Exploring the Chemical Diversity of Algerian Plants: Three New Pentacyclic Triterpenoids from Launaea acanthoclada Roots


Abstract
:1. Introduction
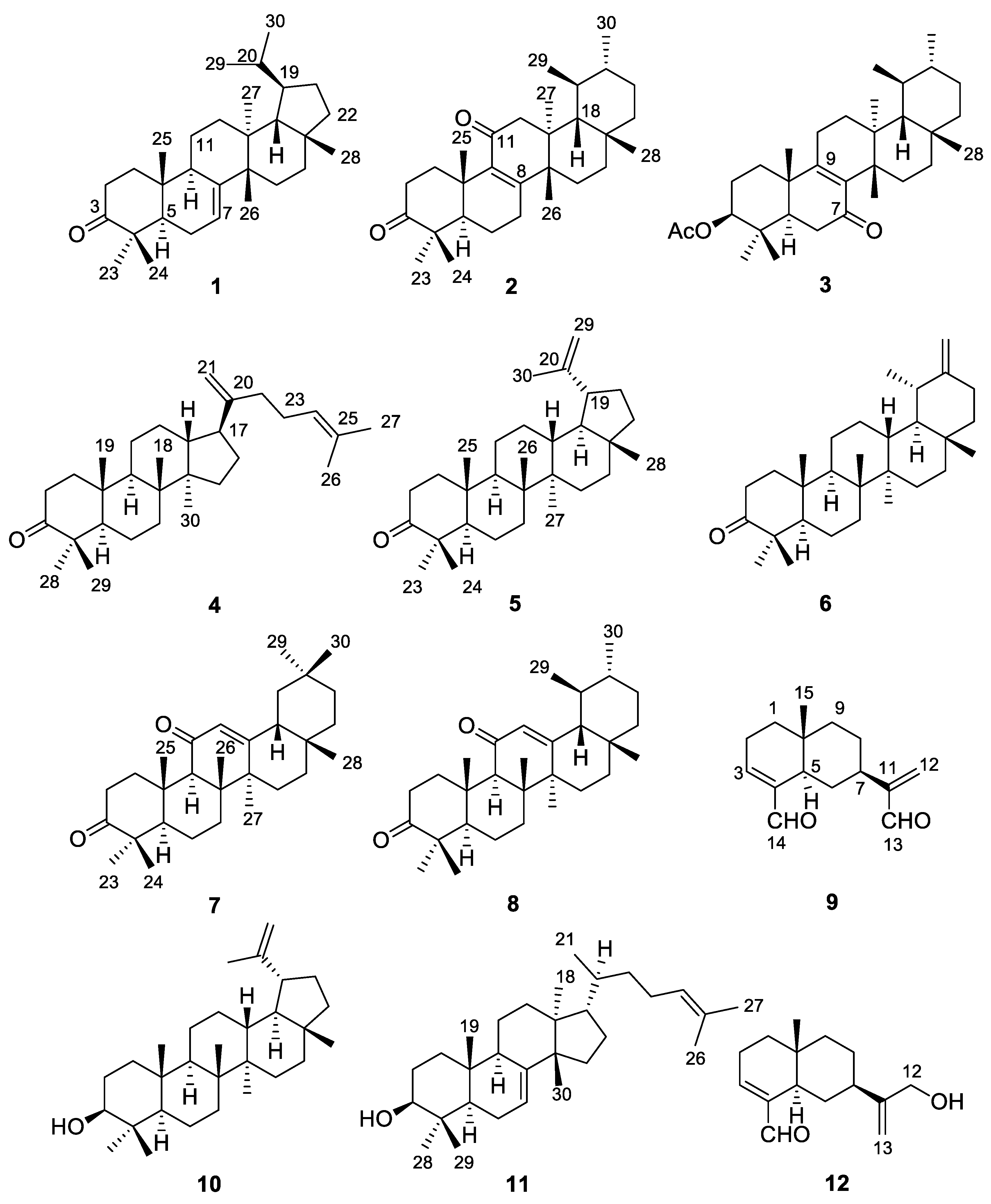
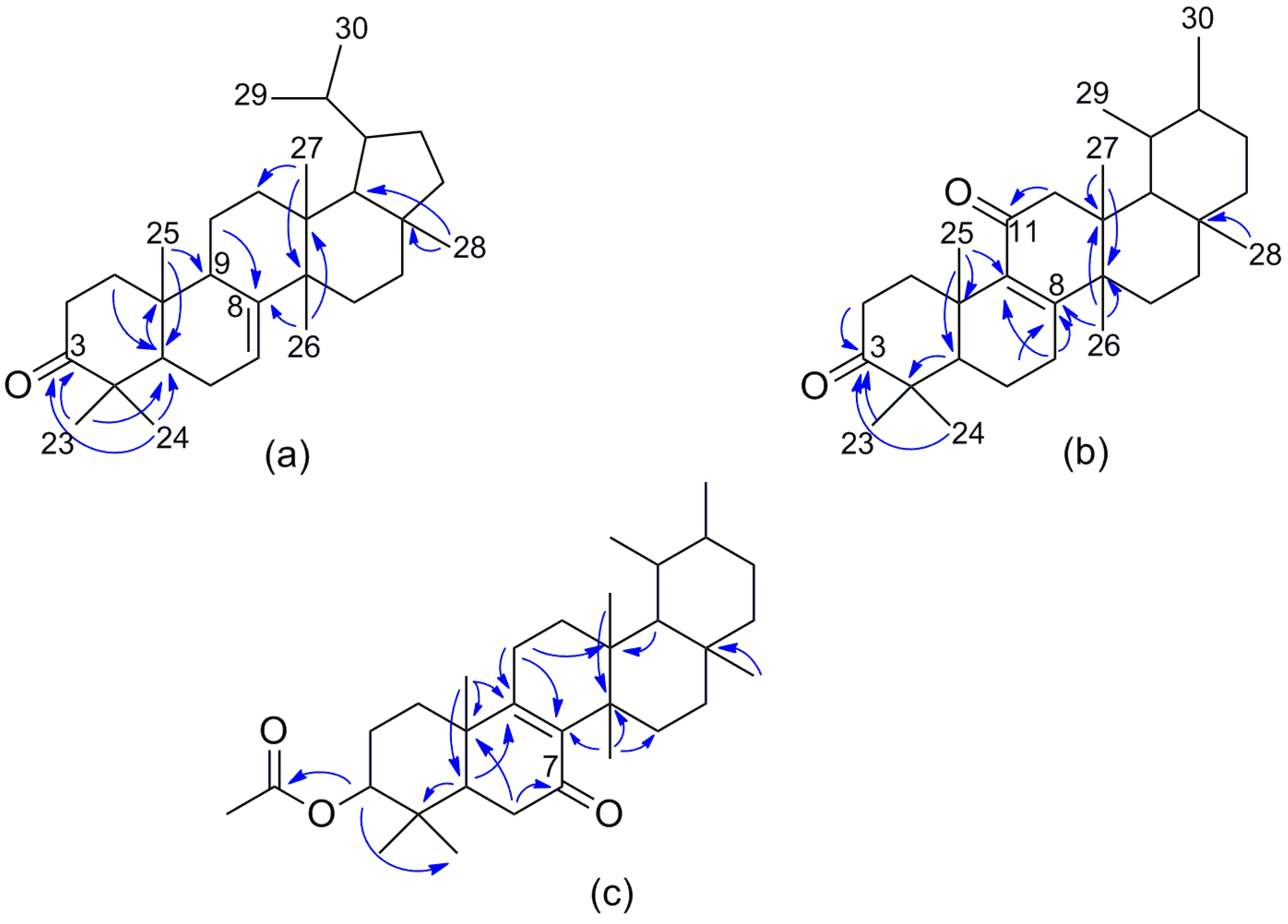
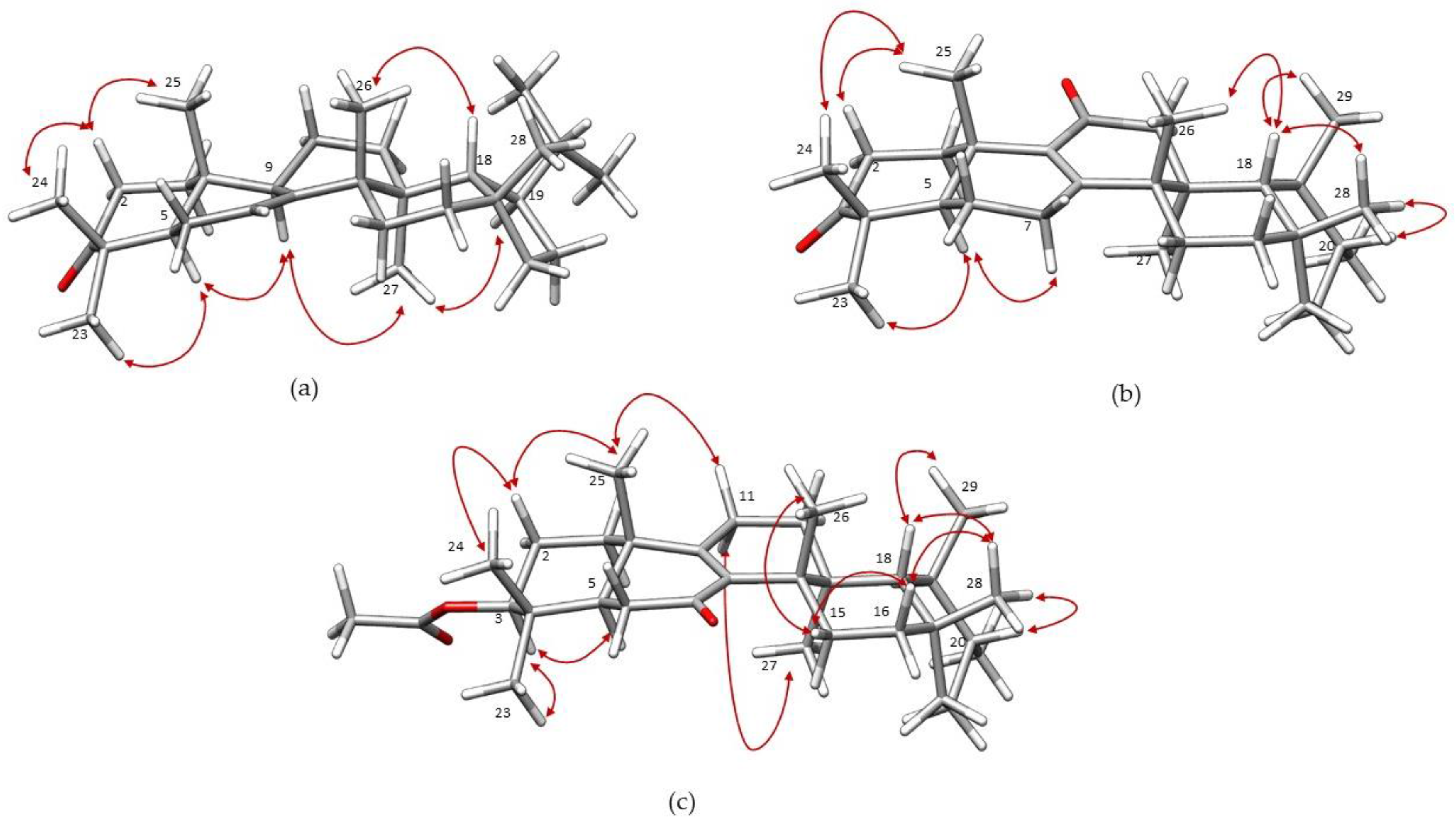
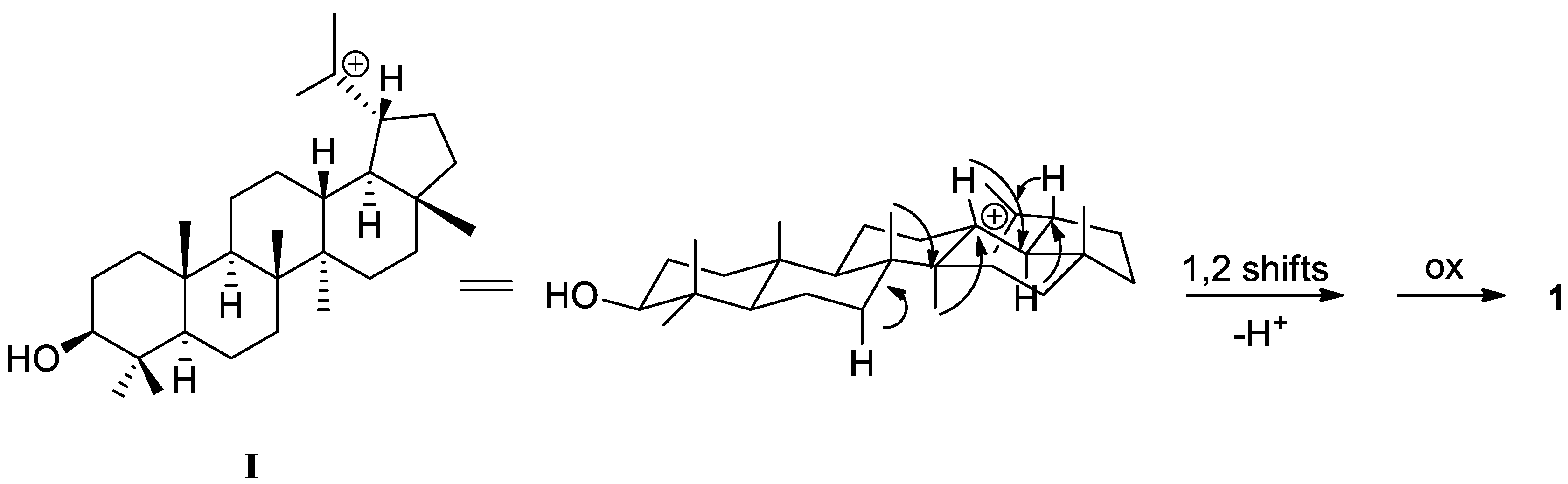
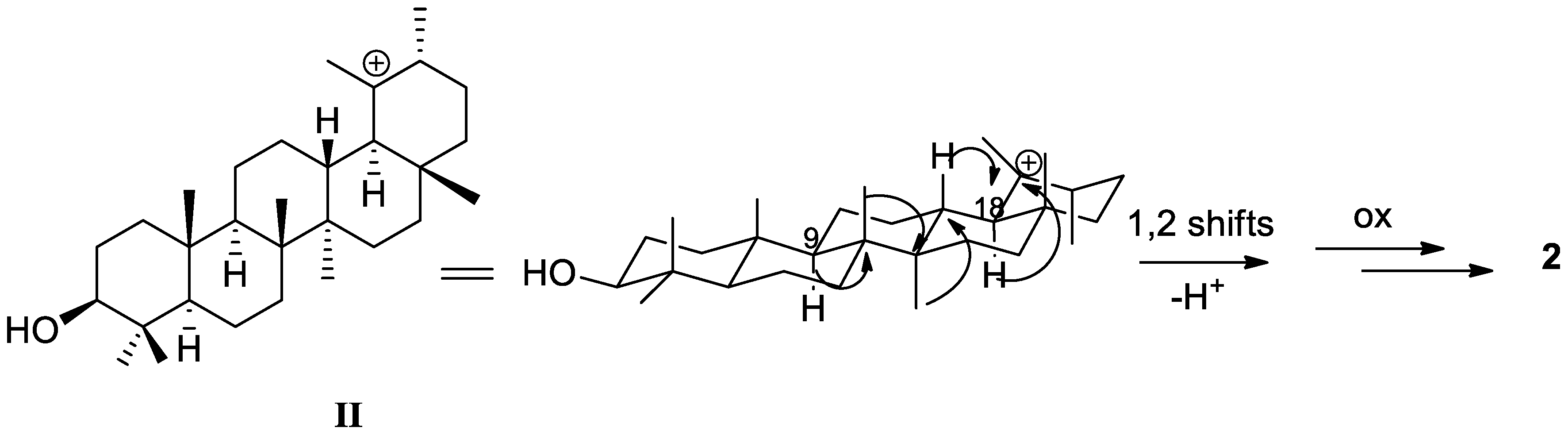
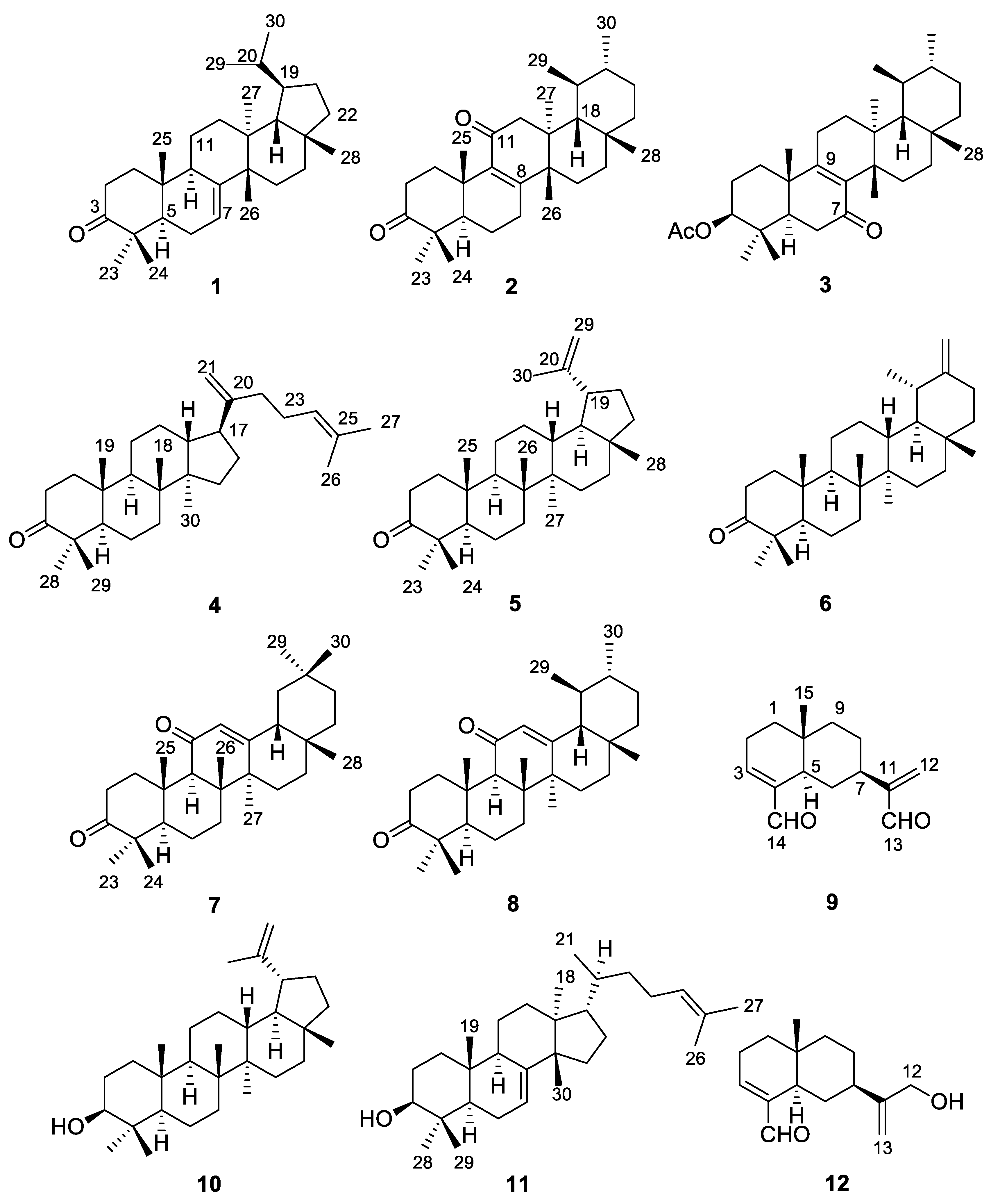
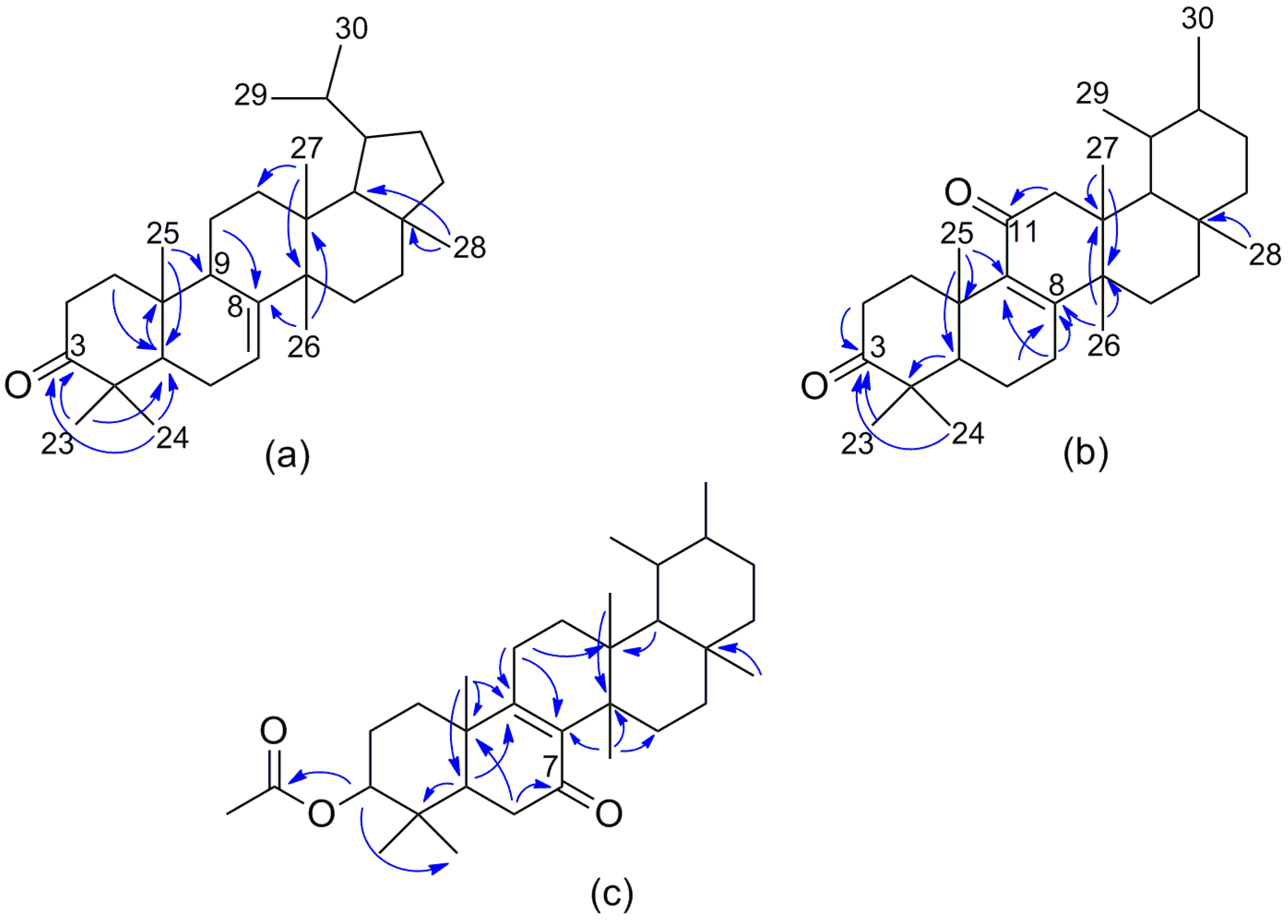
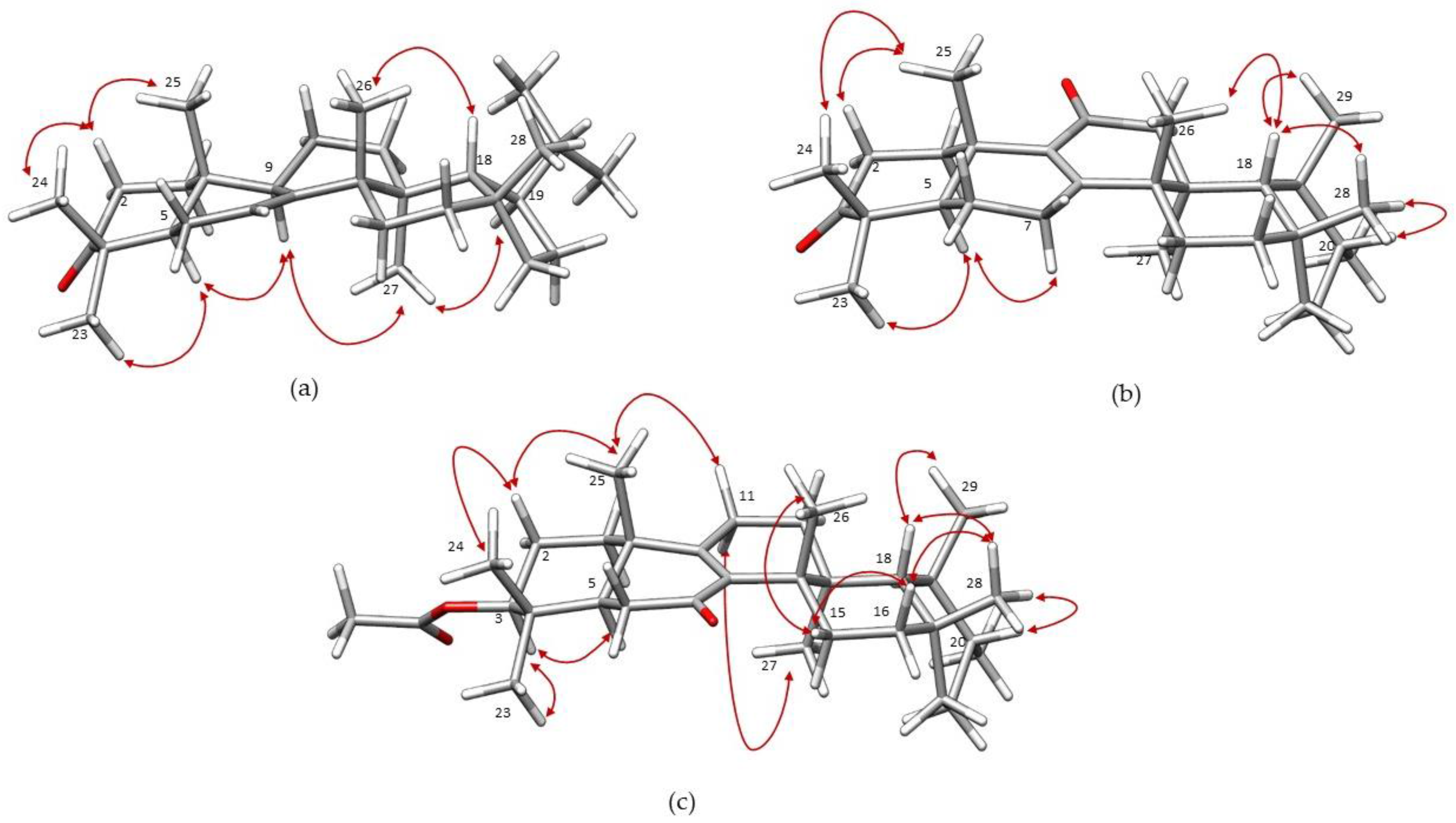
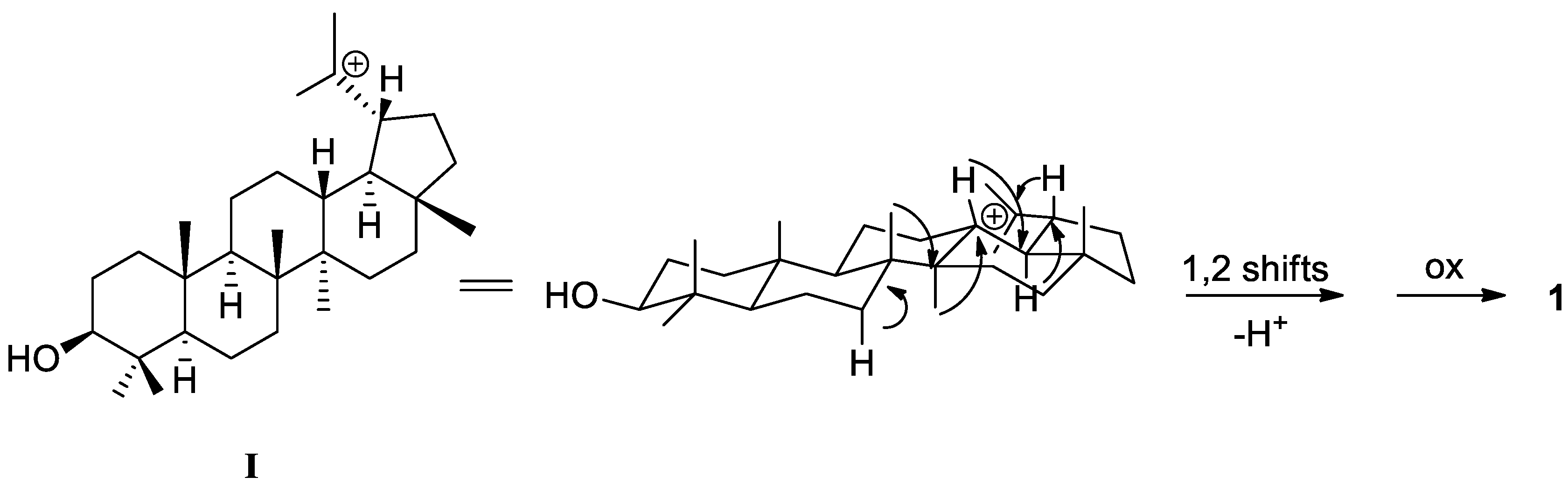
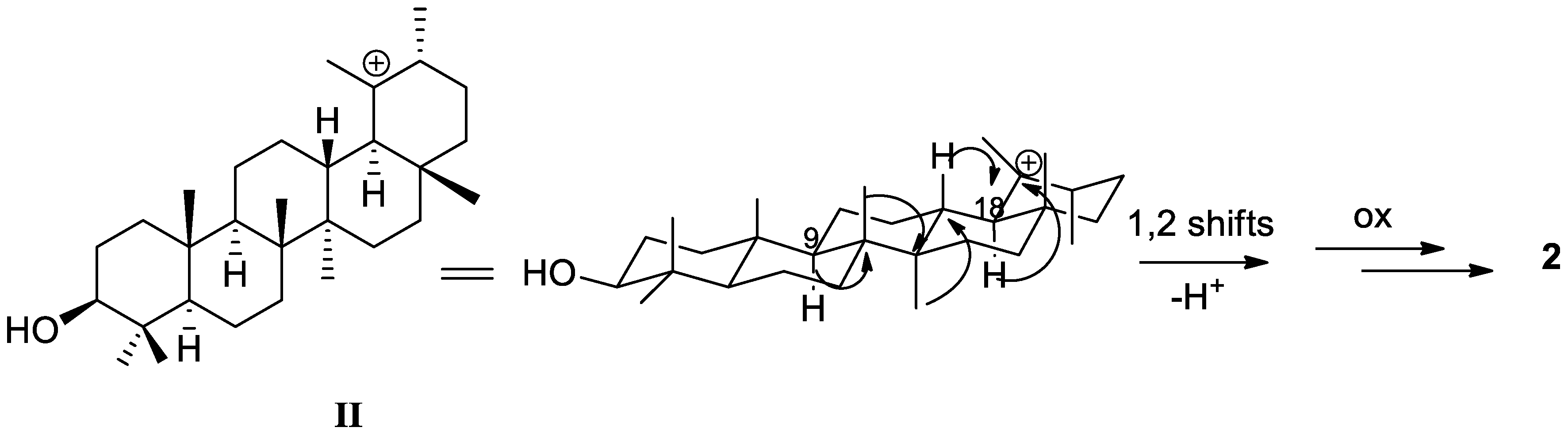
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ozenda, P. Flore et Végétation du Sahara, 3rd ed.; CNRS: Paris, French, 2004; p. 662. [Google Scholar]
- Quezel, P.; Santa, S. Nouvelle Flore de l’Algérie et des Régions Désertiques Méridionales, 1st ed.; CNRS: Paris, French, 1962–1963; Volume 1–2, p. 1162.
- Kilian, N. Revision of Launaea Cass. (Compositae, Lactuceae, Sonchinae). In Englera, 1st ed.; Botanischer Garten und Botanisches Museum: Berlin-Dahlem, Germany, 1997; Volume 17, pp. 211–217. [Google Scholar]
- Acherkouk, M.; Maatougui, A.; El Houmaiz, M.A. Communautés végétales et faciès pastoraux dans la zone de Taourirt-Tafoughalt du Maroc oriental: Écologie et inventaire floristique. Acta Bot. Malacit. 2011, 36, 125–136. [Google Scholar]
- Peñas, J.; Cabello, J.; Valle Tendero, F.; Mota, J.F. Comunidades vegetales rupícolas y subrupícolas del sudeste ibérico (Sierra de Los Filabres). Lazaroa 2001, 22, 95–107. [Google Scholar]
