Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities

Abstract

1. Introduction
2. Results
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compounds 1 and 2 to Determine the Absolute Configuration of the Monosaccharides
3.5. Anti-Inflammatory Activity Assay Against NO Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, H. Search the origin of Araceae by ecological geography. Acta Bot. Yunnanica 1996, 18, 14–42. [Google Scholar]
- El-Desouky, S.K.; Kim, K.H.; Ryu, S.Y.; Eweas, A.F.; Gamal-Eldeen, A.M.; Kim, Y.K. A new pyrrole alkaloid isolated from Arum palaestinum Boiss. and its biological activities. Arch. Pharm. Res. 2007, 30, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Yi, X.M.; Feng, J.Y.; Wang, Y.H.; He, X.J. Piperidine alkaloids from Alocasiamacrorrhiza. Phytochemistry 2017, 143, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.R.; Bliss, B.J.; Suzuki, J.Y.; Borris, R.P. Chemotaxonomy of Hawaiian Anthurium cultivars based on multivariate analysis of phenolic metabolites. J. Agric. Food Chem. 2014, 62, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Roshan, R.; Ahmed, S.; Hasan, M.M. Arisaema jacquemontii Blume (Araceae): A review of medicinal uses, phytochemistry and pharmacology. J. Pharmacogn. Phytochem. 2017, 6, 429–432. [Google Scholar]
- Wu, Y.Y.; Huang, X.X.; Zhang, M.Y.; Zhou, L.; Li, D.Q.; Cheng, Z.Y.; Li, L.Z.; Peng, Y.; Song, S.J. Chemical constituents from the tubers of Pinellia ternate (Araceae) and their chemotaxonomic interest. Biochem. Syst. Ecol. 2015, 62, 236–240. [Google Scholar] [CrossRef]
- Li, D.D.; Fang, M.L.; Liu, Q.; Li, Q.L.; Gao, Y.T. Study on the extraction of polyphenol from Scindapsus officinalis with ultrasonic wave technology optimized by central composite design-response surface method. J. Chin. Mater. Med. 2011, 34, 129–133. [Google Scholar]
- Yu, J.Q.; Song, X.Y.; Wang, D.J.; Wang, X.Y.; Wang, X. Five new chromone glycosides from Scindapsus officinalis (Roxb.) Schott. Fitoterapia 2017, 122, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Q.; Song, X.Y.; Yang, P.; Wang, X.Y.; Wang, X. Alkaloids from Scindapsus officinalis (Roxb.) Schott. and their biological activities. Fitoterapia 2018, 129, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Jia, X.C.; Luo, Q.W.; Zhang, Q.; Luo, B.; Liu, W.B.; Zhang, X.; Xu, Q.L.; Tan, J.W. Phenolics fromMikaniamicrantha and their antioxidant activity. Molecules 2017, 22, 1140. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, K.; Fujii, M.; Ida, Y.; Akita, H. Simple synthesis of β-d-Glycopyranosides using β-Glycosidase from almonds. Chem. Pharm. Bull. 2004, 52, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.C.; Wang, Y.; Yu, C.M.; Yu, D.H.; Chen, P.P.; Liu, S.M. Two new saccharide fatty acid esters from the fruit of Morindacitrifolia L. and their ABTS radical scavenging activities. Rec. Nat. Prod. 2014, 8, 25–31. [Google Scholar]
- Marzouk, M.S.; Moharram, F.A.; El Dib, R.A.; El-Shenawy, S.M.; Tawfike, A.F. Polyphenolic profile and bioactivity study of Oenotheraspeciosa Nutt. aerial parts. Molecules 2009, 14, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.Y.; Hu, L.H. Study on the chemical constituents of Daphniphyllum angustifolium. Helv. Chim. Acta 2006, 89, 884–894. [Google Scholar] [CrossRef]
- Wang, H.B.; Nair, M.G.; Strasbur, G.M.; Booren, A.M.; Gray, J.I. Novel antioxidant compounds from Tart Cherries (Prunuscerasus). J. Nat. Prod. 1999, 62, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Syntheses of optically active grasshopper ketone and dehydrovomifoliol as a synthetic support for the revised absolute configuration of (+)-abscisic acid. Tetrahedron 1974, 30, 1065–1072. [Google Scholar] [CrossRef]
- Della, G.M.; Marino, C.D.; Zarrelli, A. Isolation and phytotoxicity of apocarotenoids from Chenopodium album. J. Nat. Prod. 2004, 67, 1492–1495. [Google Scholar]
- Chae, H.S.; Yoo, H.; Kim, Y.M.; Choi, Y.H.; Lee, C.H.; Chin, Y.W. Anti-Inflammatory Effects of 6,8-Diprenyl-7,4′-dihydroxyflavanone from Sophoratonkinensis on Lipopolysaccharide-Stimulated RAW 264.7 Cells. Molecules 2016, 21, 1049. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (1–3) are available from the authors. |

 C) correlations of 1–3 (left), and key NOESY (H
C) correlations of 1–3 (left), and key NOESY (H H) correlations of 1 and 3 (right).
