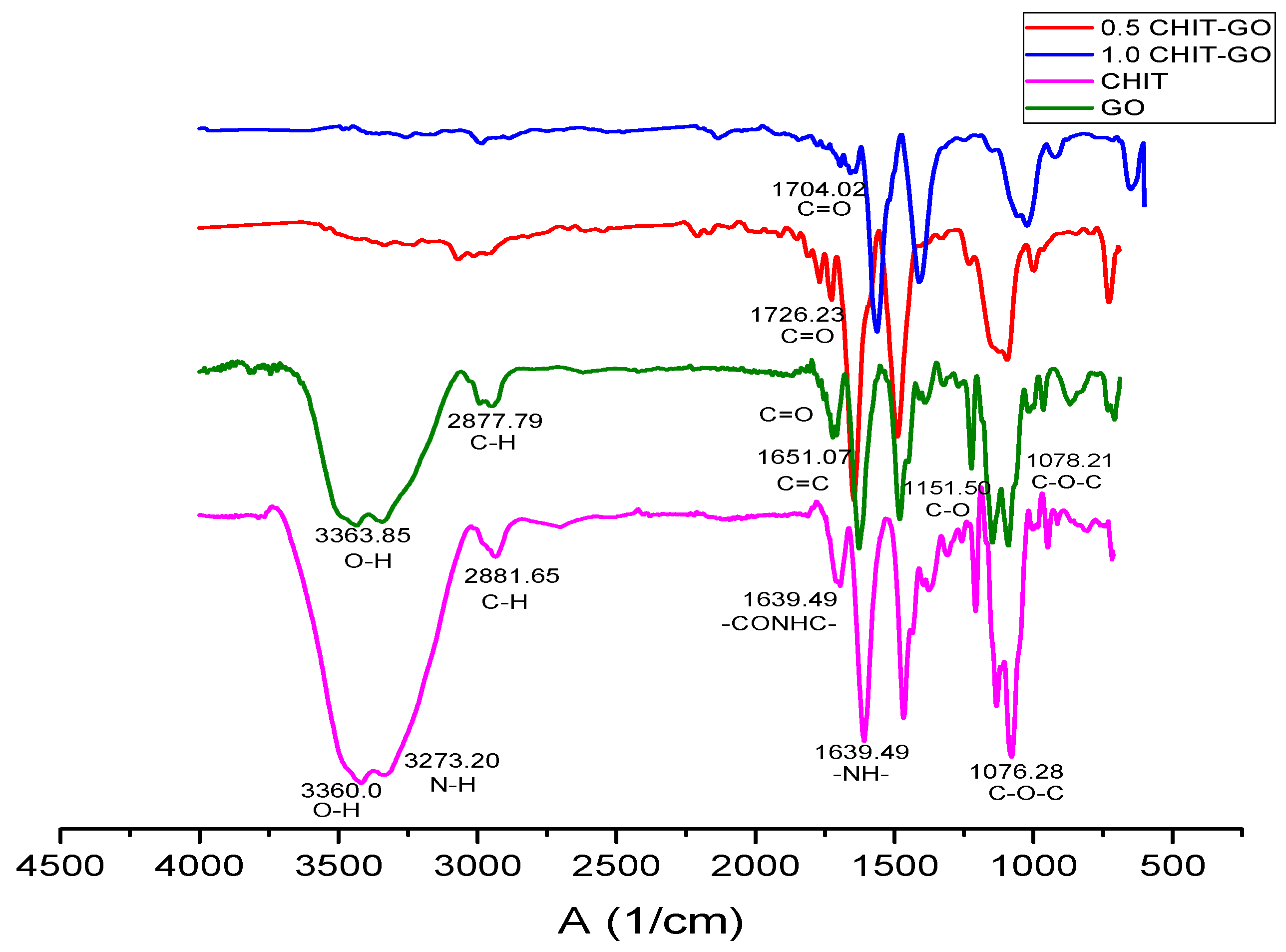
2.12. Characterization of Scaffolds by FTIR Spectroscopy
2.13. Scanning Electron Microscopy (SEM) of Scaffolds
2.14. Thermogravimetric Analysis of Scaffolds
2.15. Degradability Test of Scaffolds on Physiological Serum
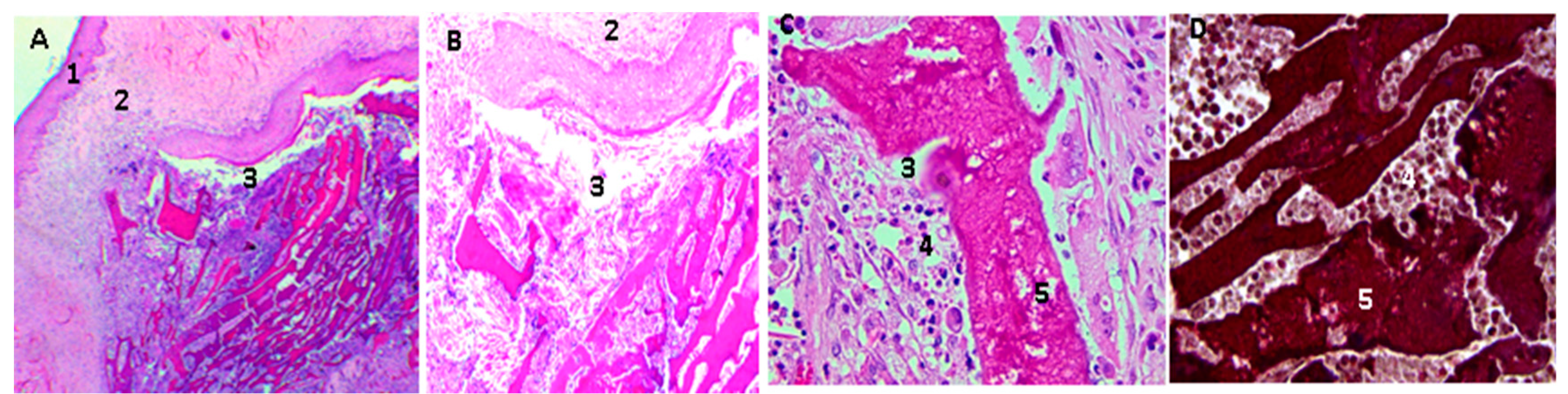
2.16. Biological Tests
3. Experimental
3.1. Materials
3.2. Synthesis
3.2.1. Extraction and Purification of CS
3.2.2. GO Synthesis
3.3.3. CS/GO Scaffold Preparation
3.3. Characterization
3.4. In Vivo Compatibility Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ho-Shui-Ling, A.; Bolander, J.; Rustom, L.E.; Johnson, A.W.; Luyten, F.P.; Picart, C. Bone regeneration strategies: Engineered scaffolds, bioactive molecules and stem cells current stage and future perspectives. Biomaterials 2018, 180, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Bray, C.C.; Walker, C.M.; Spence, D.D. Orthobiologics in Pediatric Sports Medicine. Orthop. Clin. N. Am. 2017, 48, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Barrena, E.; Rosset, P.; Lozano, D.; Stanovici, J.; Ermthaller, C.; Gerbhard, F. Bone fracture healing: Cell therapy in delayed unions and nonunions. Bone 2015, 70, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, N.; Liu, B.; Song, L.; Wen, B.; Yang, D. Aligned Porous Chitosan/graphene Oxide Scaffold for Bone Tissue Engineering. Mater. Lett. 2018, 233, 78–81. [Google Scholar] [CrossRef]
- Kerch, G. Polymer Hydration and Stiffness at Biointerfaces and Related Cellular Processes. Nanomedicine-Nanotechnology. Biol. Med. 2018, 14, 13–25. [Google Scholar]
- Yaylaci, S.U.; Sen, M.; Bulut, O.; Arslan, E.; Guler, M.O.; Tekinay, A.B. Chondrogenic Differentiation of Mesenchymal Stem Cells on Glycosaminoglycan-Mimetic Peptide Nano Fibers. ACS Biomater. Sci. Eng. 2016, 2, 871–878. [Google Scholar] [CrossRef]
- ASTM F2027-16, Standard Guide for Characterization and Testing of Raw or Starting Materials for Tissue-Engineered Medical Products; ASTM International: West Conshohocken, PA, USA, 2016.