- Cheriti, A.; Belboukhari, M.; Belboukhari, N.; Djeradi, H. Phytochemical and biological studies on Launaea Cass. genus (Asteraceae) from Algerian Sahara. Curr. Top. Phytochem. 2012, 11, 67–80. [Google Scholar]
- Cheriti, A.; Belboukhari, M. Terpenoids of the Saharan medicinal Plants Launaea Cass. Genus (Asteraceae) and Their Biological Activities. In Terpenoids and Squalene; Bates, A.R., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2015; pp. 51–70. ISBN 9781634636568. [Google Scholar]
- Bitam, F.; Ciavatta, M.L.; Manzo, E.; Dibi, A.; Gavagnin, M. First chemical characterisation of the terpenoid constituents of the Algerian plant Launaea arborescens. Phytochemistry 2008, 69, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Bouzergoune, F.; Ciavatta, M.L.; Bitam, F.; Carbone, M.; Aberkane, M.C.; Gavagnin, M. Phytochemical study of Eryngium triquetrum: Isolation of polyacetylenes and lignans. Planta Med. 2016, 82, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Boumaraf, M.; Carbone, M.; Ciavatta, M.L.; Benyahia, S.; Ameddah, S.; Menad, A.; Benayache, S.; Benayache, F.; Gavagnin, M. Exploring the bioactive terpenoid content of an Algerian plant of genus Pulicaria: The ent-series of asteriscunolides. J. Nat. Prod. 2017, 80, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Giner, R.M.; Diaz, J.; Manez, S.; Recio, M.C.; Soriano, C.; Rios, J.L. Phenolic of Spanish Launaea species. Biochem. Syst. Ecol. 1992, 20, 187–188. [Google Scholar] [CrossRef]
