Enzyme-Site Blocking Combined with Optimization of Molecular Docking for Efficient Discovery of Potential Tyrosinase Specific Inhibitors from Puerariae lobatae Radix


Abstract
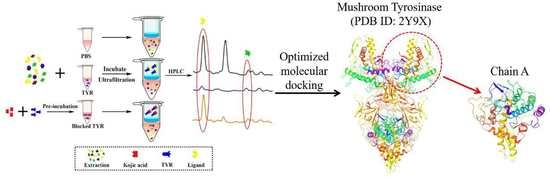
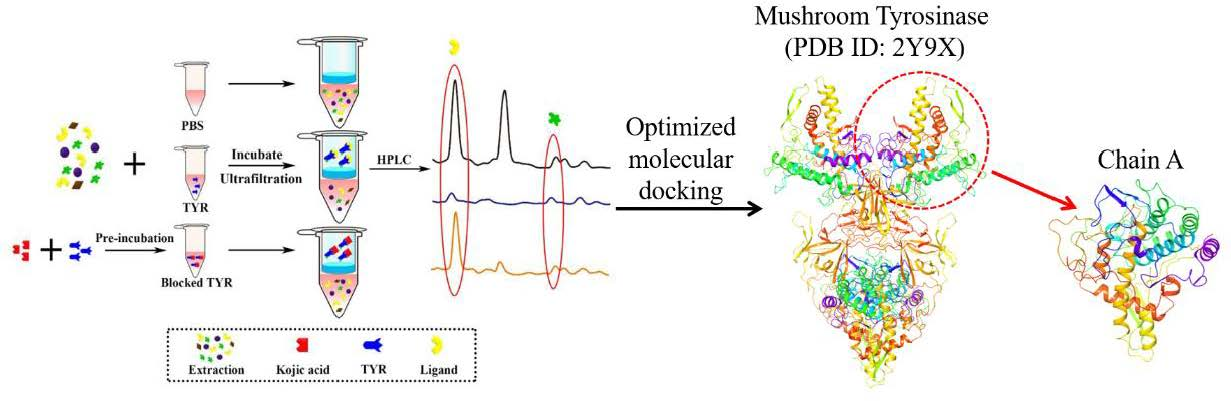
1. Introduction
2. Results and Discussions
2.1. TYR-Inhibitory Activity of the P. lobatae Radix Extract
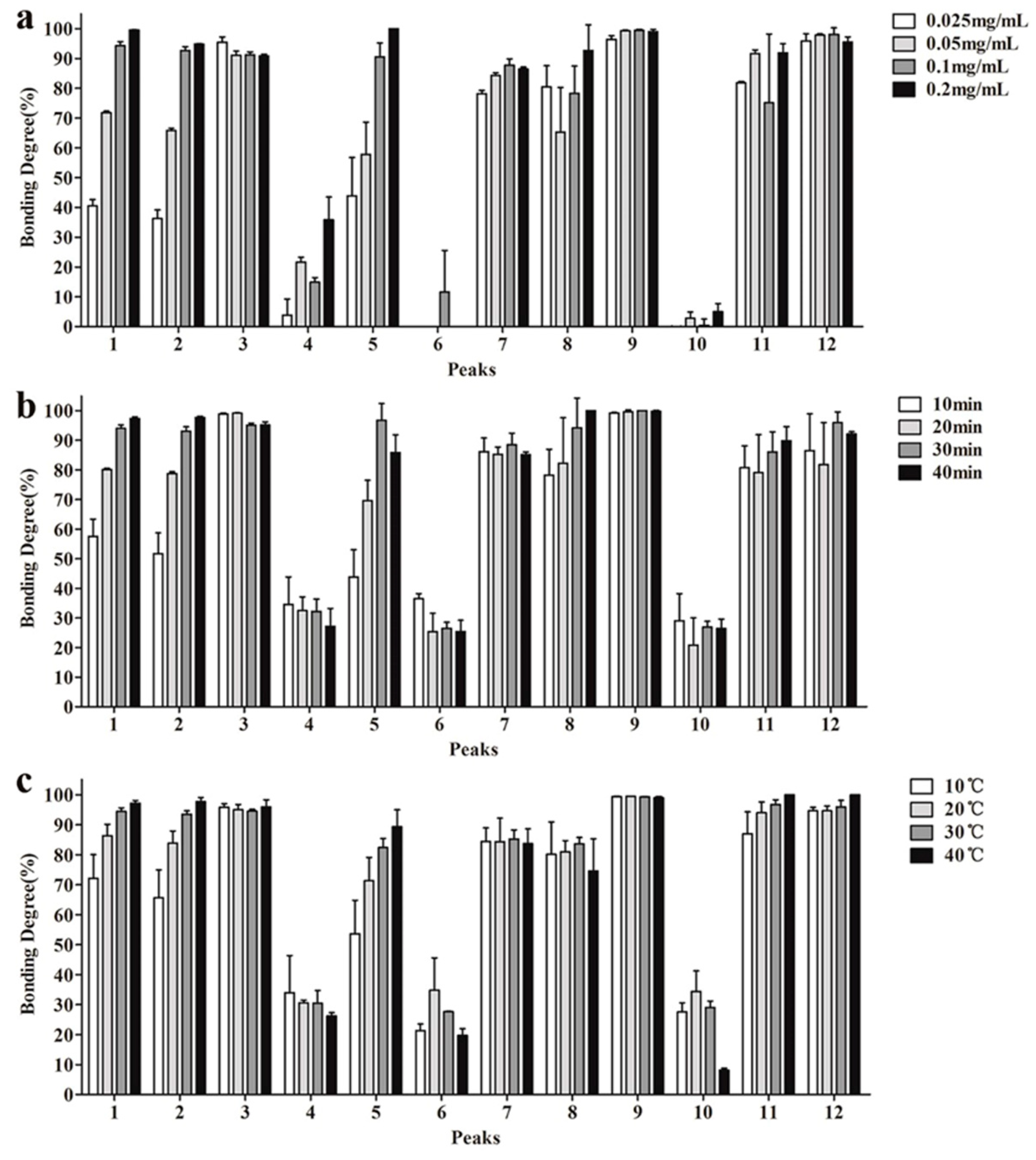
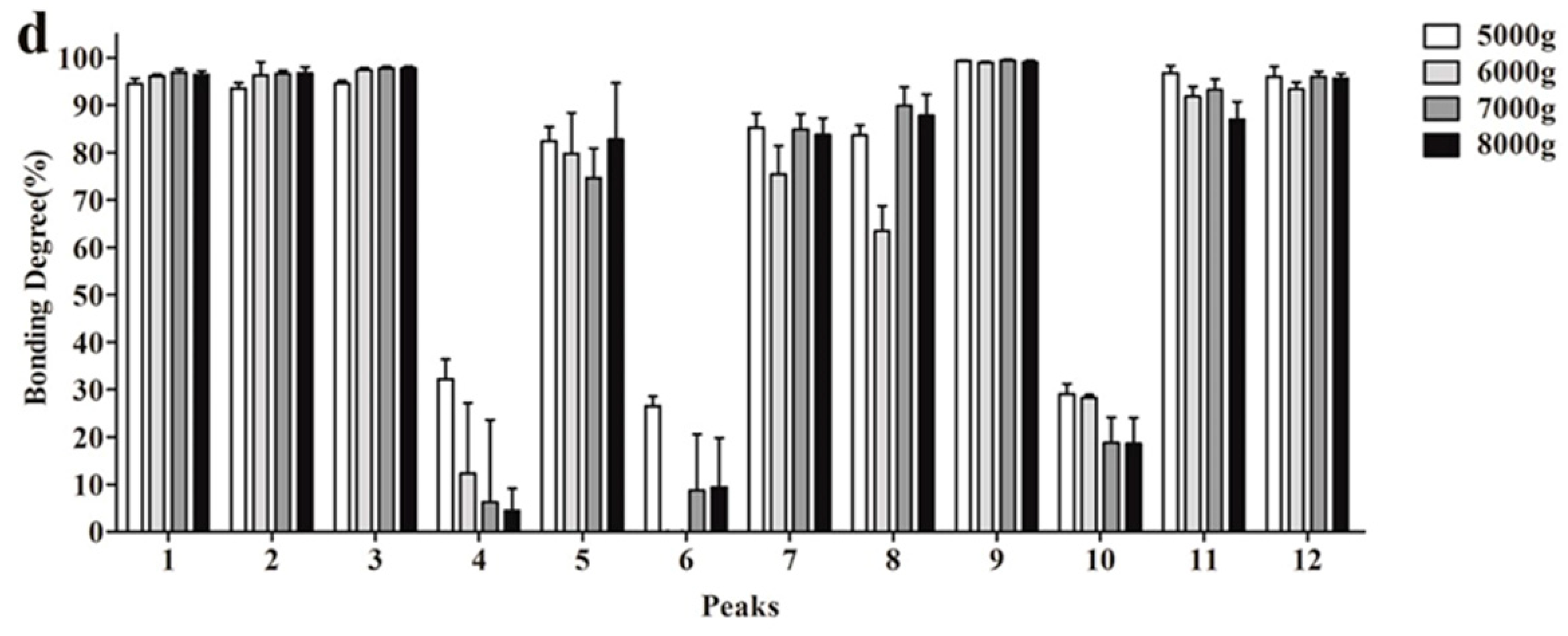
2.2. Optimization of UF Screening Parameters
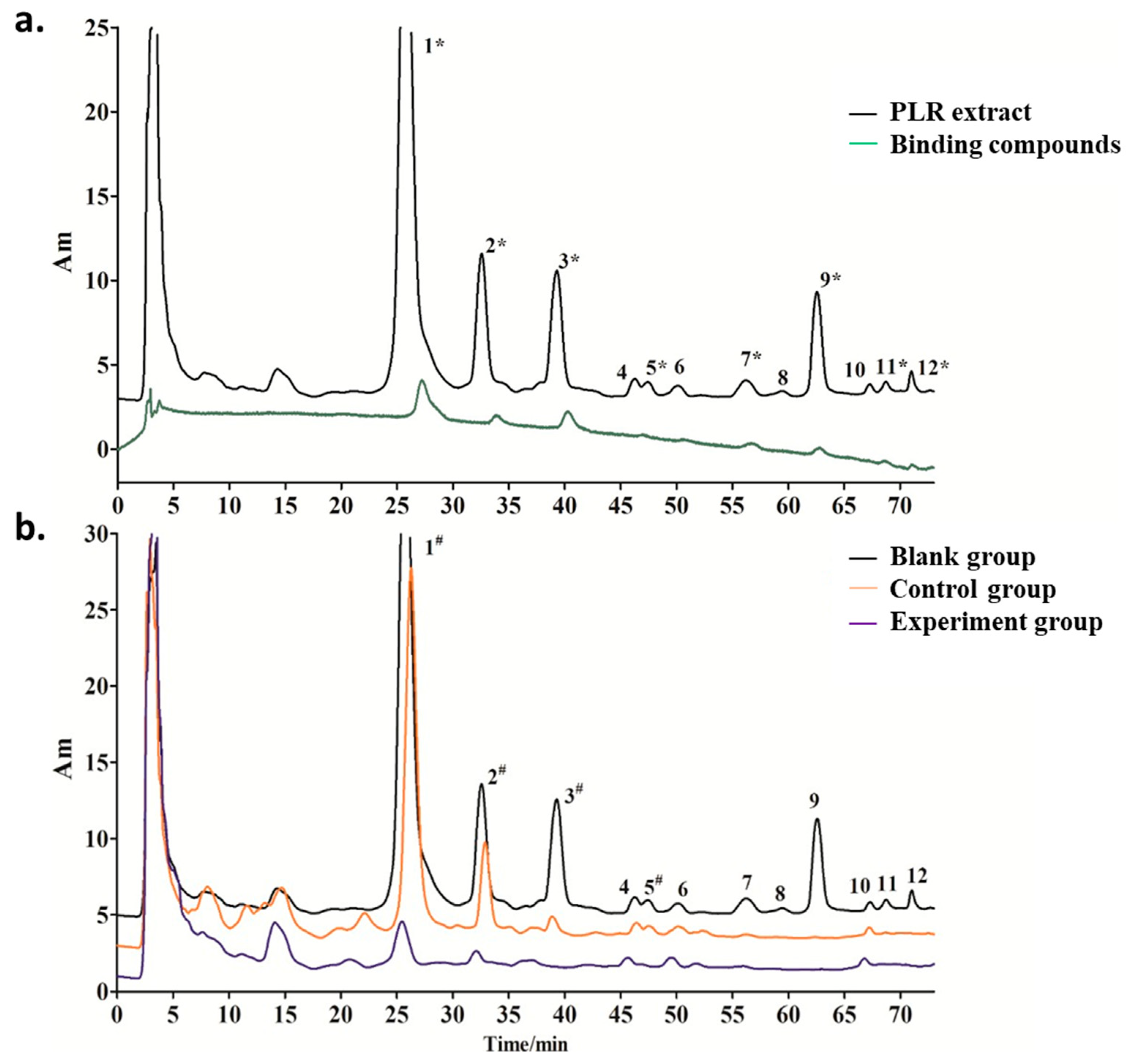
2.3. Screening of TYR Inhibitors from PLR Extract by UF-HPLC
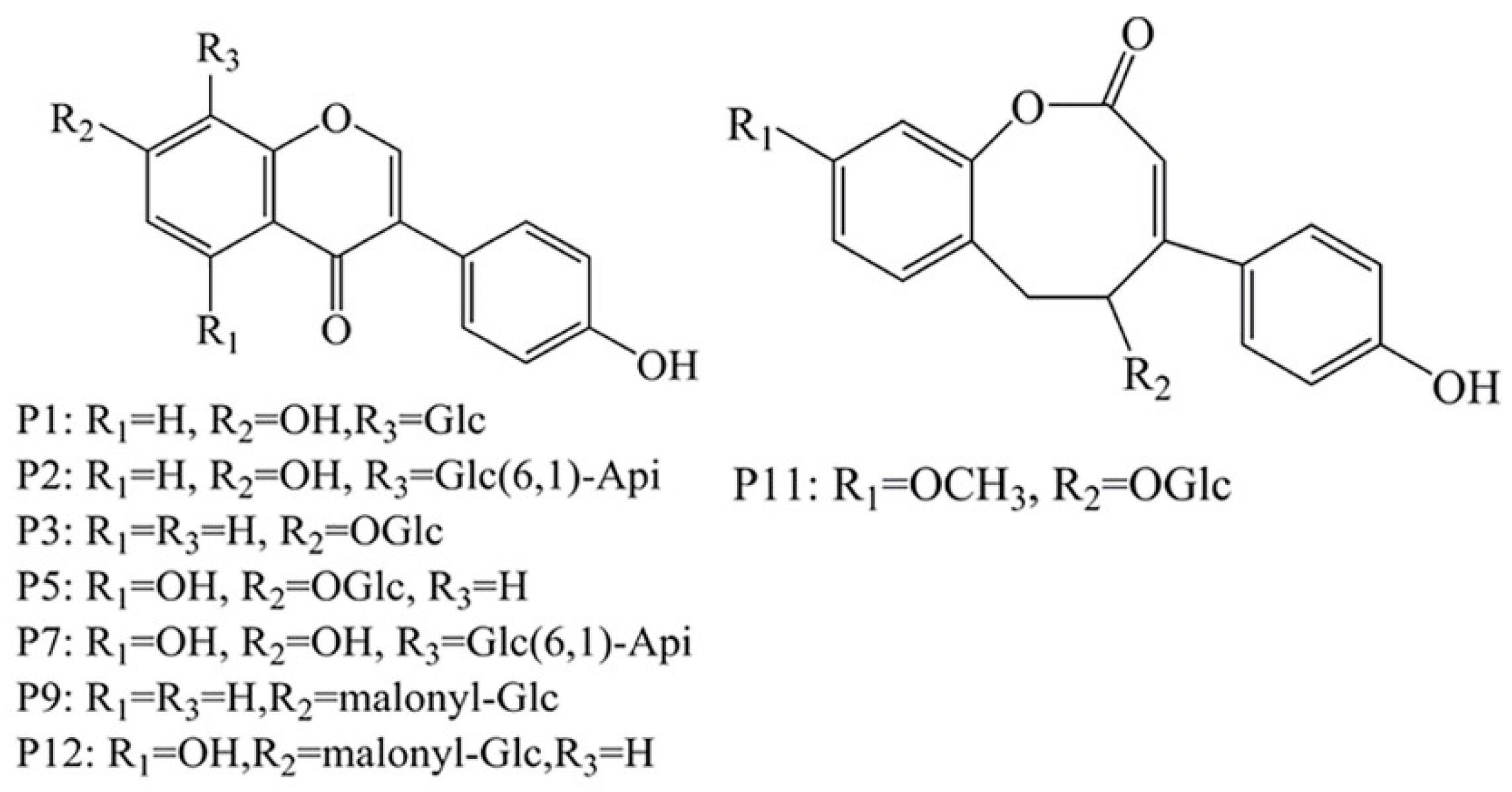
2.4. HPLC-Q-TOF MS/MS to Identify the Selected Compounds
2.5. Comparison of Four Docking Program
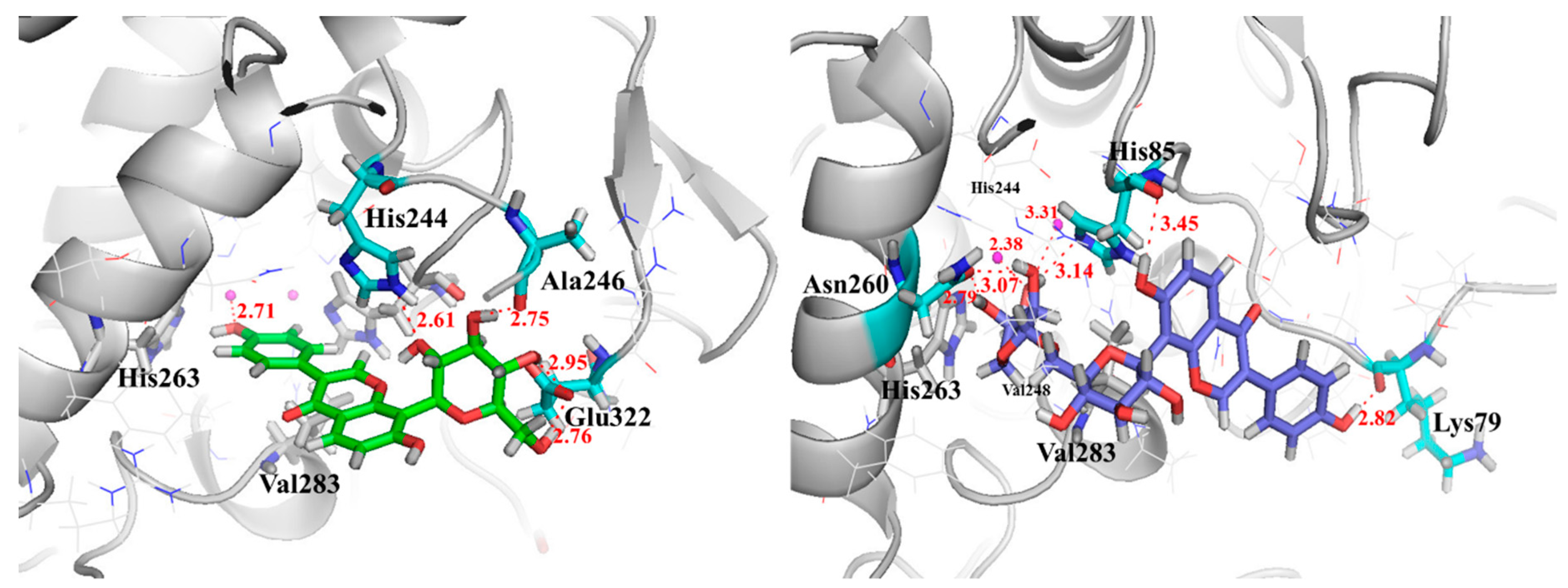
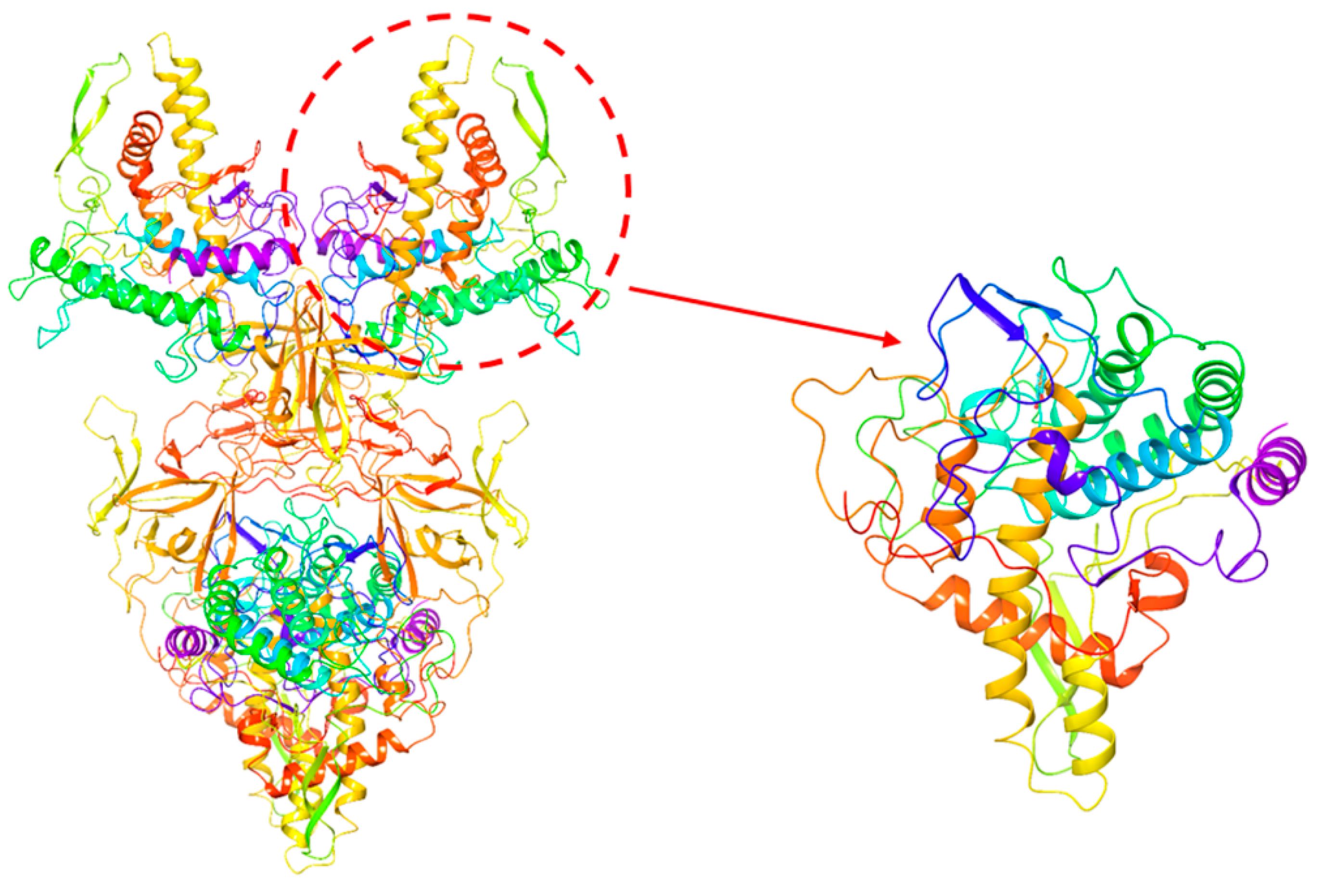
2.6. Investigation of Enzyme-Ligand Interactions by Molecular Docking
2.7. Validation of TYR Inhibitory Activity
3. Materials and Methods
3.1. Apparatus
3.2. Reagents and Materials
3.3. Preparation of Extraction and Stock Solutions
3.4. Investigation of the UF Parameters
3.5. TYR-Affinity UF
3.6. TYR-Site Blocking UF Screening
3.7. HPLC Q-TOF MS/MS Analysis
3.8. Molecular Docking
