
Development of an In Vitro Screening Platform for the Identification of Partial PPARγ Agonists as a Source for Antidiabetic Lead Compounds


Abstract
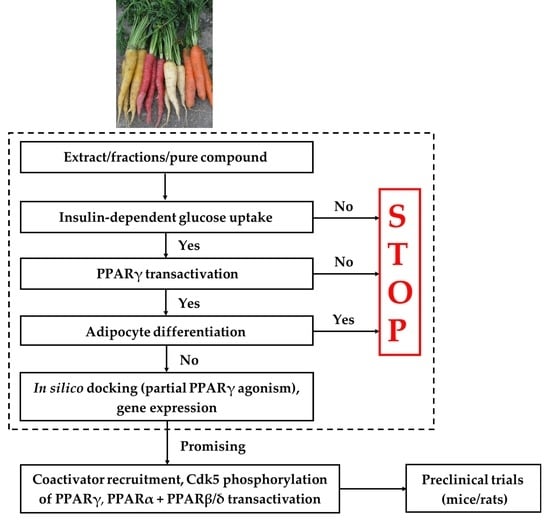
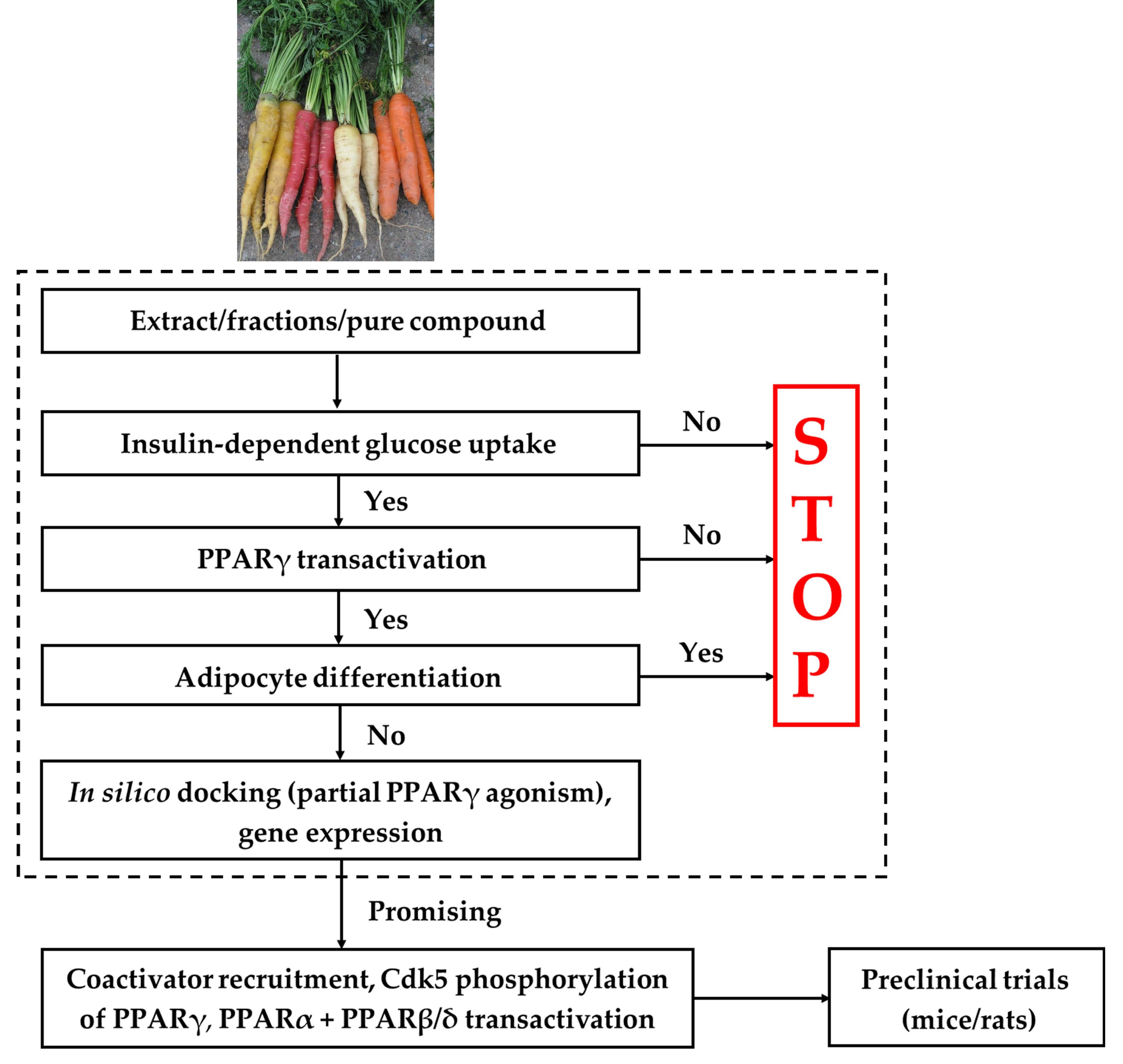{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Screening Platform
3. Insulin-Dependent and Basal GU
3.1. Identification of Potential Antidiabetic Compounds from Plants with Effect on Insulin-Dependent GU
3.1.1. Alkamides in the Roots of E. purpurea Show an Effect on Insulin-Dependent GU
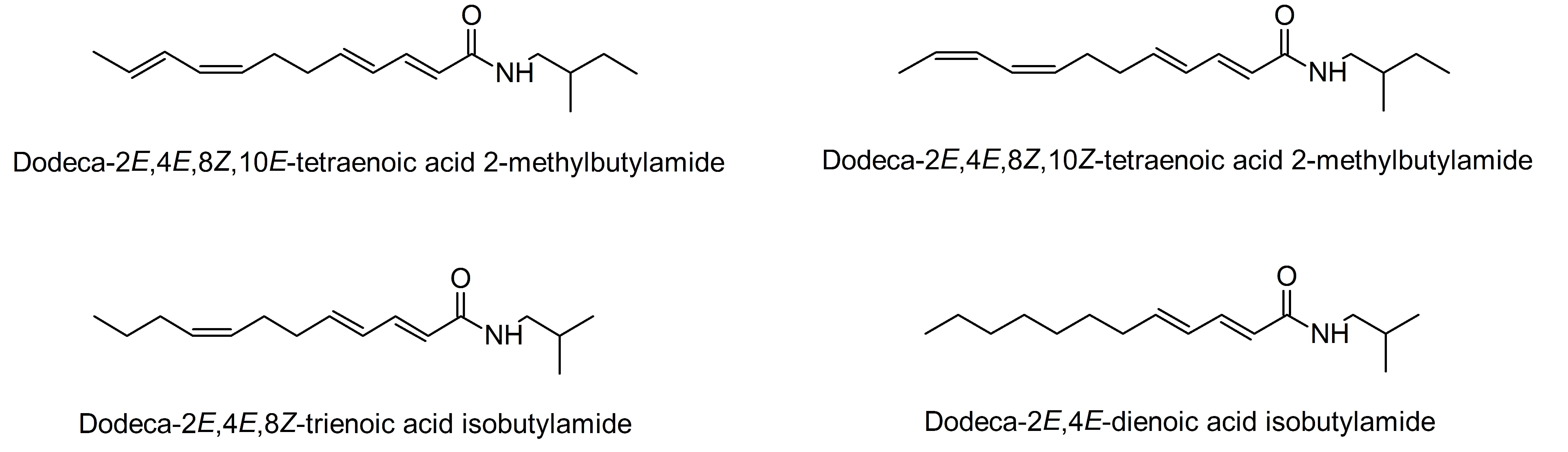
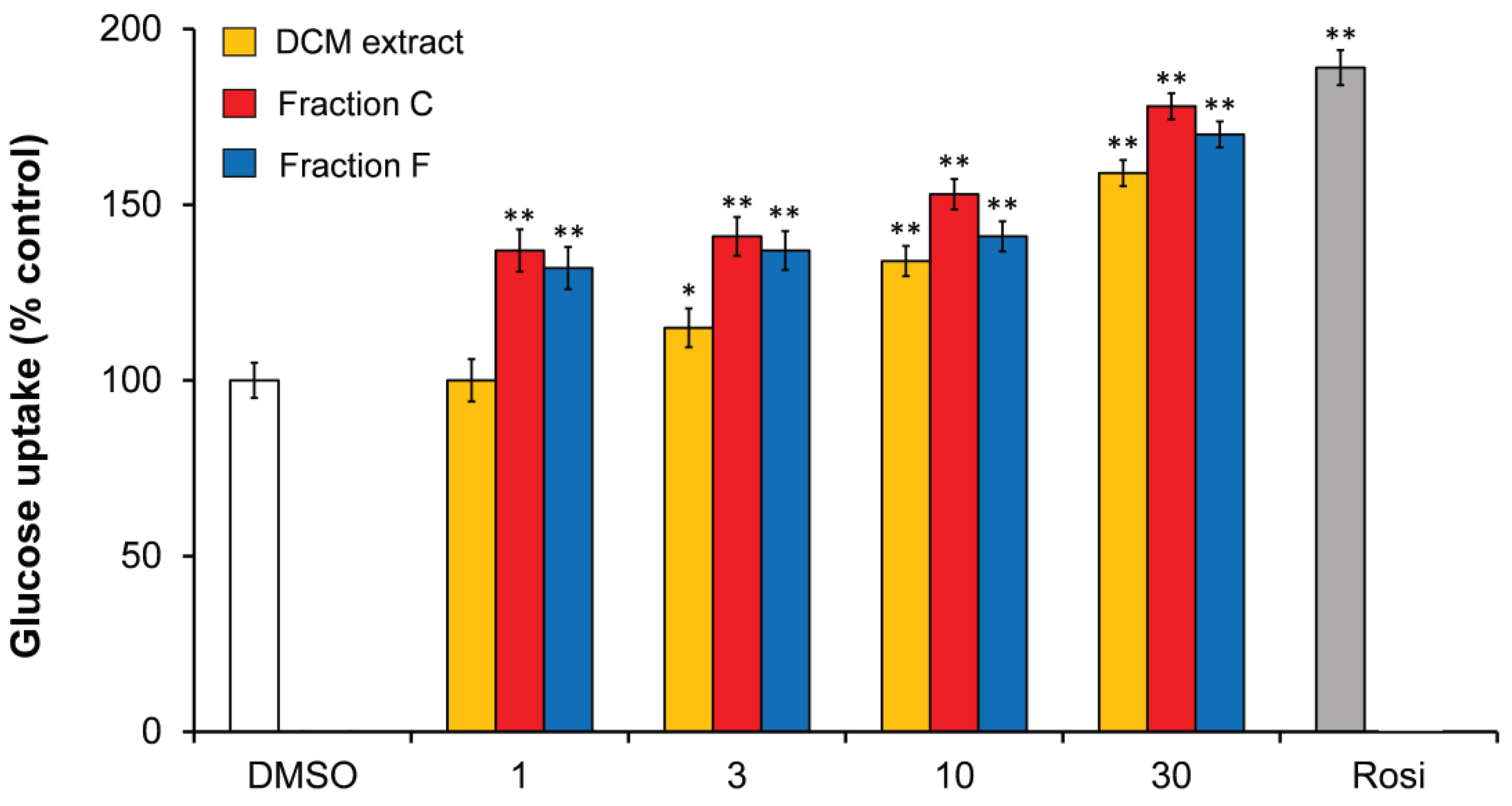
3.1.2. Polyacetylenes from Carrot Roots Show an Effect on Insulin-Dependent GU
4. PPARγ Transactivation Assay
4.1. Identification of Potential Partial PPARγ Agonists from Plants
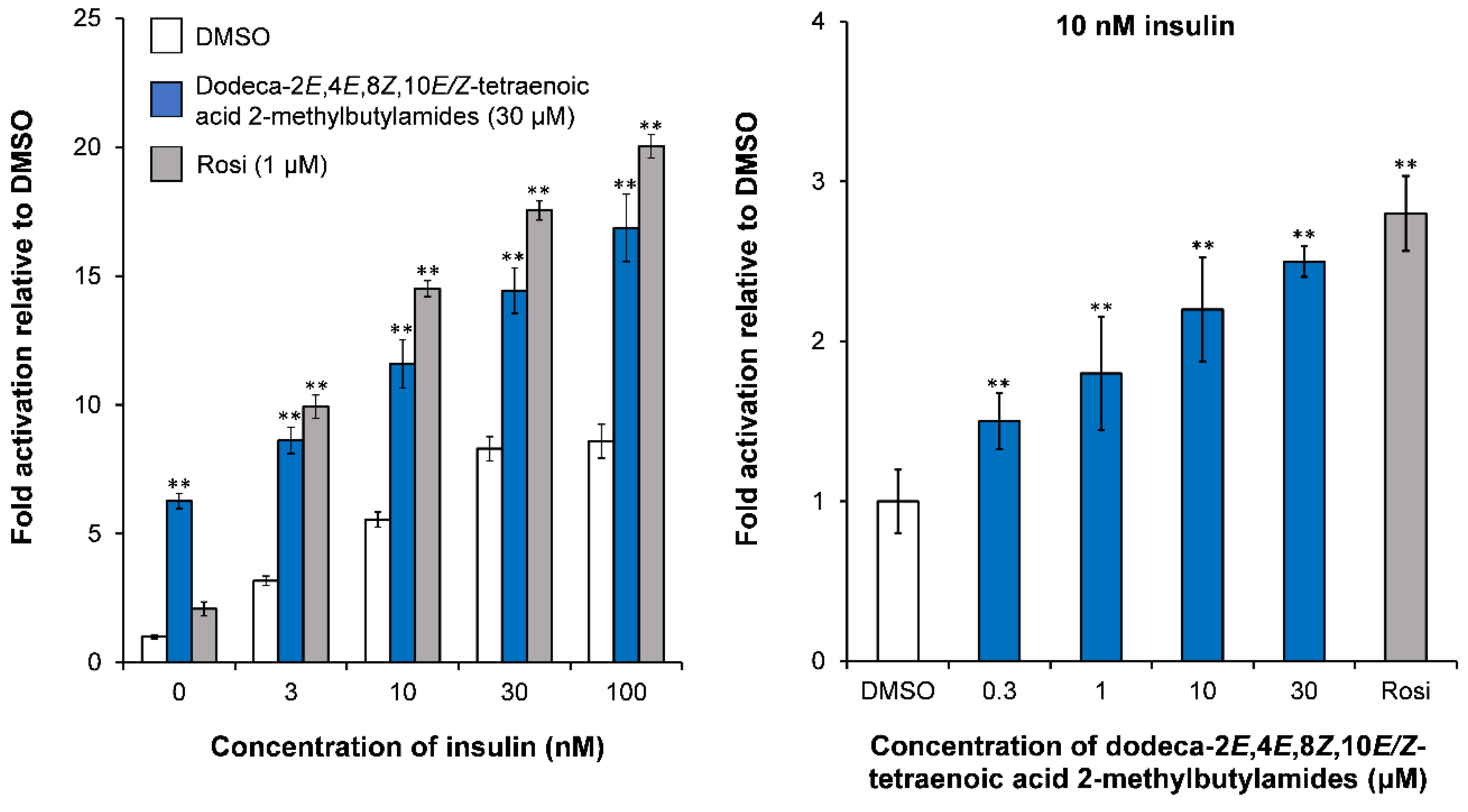
4.1.1. PPARγ Activity of Alkamides from E. purpurea
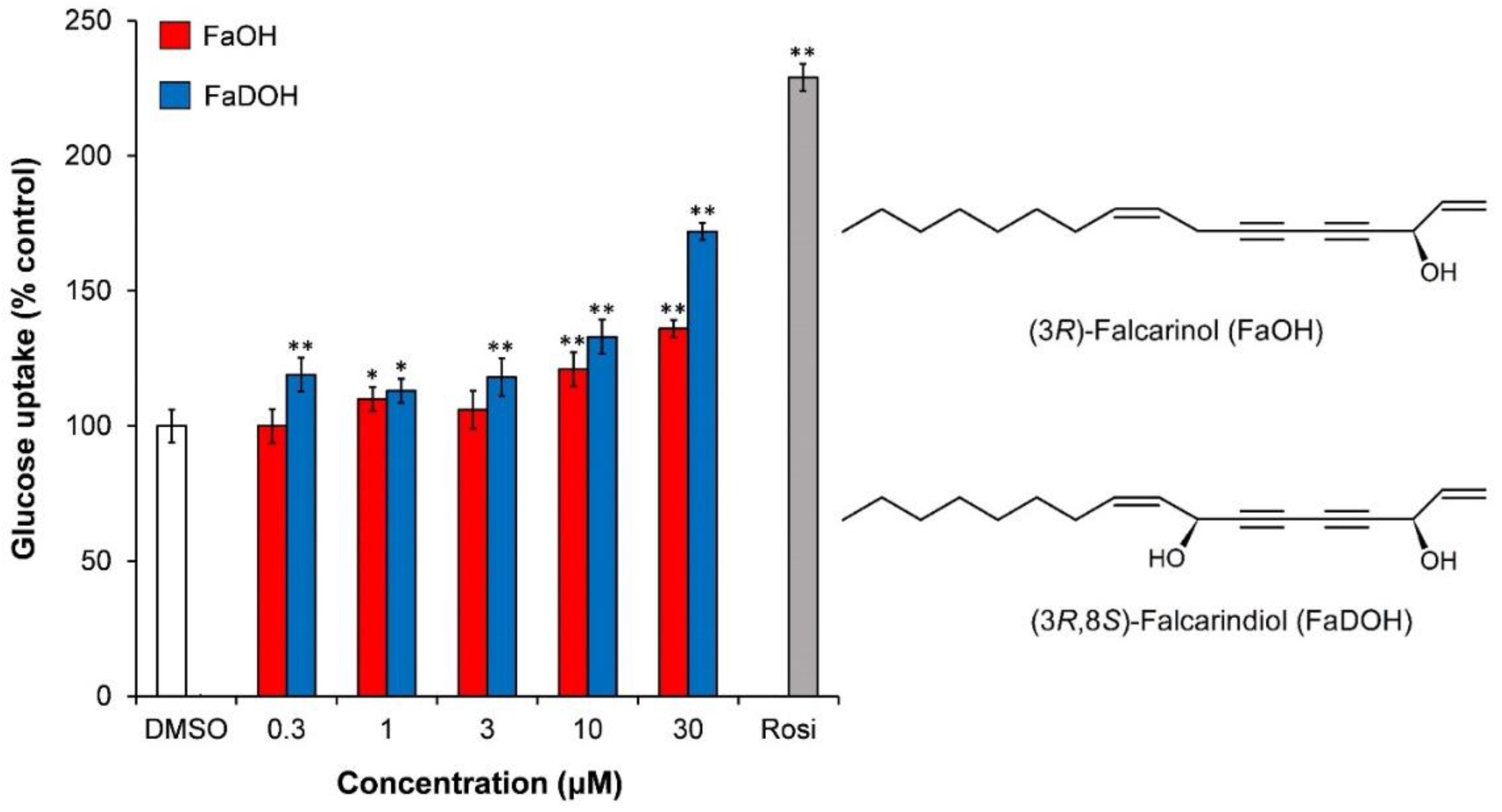
4.1.2. PPARγ Activity of FaOH and FaDOH from Carrots
5. Adipocyte Differentiation
5.1. Adipocyte Differentiation Bioassays and Gene Expression
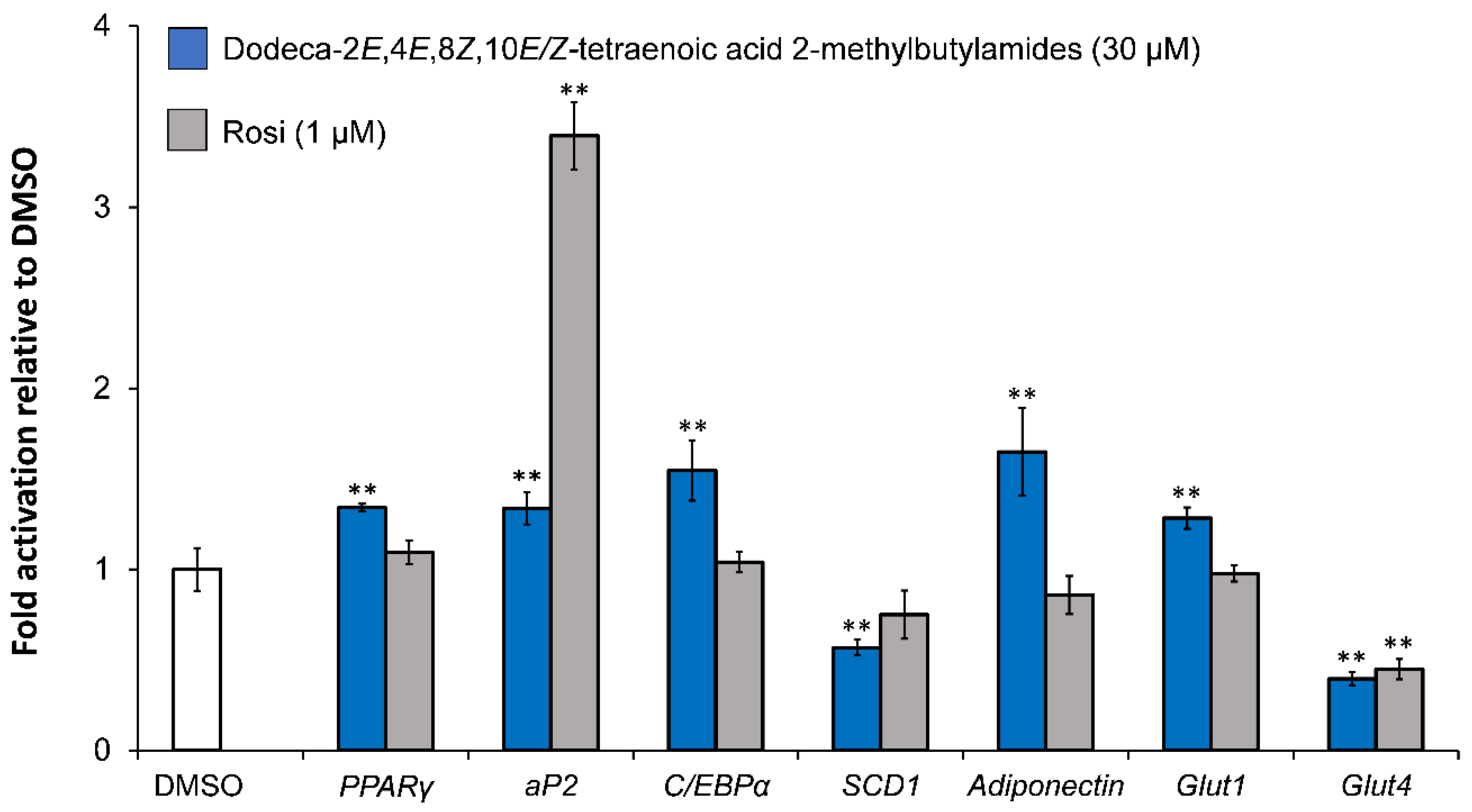
5.2. Investigation of Adipocyte Differentiation of Dodeca-2E,4E,8Z,10E/Z-Tetraenoic Acid 2-Methyl-butylamides
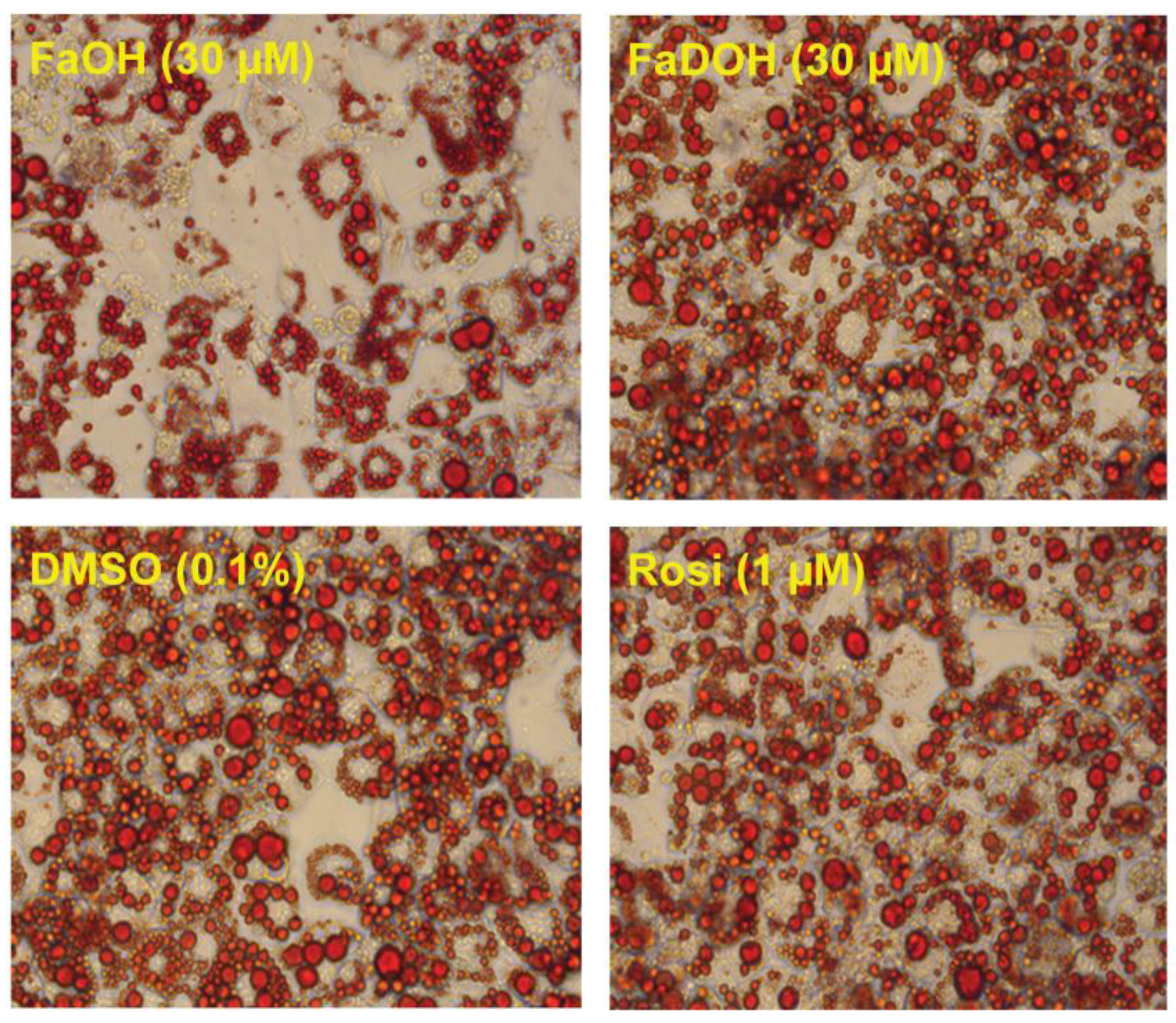
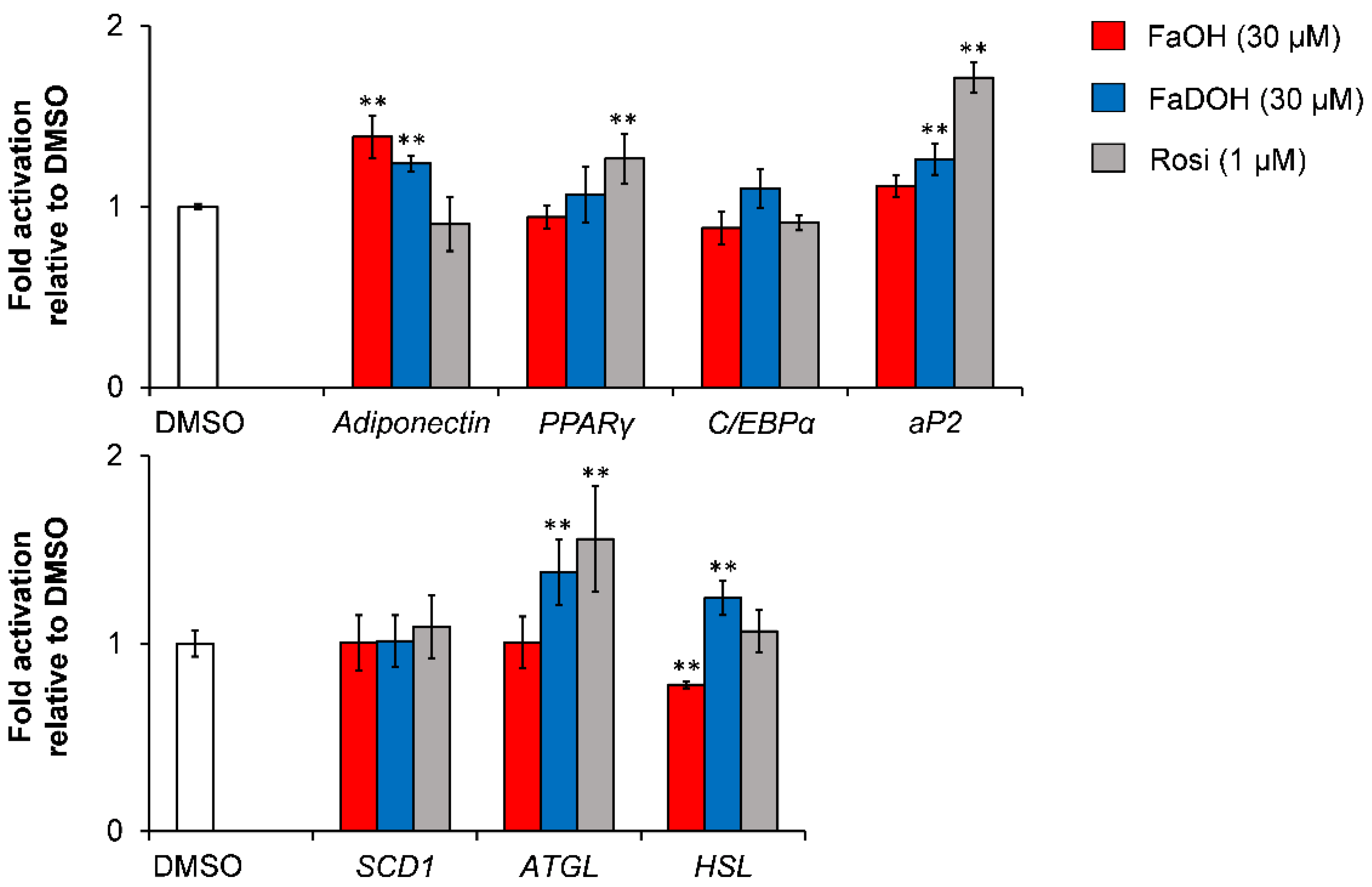
5.3. Investigation of Adipocyte Differentiation of FaOH and FaDOH
6. In Silico Screening for Identification of PPARγ Agonists
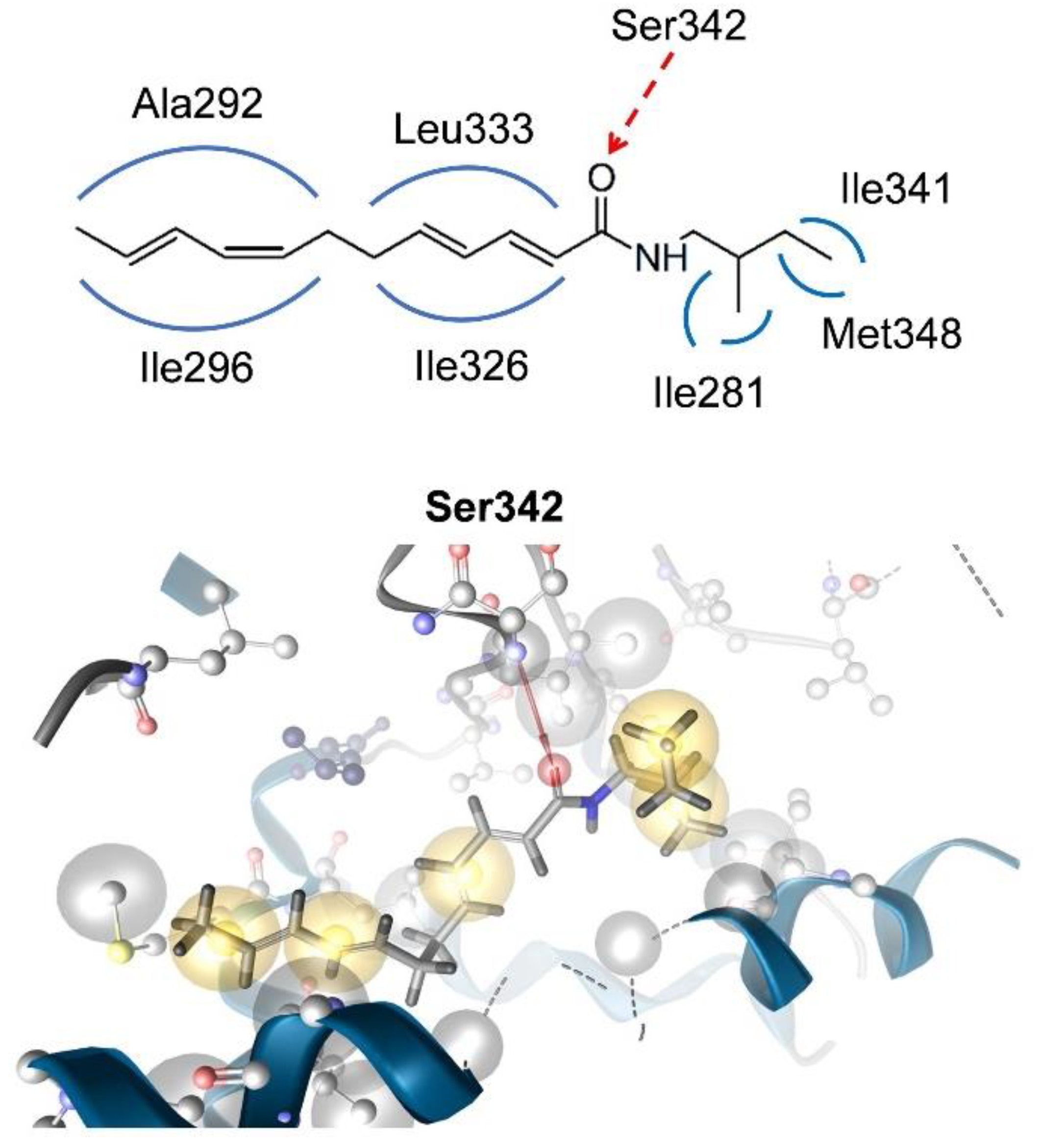
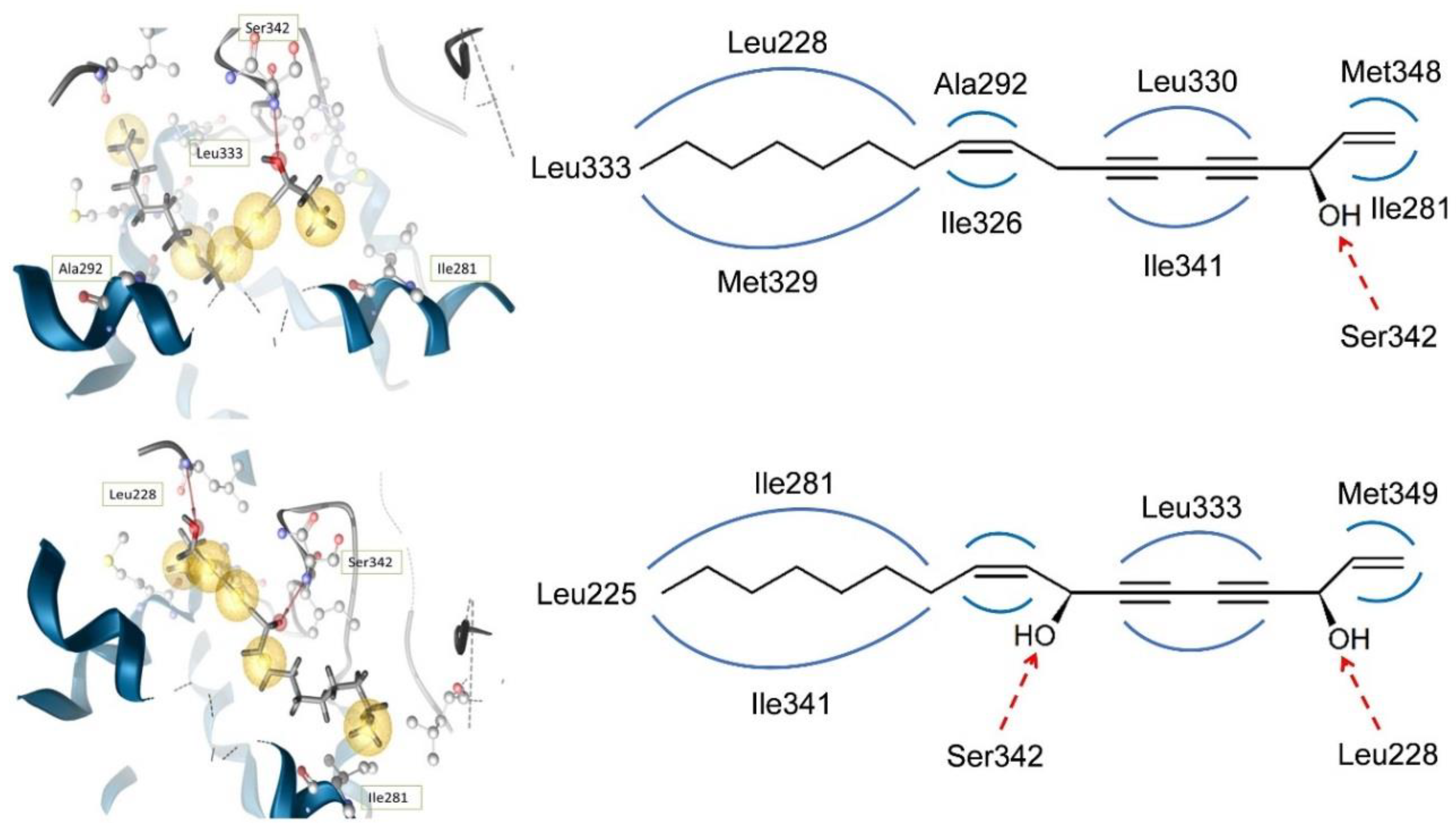
6.1. In Silico Docking for Investigation of Partial PPARγ Agonism of Natural Products
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, R.E.; Klaman, L.D. Adipose tissue as an active endocrine organ: Recent advances. Curr. Opin. Pharmacol. 2005, 5, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Mlinar, B.; Marc, J.; Janez, A.; Pfeifer, M. Molecular mechanisms of insulin resistance and associated diseases. Clin. Chim. Acta 2007, 375, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.S.; Chittiboyina, A.G.; Kurtz, T.W.; Pershadsingh, H.A.; Avery, M.A. Type 2 diabetes and oral antihyperglycemic drugs. Curr. Med. Chem. 2008, 15, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E. Oral Antihyperglycemic therapy for type 2 diabetes. JAMA 2002, 287, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Zieleniak, A.; Wójcik, M.; Woźniak, L.A. Structure and physiological