- Benmeddour, T.; Laouer, H.; Akkal, S.; Flamini, G. Chemical composition and antibacterial activity of essential oil of Launaea lanifera Pau grown in Algerian arid steppes. Asian Pac. J. Trop. Biomed. 2015, 5, 960–964. [Google Scholar] [CrossRef]
- Chapon, S.; David, S. Ètude de l’insaponifiable de l’écorce d’aulne, Alnus glutinosa. Bull. Soc. Chim. Fr. 1953, 333–334. [Google Scholar]
- Hisham, A.; Kumar, G.J.; Fujimoto, Y.; Hara, N. Salacione and salaciol, two triterpenes from Salacia beddomei. Phytochemistry 1995, 40, 1227–1231. [Google Scholar] [CrossRef]
- Hui, W.-H.; Li, M.-M. Neutral triterpenoids from Malaleuca leucadendron. Phytochemistry 1976, 15, 563. [Google Scholar] [CrossRef]
- Yanna, C.F.; Gomes, R.A.; Oliveira, M.S.; de Lucena, K.L.; do Nascimento, J.S.; Agra, M.F.; Igoli, J.O.; Gray, A.I.; de Souza, M.F.V. Phytochemical investigation of Wissadula periplocifolia (L.) C. Presl and evaluation of its antibacterial activity. Quim. Nova 2014, 37, 1491–1495. [Google Scholar]
- Mills, J.S.; Werner, A.E.A. The chemistry of dammar resin. J. Chem. Soc. (Resumed) 1955, 3132–3140. [Google Scholar] [CrossRef]
- Phongmaykin, J.; Kumamoto, T.; Ishikawa, T.; Suttisri, R.; Saifah, E. A new sesquiterpene and other terpenoid constituents of Chisocheton penduliflorus. Arch. Pharm. Res. 2008, 31, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Bandaranayake, W.M. Terpenoids of Canarium zeylanicum. Phytochemistry 1980, 19, 255–257. [Google Scholar] [CrossRef]