3.9. TYR Inhibit Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ito, S.; Wakamatsu, K. Chemistry of Mixed Melanogenesis & mdash; Pivotal Roles of Dopaquinone & dagger. Photochem. Photobiol 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.W.; Jiang, Z.S. Advances in studies on botanical inhibitors of tyrosinase. Chin. Tradit. Herb. Drugs 2004, 6, 702–705. [Google Scholar] [CrossRef]
- Parvez, S.; Kang, M.; Chung, H.S.; Bae, H. Naturally occurring tyrosinase inhibitors: Mechanism and applications in skin health, cosmetics and agriculture industries. Phytother. Res. 2010, 21, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.; Yaqiu, L. Advances in Small-Molecule Inhibitors of Protein Tyrosine Kinases. Chin. J. Org. Chem. 2011, 31, 1596–1606. [Google Scholar] [CrossRef]
- Mu, Y.; Li, L.; Hu, S.Q. Molecular inhibitory mechanism of tricin on tyrosinase. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 107, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Baek, N.; Nam, T.G. Natural, semisynthetic and synthetic tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Park, K.C. Current clinical use of depigmenting agents. Dermatologica Sinica 2014, 32, 205–210. [Google Scholar] [CrossRef]
- Yin, L.B.; Xia, Q.L.; Zhao, L.Z.; Zhang, C.F. Research progress of pharmacological effects of puerarin. Mod. Agric. Sci. Technol. 2016, 4, 68–69, 75. [Google Scholar] [CrossRef]
- Wong, K.H.; Li, G.Q.; Li, K.M.; Razmovskinaumovski, V.; Chan, K. Kudzu root: Traditional uses and potential medicinal benefits in diabetes and cardiovascular diseases. J. Ethnopharmacol. 2011, 134, 584–607. [Google Scholar] [CrossRef] [PubMed]
- Dorni, A.I.C.; Amalraj, A.; Gopi, S.; Varma, K.; Anjana, S.N. Novel cosmeceuticals from plants—An industry guided review. J. Appl. Res. Med. Aromat. Plants 2017, 7, 1–26. [Google Scholar] [CrossRef]
- Zhong, Q.Q.; Cao, G.Q.; Lu, B.L.; Liang, L.L. Inhibitory effect of active constituents from radix puerariae on tyrosinase. Nat. Prod. Res. Dev. 2008, 4, 681–684. [Google Scholar] [CrossRef]
- Zhu, J.; Yi, X.; Peng, H.; Chen, S.; Wu, Y. Drug-protein binding of Danhong injection and the potential influence of drug combination with aspirin: Insight by ultrafiltration LC–MS and molecular modeling. J. Pharm. Biomed. Anal. 2017, 134, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, S.; Tang, Y.; Liu, C.; Chen, L.; Zhang, Y. Ultrafiltration-LC–MS