functions of the human peroxisome proliferator-activated receptor γ. Arch. Immunol. Ther. Exp. 2008, 56, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.P.; Petro, A.E.; Macnaul, K.L.; Kelly, L.J.; Zhang, B.B.; Richards, K.; Elbrecht, A.; Johnson, B.A.; Zhou, G.; Doebber, T.W.; et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein γ selective modulator. Mol. Endocrinol. 2003, 17, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-talk between PPARγ and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [PubMed]
- Siersbæk, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [PubMed]
- Kroker, A.J.; Bruning, J.B. Review of the structural and dynamic mechanisms of PPARγ partial agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Blunder, M.; Liu, X.; Malainer, C.; Blazevic, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): A review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator–activated receptors and liver X receptor-α in humans: No alteration in adipose tissue of obese and NIDDM patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P. The biology of peroxisome proliferator-activated receptors: Relationship with lipid metabolism and insulin sensitivity. Diabetes 2004, 53 (Suppl. 1), S43–S50. [Google Scholar] [CrossRef]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular regulation of adipogenesis and potential anti-adipogenic bioactive molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- El-Houri, R.B.; Mortier, J.; Murgueitio, M.S.; Wolber, G.; Christensen, L.P. Identification of PPARγ agonists from natural sources using different in silico approaches. Planta Med. 2015, 81, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.C.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [PubMed]
- Burgermeister, E.; Schnoebelen, A.; Flament, A.; Benz, J.; Stihle, M.; Gsell, B.; Rufer, A.; Ruf, A.; Kuhn, B.; Märki, H.P.; et al. A novel partial agonist of peroxisome proliferator-activated receptor-γ (PPARγ) recruits PPARγ-coactivator-1α, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol. Endocrinol. 2006, 20, 809–830. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional regulation of adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Forman, B.M. Prostaglandins and fatty acids regulate transcriptional signaling via the peroxisome proliferator activated receptor nuclear receptors. Prostaglandins Other Lipid Mediat. 2000, 62, 1–13. [Google Scholar] [CrossRef]