- Hu, H.-J.; Wang, K.-W.; Wu, B.; Sun, C.-R.; Pan, Y.-J. Chemical shift assignments of two oleanane triterpenes from Euonymus hederaceus. J. Zhejiang Univ. SCI 2005, 6B, 719–721. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Andrés, L.S.; Ravelo, A.G.; Luis, J.G.; Bazzocchi, I.L.; West, J. Terpenoids from Salvia mellifera. Phytochemistry 1990, 29, 1691–1693. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; Cuatrecasas, J.; King, R.M.; Robinson, H. Neue sesquiterpene und norterpene aus vertretern der gattung Libanothamnus. Phytochemistry 1980, 19, 1145–1148. [Google Scholar] [CrossRef]
- Schulze, E.; Steiger, E. Untersuchungen über die stickstoffreien die stickstoffreien Reservestoffe der Samen von Lupinus luteus und über die Umwandlungen derselben während des Keimungsprozesses. Landw. Versuchsstat. 1889, 36, 391–476. [Google Scholar]
- Reynolds, W.F.; McLean, S.; Poplawski, J.; Enriquez, R.G.; Escobar, L.I.; Leon, I. Total assignment of 13C and 1H spectra of three isomeric triterpenol derivatives by 2D NMR: An investigation of the potential utility of 1H chemical shifts in structural investigations of complex natural products. Tetrahedron 1986, 42, 341–3428. [Google Scholar] [CrossRef]
- Itoh, T.; Tamura, T.; Matsumoto, T. Tirucalla-7,24-dienol: A new triterpene alcohol from tea seed oil. Lipids 1976, 11, 434–441. [Google Scholar] [CrossRef]