combined with semi-preparative HPLC for the simultaneous screening and isolation of lactate dehydrogenase inhibitors from Belamcanda chinensis. J. Sep. Sci. 2016, 39, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
- Song, H.P.; Zhang, H.; Fu, Y.; Mo, H.Y.; Zhang, M.; Chen, J.; Li, P. Screening for selective inhibitors of xanthine oxidase from Flos Chrysanthemum using ultrafiltration LC-MS combined with enzyme channel blocking. J. Chromatogr. B 2014, 961, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zhang, X.; Xin, T.; Wu, G. Pharmaceutical applications of affinity-ultrafiltration mass spectrometry: Recent advances and future prospects. J. Pharm. Biomed. Anal. 2016, 131, 444–453. [Google Scholar] [CrossRef]
- Song, H.P.; Chen, J.; Hong, J.-Y.; Hao, H.; Qi, L.-W.; Lu, J.; Fu, Y.; Wu, B.; Yang, H.; Li, P. A strategy for screening of high-quality enzyme inhibitors from herbal medicines based on ultrafiltration LC-MS and in silico molecular docking. Chem. Commun. 2014, 51, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964. [Google Scholar] [CrossRef] [PubMed]
- Klingaman, C.A.; Wagner, M.J.; Brown, J.R.; Klecker, J.B.; Pauley, E.H.; Noldner, C.J.; Mays, J.R. HPLC-based kinetics assay facilitates analysis of systems with multiple reaction products and thermal enzyme denaturation. Anal. Biochem. 2017, 516, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chai, C.Z.; Wang, D.W.; Yue, X.Y.; Zhu, D.N.; Yu, B.Y. HPLC-DAD-Q-TOF-MS/MS analysis and HPLC quantitation of chemical constituents in traditional Chinese medicinal formula Ge-Gen Decoction. J. Pharm. Biomed. Anal. 2013, 80, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Reppert, A.; Yousef, G.G.; Rogers, R.B.; Lila, M.A. Isolation of radiolabeled isoflavones from kudzu (Pueraria lobata) root cultures. J. Agric. Food Chem. 2008, 56, 7860–7865. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.R.; Kim, S.J.; Ham, S.H.; Cho, J.H.; Lee, Y.B.; Cho, H.Y. Simultaneous determination of puerarin and its active metabolite in human plasma by UPLC-MS/MS: Application to a pharmacokinetic study. J. Chromatogr. B 2014, 971, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, H.Y.; Zhang, Q.W.; Li, G.H.; Yang, F.Q.; Wang, Y.; Li, Y.C.; Wang, Y.T. A rapid method for simultaneous determination of 14 phenolic compounds in Radix Puerariae using microwave-assisted extraction and ultra