C) correlations of 1–3 (left), and key NOESY (HH) correlations of 1 and 3 (right).
H) correlations of 1 and 3 (right).
C) correlations of 1–3 (left), and key NOESY (HH) correlations of 1 and 3 (right).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Positions | 1 a | 2 b | 3 b | |||
|---|---|---|---|---|---|---|
| 1 | 79.4 | 144.86 | 5.10 dd (4.8,6.6) | 70.2 | ||
| 2 | 5.98 dd (11.2, 17.6) | 144.1 | 144.87 | 3.78 dd (4.2, 6.6) | 68.0 | |
| 3 | 5.09 overlapped | 114.1 | 6.80 d (1.8) | 112.9 | 127.4 | |
| 4a | 1.96 m | 22.6 | 133.4 | 2.65–2.69 m | 27.9 | |
| 4b | 2.21–2.25 m | |||||
| 5 | 1.50 m | 40.2 | 6.67 dd (1.8, 8.4) | 120.8 | 4.28 m | 65.8 |
| 6 | 5.09 overlapped | 125.3 | 7.00 d (8.4) | 115.53 | 6.72 brs | 140.1 |
| 7 | 130.8 | 3.30 d (6.6) | 39.1 | 166.7 | ||
| 8 | 1.61 s | 25.9 | 5.94 m | 137.9 | ||
| 9 | 1.54 s | 18.0 | 5.03–5.08 m | 115.49 | ||
| 10 | 1.22 s | 24.0 | ||||
| 1′ | 4.13 d (8.0) | 98.2 | 4.84 d (7.2) | 100.2 | 125.9 | |
| 2′ | 2.90 m | 74.0 | 3.23–3.28 m | 73.1 | 7.05 d (2.0) | 115.3 |
| 3′ | 3.09 t (8.4) | 77.5 | 3.23–3.28 m | 76.98 | 146.0 | |
| 4′ | 3.02 t (9.2) | 70.7 | 3.23–3.28 m | 76.78 | 148.9 | |
| 5′ | 2.95 m | 77.1 | 3.13–3.17 m | 69.7 | 6.76 d (8.4) | 116.2 |
| 6′a | 3.58 d (11.2) | 61.7 | 3.65–3.67 m | 60.8 | 7.01 dd (2.0, 8.4) | 121.9 |
| 6′b | 3.38 m | 3.42–3.45 m | ||||
| 7′ | 7.48 d (15.6) | 145.9 | ||||
| 8′ | 6.25 d (15.6) | 114.4 | ||||
| 9′ | 166.5 | |||||
| 1″ | 4.09 d (7.8) | 102.8 | ||||
| 2″ | 2.93 td (8.4, 4.2) | 73.4 | ||||
| 3″ | 3.05–3.08 m | 76.85 | ||||