- Wu, B.; Lee, J.G.; Lim, C.J.; Jia, S.D.; Kwon, S.W.; Hwang, G.; Park, J.H. Sesquiterpenoids and 2-(2-phenylethyl)-4H-chromen-4-one (=2-(2-phenylethyl)-4H-1-benzopyran-4-one) derivatives from Aquilaria malaccensis Agarwood. Helv. Chim. Acta 2012, 95, 636–642. [Google Scholar] [CrossRef]
- Xu, R.; Fazio, G.C.; Matsuda, S.P.T. On the origins of triterpenoid skeletal diversity. Phytochemistry 2004, 65, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, K.; Hashimoto, Y.; Qiang, W.; Zhenwen, X. Separation of the pentacyclic triterpenes tylolupenols A and B from Tylophora kerrii. Phytochemistry 1985, 24, 2051–2054. [Google Scholar] [CrossRef]
- Konda, Y.; Urano, M.; Harigaya, Y.; Takayanagi, H.; Ogura, H.; Li, X.; Lou, H.; Onda, M. Novel triterpenes, hancolupenone and hancolupenol, from Cynanchum hancokianum. Chem. Pharm. Bull. 1990, 38, 2899–2901. [Google Scholar] [CrossRef]
- Lou, H.; Li, X.; Onda, M.; Konda, Y.; Urano, M.; Harigaya, Y.; Takayanagi, H.; Ogura, H. Stereochemistry of novel triterpenes from Cynanchum hancokianum. Chem. Pharm. Bull. 1991, 39, 2271–2276. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogura, H.; Konda, Y.; Urano, M.; Harigaya, Y.; Li, X.; Lou, H.; Onda, M. The crystal and molecular structures of hancokinol and hancolupenone from Cynanchum hancokianum (Maxim.) Al. Iljinski. (Asclepiadaceae). Chem. Pharm. Bull. 1991, 39, 1234–1237. [Google Scholar] [CrossRef]