- Choi, S.S.; Park, J.; Choi, J.H. Revisiting PPARγ as a target for the treatment of metabolic disorders. BMB Rep. 2014, 47, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Kota, B.P.; Razmovski, V.; Roufogalis, B.D. Herbal or natural medicines as modulators of peroxisome proliferator-activated receptors and related nuclear receptors for therapy of metabolic syndrome. Basic Clin. Pharmacol. Toxicol. 2005, 96, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.B.; Petersen, R.K.; Petersen, S.; Kristiansen, K.; Christensen, L.P. Activation of PPARγ by metabolites from the flowers of purple coneflower (Echinacea purpurea). J. Nat. Prod. 2009, 72, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.B.; Petersen, R.K.; Kristiansen, K.; Christensen, L.P. Identification of bioactive compounds from flowers of black elder (Sambucus nigra L.) that activate the human peroxisome proliferator-activated receptor (PPAR) γ. Phytother. Res. 2010, 24 (Suppl. 2), S129–S132. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, G.; Muthukumaran, P.; Sarath Kumar, B.; Muthusamy, V.S.; Lakshmi, B.S. Selective Inhibition of PTP1B by vitalboside A from Syzygium cumini enhances insulin sensitivity and attenuates lipid accumulation via partial agonism to PPARγ: In vitro and in silico investigation. Chem. Biol. Drug Des. 2016, 88, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, N.; Al Bratty, M.; Akhtar Javed, S.; Ahsan, W.; Alhazmi, H.A. Targeting peroxisome proliferator-activated receptors using thiazolidinediones: Strategy for design of novel antidiabetic drugs. Int. J. Med. Chem. 2017, 2017, 1069718. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Charbonnel, B.; Staels, B. Thiazolidinediones and PPARγ agonists: Time for a reassessment. Trends Endocrinol. Metab. 2012, 23, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.J.; Lin, Y.; Chen, Y.E.; Vance, D.E.; Leiter, E.H. Adverse hepatic and cardiac responses to rosiglitazone in a new mouse model of type 2 diabetes: Relation to dysregulated phosphatidylcholine metabolism. Vasc. Pharmacol. 2006, 45, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARγ using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Guasch, L.; Sala, E.; Castell-Auví, A.; Cedó, L.; Liedl, K.R.; Wolber, G.; Muehlbacher, M.; Mulero, M.; Pinent, M.; Ardévol, A.; et al. Identification of PPARgamma partial agonists of natural origin (I): Development of a virtual screening procedure and in vitro validation. PLoS ONE 2012, 7, e50816. [Google Scholar] [CrossRef] [PubMed]