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D Bioactive Composite Scaffolds for Bone Tissue Engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef] [PubMed]
- Hermenean, A.; Codreanu, A.; Herman, H.; Balta, C.; Rosu, M.; Mihali, C.V.; Ivan, A.; Dinescu, S.; Ionita, M.; Costache, M. Chitosan-Graphene Oxide 3D Scaffolds as Promising Tools for Bone Regeneration in Critical-Size Mouse Calvarial Defects. Sci. Rep. 2017, 7, 16641. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.F.; Billups, W.E. Synthesis of Soluble Graphite and Graphene. Acc. Chem. Res. 2013, 46, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.W.; Piner, R.D.; An, J.; Ruoff, R.S. Mechanical Properties of Monolayer Graphene Oxide. ACS Nano 2010, 4, 6557–6564. [Google Scholar] [CrossRef] [PubMed]
- Grande, C.; Mangadlao, J.; Fan, J.; De Leon, A.; Delgado-Ospina, J.; Rojas, J.; Rodrigues, D.; Advincula, R. Chitosan Cross-Linked Graphene Oxide Nanocomposite Films with Antimicrobial Activity for Application in Food Industry. Symp. Macromol. 2017, 374, 1600114. [Google Scholar] [CrossRef]
- Fan, J.; Grande, C.D.; Rodrigues, D.F. Biodegradation of Graphene Oxide-Polymer Nanocomposite Films in Wastewater. Environ. Sci. Nano 2017, 4, 1808–1816. [Google Scholar] [CrossRef]
- Pan, Y.; Wu, T.; Bao, H.; Li, L. Green Fabrication of Chitosan Films Reinforced with Parallel Aligned Graphene Oxide. Carbohydr. Polym. 2011, 83, 1908–1915. [Google Scholar] [CrossRef]
- Motamedian, S.R.; Hosseinpour, S.; Ahsaie, M.G.; Khojasteh, A. Smart Scaffolds in Bone Tissue Engineering: A Systematic Review of Literature. World J. Stem Cells 2015, 7, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Kurmirska, J.; Czerwicka, M.; Kaczynski, Z.; Bychowska, A.; Brzozowski, K.; Thoming, J.; Stepnowski, P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drug 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [PubMed]
- Correa, L.S.; Godoy, J.E.; Zuluaga, F.; Valencia, C. Elaboración de Andamios Porosos Osteoinductivos de Poli (Ácido l-Láctico)/Quitosano Para La Regeneración de Tejido Óseo. Rev. Col. Mat. 2015, 6, 34–53. [Google Scholar]
- Sadighara, P.; Moghadam, H.T.; Eskandari, S.; Salehi, A. Optimization of Extraction of Chitosan and Carotenoids from Shrimp Waste. Int. J. Fish. Aquat. Stud. 2015, 2, 51–54. [Google Scholar]
- Murillo, W.; Dávila, L.R. Glucanos En Hongos Superiores, Polisacaridos Con Un Interesante Espectro de Actividad Biológica. Química e Ind. 2017, 29, 25–30. [Google Scholar]
- Trevor, A.; Katzung, B. Toxicology. In Pharmacology: Examination and Board Review, 11th ed.; McGraw Hill: San Francisco, CA, USA, 2015; p. 495. ISBN 0071789235. [Google Scholar]
- Wang, H.; Qian, C.; Roman, M. Effects of pH and Salt Concentration on the Formation and Properties of Chitosan-Cellulose Nanocrystal Polyelectrolyte—Macroion Complexes. Biomacromolecules 2011, 12, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, N.; Jackson, T.L.; Elsaid, Z.; Alqathama, A.; Somavarapu, S. PLGA Microparticles Entrapping Chitosan-Based Nanoparticles for the Ocular Delivery of Ranibizumab. Mol. Pharm. 2016, 13, 2923–2940. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, F.; Gómez, F. Introducción: Antecedentes históricos. In Introducción a La Química de Polímeros, 3rd ed.; Programa Editorial Universidad del Valle: Cali, Colombia, 2015; pp. 26–30. ISBN 978-958-765-622-9. [Google Scholar]
- Novoa-carballal, R.; Fernandez-megia, E.; Riguera, R. Dynamics of Chitosan by 1H NMR Relaxation. Biomacromolecules 2010, 110, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.; Whiteley, C.G.; Lee, C. Hydrophobically Modified Chitosan-Grafted Magnetic Nanoparticles for Bacteria Removal. Ind. Eng. Chem. 2015, 54, 9270–9277. [Google Scholar] [CrossRef]
- USP-NF35, Official Monographs/Chitosan GPC; The United States Pharmacopeial Convention (USP): Rockville, MD, USA, 2017; pp. 7601–7605.