- Shiojima, K.; Masuda, K.; Suzuki, H.; Lin, T.; Ooishi, Y.; Ageta, H. Composite constituents: Forty-two triterpenoids including eight novel compounds isolated from Picris hieracioides subsp. japonica. Chem. Pharm. Bull. 1995, 43, 1634–1639. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Das, B.; Pakrashi, C.S.; McPhail, D.R.; McPhail, A.T. X-ray crystal structure of swertanone, a triterpene of new skeletal type from Swerfia chirata Buch-Ham. J. Chem. Soc. Chem. Commun. 1989, 438–440. [Google Scholar] [CrossRef]
- Lahey, F.N.; Leeding, M.V. A New triterpene alcohol, bauerenol. Proc. Chem. Soc. 1958, 342–343. [Google Scholar]
- Fukuoka, M.; Natori, S. Oxidation of bauerenol derivatives with chromium trioxide: Confirmation of the structure of bauerenol. Chem. Pharm. Bull. 1972, 20, 974–979. [Google Scholar] [CrossRef]
- De Paiva Campello, J.; Marsaioli, A.J. Terebenthifolic acid and bauerenone: New triterpenoid ketones from Schinus terebenthifolius. Phytochemistry 1975, 14, 2300–2302. [Google Scholar] [CrossRef]
- Cerda-García-Rojas, C.; Hernández-Vidal, H.H.; Joseph-Nathan, P. 13C NMR assignments of D:C-friedours-7-ene derivatives. Evidence of an abnormal methyl group chemical shift. Magn. Res. Chem. 1996, 34, 777–781. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Das, B.; Masuda, K.; Arai, Y.; Shiojima, K. Peracid induced oxidative rearrangements of triterpenoids: Products of new skeletons from bauerenyl acetate. Tetrahedron 1998, 54, 6065–6078. [Google Scholar] [CrossRef]
- Vouffo, B.; Krohn, K.; Kouam, S.F.; Hussain, H.; Dongo, E.; Meier, K.; Schulz, B. Dinklagenonoate: A new isobauerane-type triterpenoid and other minor constituents from the twigs of Dorstenia dinklagei. Biochem. Syst. Ecol. 2008, 36, 655–658. [Google Scholar] [CrossRef]