- Guasch, L.; Sala, E.; Mulero, M.; Valls, C.; Salvadó, M.J.; Pujadas, G.; Garcia-Vallvé, S. Identification of PPARgamma partial agonists of natural origin (II): In silico prediction in natural extracts with known antidiabetic activity. PLoS ONE 2013, 8, e55889. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Vargas, D. Diverse coactivator recruitment through differential PPARγ nuclear receptor agonism. Genet. Mol. Biol. 2013, 36, 134–139. [Google Scholar] [CrossRef] [PubMed]
- El-Houri, R.B.; Kotowska, D.; Olsen, L.C.; Bhattacharya, S.; Christensen, L.P.; Grevsen, K.; Oksbjerg, N.; Færgeman, N.; Kristiansen, K.; Christensen, K.B. Screening for bioactive metabolites in plant extracts modulating glucose uptake and fat accumulation. Evid. Based Complement. Altern. Med. 2014, 2014, 156398. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.B.; Minet, A.; Svenstrup, H.; Grevsen, K.; Zhang, H.; Schrader, E.; Rimbach, G.; Wein, S.; Wolffram, S.; Kristiansen, K.; et al. Identification of plant extracts with potential antidiabetic properties: Effect on human peroxisome proliferator-activated receptor (PPAR), adipocyte differentiation and insulin-stimulated glucose uptake. Phytother. Res. 2009, 23, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Yeh, G.Y.; Eisenberg, D.M.; Kaptchuk, T.J.; Phillips, R.S. Systematic review of herbs and dietary supplements for glycemic control in diabetes. Diabetes Care 2003, 26, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Marles, R.J.; Farnsworth, N.R. Antidiabetic plants and their active constituents. Phytomedicine 1995, 2, 137–189. [Google Scholar] [CrossRef]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Takahashi, S.; Tanaka, T.; Kodama, T.; Sakai, J. Peroxisome proliferator-activated receptor δ (PPARδ), a novel target site for drug discovery in metabolic syndrome. Pharmacol. Res. 2006, 53, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Eddouks, M.; Bidi, A.; El Bouhali, B.; Hajji, L.; Zeggwagh, N.A. Antidiabetic plants improving insulin sensitivity. J. Pharm. Pharmacol. 2014, 66, 1197–1214. [Google Scholar] [CrossRef] [PubMed]