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Losic, D. From Graphene Oxide to Reduced Graphene Oxide: Impact on the Physiochemical and Mechanical Properties of Graphene—Cement Composites. Appl Mater. Interfaces. 2017, 9, 43275–43286. [Google Scholar]
- Lo, D.; Lo, M.; Garc, J.L.; Vela, M.M. Evolution of the Raman Spectrum with the Chemical Composition of Graphene Oxide. J. Phys. Chem. 2017, 121, 20489–20497. [Google Scholar]
- Shen, J.; Hu, Y.; Shi, M.; Lu, X.; Qin, C.; Li, C.; Ye, M. Fast and Facile Preparation of Graphene Oxide and Reduced Graphene Oxide Nanoplatelets. Chem. Mater. 2009, 21, 3514–3520. [Google Scholar] [CrossRef]
- Mohandesa, F.; Salavati-Niasari, M. Freeze-Drying Synthesis, Characterization and in Vitro Bioactivity of Chitosan/graphene Oxide/hydroxyapatite Nanocomposite. RSC Adv. 2014, 4, 25993–26001. [Google Scholar] [CrossRef]
- Zuluaga, F.; Yates, B.; Insuasty, B. Aplicaciones de la espectroscopia de infrarrojo al análisis orgánico cualitativo. In Análisis Orgánico Clásico Y Espectral, 3rd ed.; Programa Editorial Universidad del Valle: Cali, Colombia, 2015; p. 103. ISBN 978-958-670-167-9. [Google Scholar]
- Zhang, P.; Wu, H.; Wu, H.; Zhongwen, L.; Jing, X.; Chen, X. RGD-Conjugated Copolymer Incorporated into Composite of Poly (Lactide-Co-Glycotide) and Poly (l-Lactide)-Grafted Nanohydroxyapatite for Bone Tissue Engineering. Biomacromolecules 2011, 12, 2667–2680. [Google Scholar] [CrossRef] [PubMed]
- Depan, D.; Girase, B.; Shah, J.S.; Misra, R.D.K. Structure-process-property Relationship of the Polar Graphene Oxide-Mediated Cellular Response and Stimulated Growth of Osteoblasts on Hybrid Chitosan Network Structure Nanocomposite Scaffolds. Acta Biomater. 2011, 7, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.F.B.; Pinto, R.J.B.; Serra, V.I.R.C.V.; Silvestre, A.J.D.; Trindade, T.; Grac, M.; Neves, P.M.S.; Cavaleiro, J.A.S.; Daina, S.; Sadocco, P.; et al. Fluorescent Bioactive Corrole Grafted-Chitosan Films. Biomacromol. 2016, 17, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Yan, L. Supramolecular Hydrogel of Chitosan in the Presence of Graphene Oxide Nanosheets as 2D Cross-Linkers. ACS Sustain. Chem. Eng. 2014, 2, 296–300. [Google Scholar] [CrossRef]
- Thangavel, P.; Kannan, R.; Ramachandran, B.; Moorthy, G.; Suguna, L. Development of Reduced Graphene Oxide (rGO)-Isabgol Nanocomposite Dressings for Enhanced Vascularization and Accelerated Wound Healing in Normal and Diabetic Rats. J. Colloid Interface Sci. 2018, 517, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Awaja, F.; Tripathi, M.; Coraça-huber, D.; Speranza, G. Biocompatibility of Different Graphene Oxide Coatings on Polymers. Materialia 2018, 2, 9–18. [Google Scholar] [CrossRef]