- Kikuchi, T.; Tanaka, A.; Uriuda, M.; Yamada, T.; Tanaka, R. Three novel triterpenoids from Taraxacum officinale roots. Molecules 2016, 21, 1121. [Google Scholar] [CrossRef] [PubMed]
- Talapatra, S.K.; Sengupta, S.; Talapatra, B. A new pentacyclic triterpene alcohol from Evodia franxinifolia Hook F. Tetrahedron Lett. 1968, 57, 5963–5968. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–12 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| 13C | 1H (J, Hz) | 13C | 1H (J, Hz) | 13C | 1H (J, Hz) | |
| 1a | 38.3, CH2 | 1.99, m | 35.2, CH2 | 2.79, ddd (13.5, 7.6, 4.5) | 33.9, CH2 | 2.30, m |
| 1b | 1.43, m | 1.44, m | 1.84, m | |||
| 2a | 35.0, CH2 | 2.74, ddd (14.4, 14.4, 5.5) | 34.8, CH2 | 2.55, m | 25.1, CH2 | 1.71, m |
| 2b | 2.24, m | 2.48, m | 1.54, m | |||
| 3 | 216.9, C | 217.5, C | 79.8, CH | 4.52, dd (11.6, 4.1) | ||
| 4 | 47.7, C | 47.3, C | 37.6, C | |||
| 5 | 51.9, CH | 1.69, dd (9.9, 7.9) | 51.6, CH | 1.60, m | 47.4, CH | 1.72, dd (12.5, 6.8) |
| 6a | 24.4, CH2 | 2.11 m | 21.6, CH2 | 1.72, m | 36.4, CH2 | 2.41, dd (18.6, 6.8) |
| 6b | 1.48, m | 2.36, dd (18.6, 12.5) | ||||
| 7a | 117.2, CH | 5.53 dd (6.4, 3.2) | 30.5, CH2 | 2.45, m | 198.4, C | |
| 7b | 2.14, m | |||||
| 8 | 145.4, C | 164.1, C | 139.3, C | |||
| 9 | 47.9, CH | 2.24, m | 139.5, C | 164.4, C | ||
| 10 | 35.4, C | 37.1, C | 39.2, C | |||