| 4″ | 3.10–3.13 m | 76.81 | ||||
| 5″ | 3.01–3.05 m | 70.1 | ||||
| 6″a | 3.65–3.67 m | 61.1 | ||||
| 6″b | 3.42–3.45 m | |||||
| 1‴a | 3.74 d (13.2) | 68.5 | ||||
| 1‴b | 3.40 d (13.2) | |||||
| 2‴ | 31.2 | |||||
| 3‴ | 1.23–1.32 ov | 25.2 | ||||
| 4‴a | 1.49–1.53 m | 29.2 | ||||
| 4‴b | 1.23–1.32 ov | |||||
| 5‴ | 1.23–1.32 ov | 21.9 | ||||
| 6‴ | 0.87 t (7.2) | 13.9 | ||||
| OMe | 3.74 s (OMe-2) | 55.6 | 3.69 s | 52.3 |
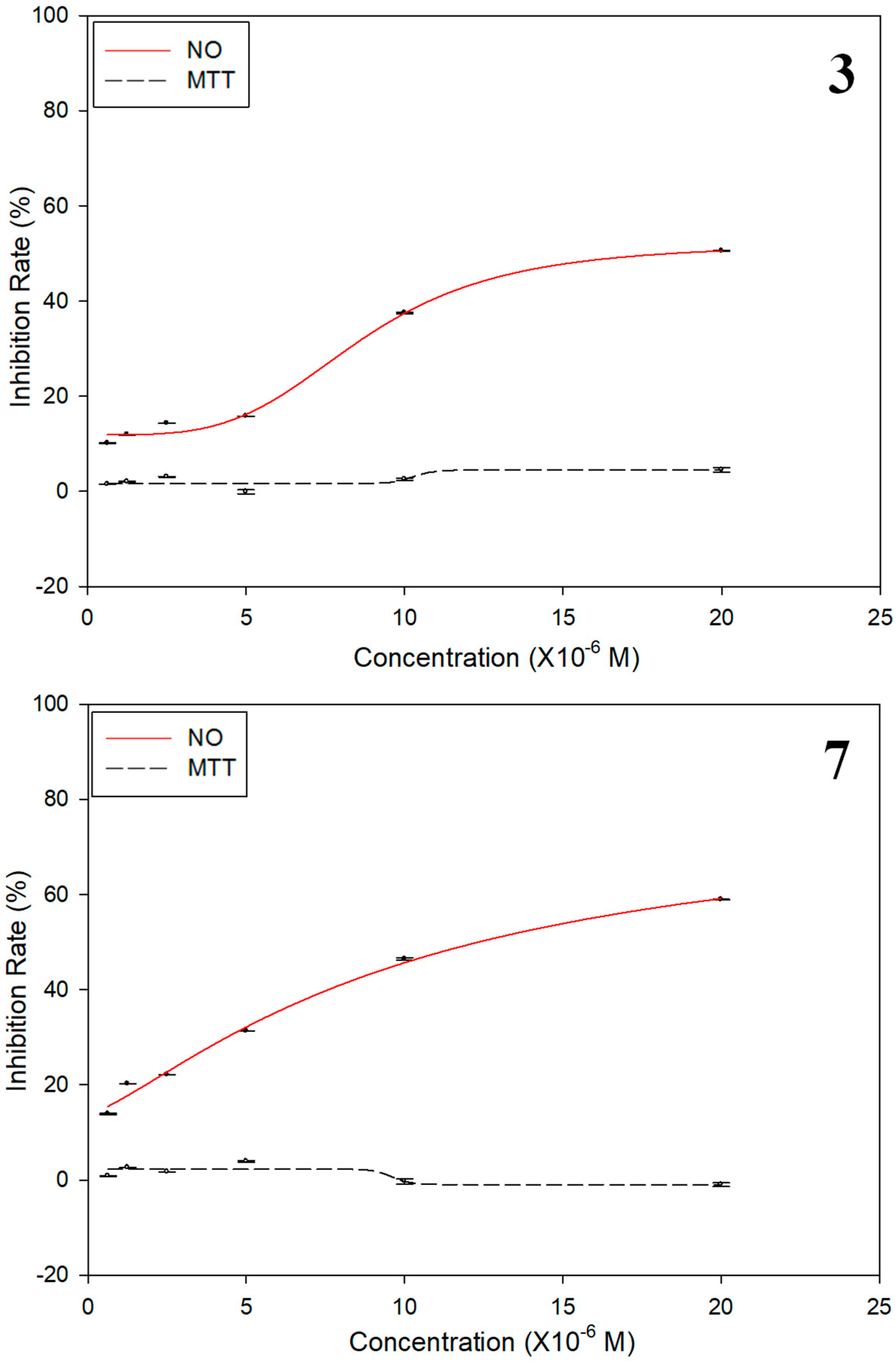
| Compounds | IC50 (μM) | Inhibition Rate (%) b |
|---|---|---|
| NO | Cytotoxicity | |
| 1 | >100 | 1.8 |
| 2 | >100 | −0.5 |
| 3 | 12.2 ± 0.8 | 4.7 |
| 4 | >100 | 1.1 |
| 5 | >100 | 4.5 |
| 6 | >100 | 1.2 |
| 7 | 18.9 ± 0.3 | 2.4 |
| Dexamethasone | 2.2 ± 0.02 | NT c |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Geng, Y.; Wang, X.; Song, X.; Wang, X.; Yu, J. Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules 2018, 23, 2577. https://doi.org/10.3390/molecules23102577
Dong H, Geng Y, Wang X, Song X, Wang X, Yu J. Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules. 2018; 23(10):2577. https://doi.org/10.3390/molecules23102577
Chicago/Turabian StyleDong, Hongjing, Yanling Geng, Xueyong Wang, Xiangyun Song, Xiao Wang, and Jinqian Yu. 2018. "Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities" Molecules 23, no. 10: 2577. https://doi.org/10.3390/molecules23102577
APA StyleDong, H., Geng, Y., Wang, X., Song, X., Wang, X., & Yu, J. (2018). Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules, 23(10), 2577. https://doi.org/10.3390/molecules23102577