- Cruz, E.A.; Morales, S.G.; Arce, S.; Valencia, C.H. Biocompatibilidad Del Fosfato Tricálcico Con Quitosano Para Su Uso En Regeneración Ósea. Univ. Odontol. 2015, 34, 109–116. [Google Scholar]
- Figueiredo, A.; Coimbra, P.; Cabrita, A.; Guerra, F.; Figueiredo, M. Comparison of a Xenogeneic and an Alloplastic Material Used in Dental Implants in Terms of Physico-Chemical Characteristics and in Vivo in Fl Ammatory Response. Mater. Sci Eng. 2013, 33, 3506–3513. [Google Scholar] [CrossRef] [PubMed]
- Broon, J.N.; Martinex, P.; Ramirez, G.L.; Tinajero, C.; Lgunas, L.H. Inflammatory Response of Bioceramic Material to Dentin Tube Implantation in Rats’ Subcutanous Tissue. Rev. Odon Mex. 2016, 20, e170–e174. [Google Scholar]
- Balanta, D.J.; Zuluaga, F.; Valencia, C.H. Evaluation of Biocompatibility of Chitosan Films from the Mycelium of Aspergillus Niger in Connective Tissue of Rattus Norvegicus. J. Mol. Genet. Med. 2015, 9, 1000174. [Google Scholar]
- José, M.; Brochado, J.; Helena, M.; Margarida, M. In Situ Forming Chitosan Hydrogels: Preliminary Evaluation of the in Vivo in Fl Ammatory Response. Mater. Sci. Eng. 2017, 75, 279–285. [Google Scholar]
- Katsumiti, A.; Tomovska, R.; Cajaraville, M.P. Intracellular Localization and Toxicity of Graphene Oxide and Reduced Graphene Oxide Nanoplatelets to Mussel Hemocytes in Vitro. Aquat. Toxicol. 2017, 188, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, G.; Agati, M.D.; Mahmud, A.; Sitharaman, B. Toxicology of Graphene-Based Nanomaterials. Adv. Drug Deliv. Rev. 2016, 105, 109–144. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencia, C.; Valencia, C.H.; Zuluaga, F.; Valencia, M.E.; Mina, J.H.; Grande-Tovar, C.D. Synthesis and Application of Scaffolds of Chitosan-Graphene Oxide by the Freeze-Drying Method for Tissue Regeneration. Molecules 2018, 23, 2651. https://doi.org/10.3390/molecules23102651
Valencia C, Valencia CH, Zuluaga F, Valencia ME, Mina JH, Grande-Tovar CD. Synthesis and Application of Scaffolds of Chitosan-Graphene Oxide by the Freeze-Drying Method for Tissue Regeneration. Molecules. 2018; 23(10):2651. https://doi.org/10.3390/molecules23102651
Chicago/Turabian StyleValencia, Cesar, Carlos H. Valencia, Fabio Zuluaga, Mayra E. Valencia, José H. Mina, and Carlos David Grande-Tovar. 2018. "Synthesis and Application of Scaffolds of Chitosan-Graphene Oxide by the Freeze-Drying Method for Tissue Regeneration" Molecules 23, no. 10: 2651. https://doi.org/10.3390/molecules23102651
APA StyleValencia, C., Valencia, C. H., Zuluaga, F., Valencia, M. E., Mina, J. H., & Grande-Tovar, C. D. (2018). Synthesis and Application of Scaffolds of Chitosan-Graphene Oxide by the Freeze-Drying Method for Tissue Regeneration. Molecules, 23(10), 2651. https://doi.org/10.3390/molecules23102651