- Kotowska, D.; El-Houri, R.B.; Borkowski, K.; Petersen, R.K.; Fretté, X.C.; Wolber, G.; Grevsen, K.; Christensen, K.B.; Christensen, L.P.; Kristiansen, K. Isomeric C12-alkamides from the roots of Echinacea purpurea improve basal and insulin-dependent glucose uptake in 3T3-L1 adipocytes. Planta Med. 2014, 80, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- El-Houri, R.B.; Kotowska, D.; Christensen, K.B.; Bhattacharya, S.; Oksbjerg, N.; Wolber, G.; Kristiansen, K.; Christensen, L.P. Polyacetylenes from carrots (Daucus carota) improve glucose uptake in vitro in adipocytes and myotubes. Food Funct. 2015, 6, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.T.; Kanzaki, M.; Pessin, J.E. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr. Rev. 2004, 25, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Leney, S.E.; Tavaré, J.M. The molecular basis of insulin-stimulated glucose uptake: Signalling, trafficking and potential drug targets. J. Endocrinol. 2009, 203, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.; Boguslavsky, S.; Klip, A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 2011, 50, 3048–3061. [Google Scholar] [CrossRef] [PubMed]
- Sayem, A.S.M.; Arya, A.; Karimian, H.; Krishnasamy, N.; Ashok Hasamnis, A.; Hossain, C.F. Action of phytochemicals on insulin signaling pathways accelerating glucose transporter (GLUT4) protein translocation. Molecules 2018, 23, 258. [Google Scholar] [CrossRef] [PubMed]
- Karnieli, E.; Armoni, M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: From physiology to pathology. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E38–E45. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Nguyen, M.T.; Yoshizaki, T.; Favelyukis, S.; Patsouris, D.; Imamura, T.; Verma, I.M.; Olefsky, J.M. Suppression of PPAR-γ attenuates insulin-stimulated glucose uptake by affecting both GLUT1 and GLUT4 in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E219–E227. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, T.; Steger, D.J.; Lefterova, M.I.; Schupp, M.; Lazar, M.A. Adipocyte-specific expression of murine resistin is mediated by synergism between peroxisome proliferator-activated receptor γ and CCAAT/enhancer-binding proteins. J. Biol. Chem. 2009, 284, 6116–6125. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Lorenz, K.; Braithwaite, S.S.; Colca, J.R.; Palazuk, B.J.; Hotamisligil, G.S.; Spiegelman, B.M. Altered gene expression for tumor necrosis factor-α and its receptors during drug and dietary modulation of insulin resistance. Endocrinology 1994, 134, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Kawano, J.; Arora, R. The role of adiponectin in obesity, diabetes, and cardiovascular disease. J. Cardiometab. Syndr. 2009, 4, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Stumvoll, M. Adiponectin—Its role in metabolism and beyond. Horm. Metab. Res. 2002, 34, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Lee, W.J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.L.; Chen, C.L.; Tai, T.Y.; Chuang, L.M. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J. Clin. Endocrinol. Metab. 2001, 86, 3815–3819. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Koppen, A.; Kalkhoven, E. Brown vs white adipocytes: The PPARγ coregulator story. FEBS Lett. 2010, 584, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Chen, J.D. The SRC family of nuclear receptor coactivators. Gene 2000, 245, 1–11. [Google Scholar] [CrossRef]
- Zhang, F.; Lavan, B.E.; Gregoire, F.M. Selective modulators of PPAR-γ activity: Molecular aspects related to obesity and side effects. PPAR Res. 2007, 2007, 32696. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Géhin, M.; Annicotte, J.; Rocchi, S.; Champy, M.F.; O’Malley, B.W.; Chambon, P.; Auwerx, J. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 2002, 111, 931–941. [Google Scholar] [CrossRef]
- Louet, J.F.; Chopra, A.R.; Sagen, J.V.; An, J.; York, B.; Tannour-Louet, M.; Saha, P.K.; Stevens, R.D.; Wenner, B.R.; Ilkayeva, O.R.; et al. The coactivator SRC-1 is an essential coordinator of hepatic glucose production. Cell Metab. 2010, 12, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lin, J.D. PGC-1 coactivators in the control of energy metabolism. Acta Biochim. Biophys. Sin. 2011, 43, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Ge, K.; Guermah, M.; Yuan, C.X.; Ito, M.; Wallberg, A.E.; Spiegelman, B.M.; Roeder, R.G. Transcription coactivator TRAP220 is required for PPARγ 2-stimulated adipogenesis. Nature 2002, 417, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Leonardsson, G.; Steel, J.H.; Christian, M.; Pocock, V.; Milligan, S.; Bell, J.; So, P.W.; Medina-Gomez, G.; Vidal-Puig, A.; White, R.; et al. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. USA 2004, 101, 8437–8442. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Blunder, M.; Fakhrudin, N.; Liu, X.; Noha, S.M.; Malainer, C.; Kramer, M.P.; Cocic, A.; Kunert, O.; Schinkovitz, A.; et al. Polyacetylenes from Notopterygium incisum—New selective partial agonists of peroxisome proliferator-activated receptor-gamma. PLoS ONE 2013, 8, e61755. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Kim, Y.J.; Yang, S.J.; Lee, Y.; Lee, M. Nutrigenomic functions of PPARs in obesogenic environments. PPAR Res. 2016, 2016, 4794576. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Liu, W.; Kuang, S. Fatty acid binding protein 4 expression marks a population of adipocyte progenitors in white and brown adipose tissues. FASEB J. 2013, 27, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Nicholson, A.C.; Hajjar, D.P.; Gotto, A.M., Jr.; Han, J. Adipogenic differentiating agents regulate expression of fatty acid binding protein and CD36 in the J744 macrophage cell line. J. Lipid Res. 2003, 44, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Yao-Borengasser, A.; Rassouli, N.; Varma, V.; Bodles, A.M.; Rasouli, N.; Unal, R.; Phanavanh, B.; Ranganathan, G.; McGehee, R.E., Jr.; Kern, P.A. Stearoyl-coenzyme A desaturase 1 gene expression increases after pioglitazone treatment and is associated with peroxisomal proliferator-activated receptor-γ responsiveness. J. Clin. Endocrinol. Metab. 2008, 93, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Ralston, J.C.; Badoud, F.; Cattrysse, B.; McNicholas, P.D.; Mutch, D.M. Inhibition of stearoyl-CoA desaturase-1 in differentiating 3T3-L1 pre-adipocytes up-regulates elongase 6 and down-regulates genes affecting triacylglycerol synthesis. Int. J. Obes. 2014, 38, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Ntambi, J.M. The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann. N. Y. Acad. Sci. 2011, 1243, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Jazurek, M.; Dobrzyn, A. Stearoyl-CoA desaturase and insulin signaling--what is the molecular switch? Biochim. Biophys. Acta 2010, 1797, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Yu, Z.; Patel, S.; Jue, D.; Liu, L.F.; Kraemer, F.B. Hormone-sensitive lipase modulates adipose metabolism through PPARγ. Biochim. Biophys. Acta 2011, 1811, 9–16. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Sherman, A.; Tsao, P.; Gonzalez, O.; Yee, G.; Lamendola, C.; Reaven, G.M.; Cushman, S.W. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia 2007, 50, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Hallenborg, P.; Jørgensen, C.; Petersen, R.K.; Feddersen, S.; Araujo, P.; Markt, P.; Langer, T.; Furstenberger, G.; Krieg, P.; Koppen, A.; et al. Epidermis-type lipoxygenase 3 regulates adipocyte differentiation and peroxisome proliferator-activated receptor γ activity. Mol. Cell. Biol. 2010, 30, 4077–4091. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Christensen, K.B.; Olsen, L.C.; Christensen, L.P.; Grevsen, K.; Færgeman, N.J.; Kristiansen, K.; Young, J.F.; Oksbjerg, N. Bioactive components from flowers of Sambucus nigra L. increase glucose uptake in primary porcine myotube cultures and reduce fat accumulation in Caenorhabditis elegans. J. Agric. Food Chem. 2013, 61, 11033–11040. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M. PPAR-gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 47, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Hamm, J.K.; Verhagen, L.A.W.; Peroni, O.; Katic, M.; Flier, J.S. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006, 55, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.S.; Chalmers, M.J.; Novick, S.; Kuruvilla, D.S.; Chang, M.R.; Kamenecka, T.M.; Rance, M.; Johnson, B.A.; Burris, T.P.; Griffin, P.R.; et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 2012, 20, 139–150. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porskjær Christensen, L.; Bahij El-Houri, R. Development of an In Vitro Screening Platform for the Identification of Partial PPARγ Agonists as a Source for Antidiabetic Lead Compounds. Molecules 2018, 23, 2431. https://doi.org/10.3390/molecules23102431
Porskjær Christensen L, Bahij El-Houri R. Development of an In Vitro Screening Platform for the Identification of Partial PPARγ Agonists as a Source for Antidiabetic Lead Compounds. Molecules. 2018; 23(10):2431. https://doi.org/10.3390/molecules23102431
Chicago/Turabian StylePorskjær Christensen, Lars, and Rime Bahij El-Houri. 2018. "Development of an In Vitro Screening Platform for the Identification of Partial PPARγ Agonists as a Source for Antidiabetic Lead Compounds" Molecules 23, no. 10: 2431. https://doi.org/10.3390/molecules23102431
APA StylePorskjær Christensen, L., & Bahij El-Houri, R. (2018). Development of an In Vitro Screening Platform for the Identification of Partial PPARγ Agonists as a Source for Antidiabetic Lead Compounds. Molecules, 23(10), 2431. https://doi.org/10.3390/molecules23102431