high performance liquid chromatography coupled with diode array detection and time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.C.; Mun, G.S. Dynamics of phytoestrogen, isoflavonoids and its isolation from stems of Pueraria lobata (Willd.) Ohwi growing in Democratic People’s Republic of Korea. J. Food Drug Anal. 2015, 23, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Li, H.; Dong, J.; Wang, H.; Hashi, Y.; Chen, S. Identification of isoflavonoids in Radix Puerariae for quality control using on-line high performance liquid chromatography-diode array detector-electrospray ionization-mass spectrometry coupled with post-column derivatization. Food Res. Int. 2012, 48, 528–537. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, F.Q.; Li, C.H.; Zhang, Q.; Xia, Z.N.; Yang, F.Q. Determination of eight isoflavones in Radix Puerariae by capillary zone electrophoresis with an ionic liquid as an additive. Anal. Methods 2015, 7, 1098–1103. [Google Scholar] [CrossRef]
- Zheng, Z.P.; Lan, S.; Qin, C.; Yuan, K.; Chen, J. Research progress in natural tyrosinase inhibitors: Types and structure-activity relationships. Sci. Technol. Food Ind. 2014, 35, 374–377. [Google Scholar] [CrossRef]
- Yang, H.; Han, K.; Wan, X.C.; Fang, C.B. Free radical scavenging capacities of isoflavonoid glycosides isolated from Pueraria Radix. J. Anhui Agric. Univ. 2011, 38, 151–155. [Google Scholar]
- SYBYL, version 7.1; Tripos Inc.: St. Louis, MO, USA, 2005.
- Maestro; Version 9.0; Schrödinger, LLC.: New York, NY, USA, 2011.
- GOLD; Version 4.0; Astex Technology: Cambridge, MA, USA, 2001.
- Discovery, Studio; Accelrys Inc.: San Diego, CA, USA, 2008.
Sample Availability: Samples of the compounds are available from commercial sources indicated in the methods section of the manuscript. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformation | RMSD (Å) | |||
|---|---|---|---|---|
| Glide | Gold | Cdocker | LibDock | |
| 1 | 1.2840 | 1.6428 | 1.4536 | 3.1159 |
| 2 | 1.2699 | 1.5603 | 1.3451 | 2.9661 |
| 3 | 1.4672 | 1.6947 | 1.3515 | 2.9537 |
| Peak | Compound | Score | Energy | IC50 ± SD (μM) |
|---|---|---|---|---|
| 1 | Puerarin | −6.21 | −53.34 | 9.83 ± 1.45 |
| 2 | Mirificin | −6.04 | −51.65 | 12.66 ± 2.77 |
| 3 | Daidzin | −5.17 | −40.65 | >500 |
| 5 | Genistin | −5.48 | −42.43 | >500 |
| control group | Kojic acid | −5.87 | −47.31 | 46.91 ± 3.21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zhu, Y.; Wang, T.; Qi, J.; Liu, X. Enzyme-Site Blocking Combined with Optimization of Molecular Docking for Efficient Discovery of Potential Tyrosinase Specific Inhibitors from Puerariae lobatae Radix. Molecules 2018, 23, 2612. https://doi.org/10.3390/molecules23102612
Liu H, Zhu Y, Wang T, Qi J, Liu X. Enzyme-Site Blocking Combined with Optimization of Molecular Docking for Efficient Discovery of Potential Tyrosinase Specific Inhibitors from Puerariae lobatae Radix. Molecules. 2018; 23(10):2612. https://doi.org/10.3390/molecules23102612
Chicago/Turabian StyleLiu, Haichun, Yitian Zhu, Ting Wang, Jin Qi, and Xuming Liu. 2018. "Enzyme-Site Blocking Combined with Optimization of Molecular Docking for Efficient Discovery of Potential Tyrosinase Specific Inhibitors from Puerariae lobatae Radix" Molecules 23, no. 10: 2612. https://doi.org/10.3390/molecules23102612
APA StyleLiu, H., Zhu, Y., Wang, T., Qi, J., & Liu, X. (2018). Enzyme-Site Blocking Combined with Optimization of Molecular Docking for Efficient Discovery of Potential Tyrosinase Specific Inhibitors from Puerariae lobatae Radix. Molecules, 23(10), 2612. https://doi.org/10.3390/molecules23102612