| 11a | 16.6, CH2 | 1.62, m | 198.0, C | 23.6, CH2 | 2.29, m | |
| 11b | 1.54, m | 2.14, m | ||||
| 12a | 32.4, CH2 | 1.54, m | 49.5, CH2 | 2.26, s | 29.6, CH2 | 1.50, m |
| 12b | 1.40, m | |||||
| 13 | 37.7, C | 39.6, C | 38.4, C | |||
| 14 | 40.4, C | 43.1, C | 40.4, C | |||
| 15a | 28.8, CH2 | 1.32, m | 26.1, CH2 | 1.14, m | 23.8, CH2 | 1.38, m |
| 15b | 1.60, m | |||||
| 16a | 34.7, CH2 | 1.49, m | 36.6, CH2 | 1.57, m | 37.8, CH2 | 1.55, m |
| 16b | 1.27, m | 1.17, m | ||||
| 17 | 40.6, C | 32.3, C | 31.5, C | |||
| 18 | 56.4, CH | 1.50, m | 52.2, CH | 1.42, brs | 51.5, CH | 1.33, brd (2.3) |
| 19 | 49.7, CH | 1.58, m | 36.2, CH | 1.02, m | 36.0, CH | 1.03, m |
| 20 | 35.0, CH | 1.55, m | 31.0, CH | 1.40, m | 33.1, CH | 1.59, m |
| 21a | 28.6, CH2 | 1.77, m | 28.6, CH2 | 1.60, m | 27.9, CH2 | 1.67, m |
| 21b | 1.51, m | 1.31,m | ||||
| 22a | 38.6, CH2 | 1.75, m | 31.5, CH2 | 1.26, m | 31.8, CH2 | 1.55, m |
| 22b | 1.17, m | 1.26, m | ||||
| 23 | 24.7, CH3 | 1.04, s | 27.6, CH3 | 1.11, s | 29.6, CH3 | 0.87, s |
| 24 | 21.5, CH3 | 1.12, s | 21.8, CH3 | 1.08, s | 16.0, CH3 | 0.95, s |
| 25 | 12.7, CH3 | 0.99, s | 19.8, CH3 | 1.27, s | 18.5, CH3 | 1.01, s |
| 26 | 23.5, CH3 | 1.00, s | 22.0, CH3 | 1.16, s | 21.7, CH3 | 1.22, s |
| 27 | 23.2, CH3 | 0.91, s | 18.1, CH3 | 1.01, s | 15.4, CH3 | 0.84, s |
| 28 | 33.1, CH3 | 0.92, s | 38.3, CH3 | 1.09, s | 38.1, CH3 | 1.06, s |
| 29 | 22.0, CH3 | 0.88, d (6.3) | 25.7, CH3 | 1.03, brs | 27.1, CH3 | 0.99, brs |
| 30 | 23.2, CH3 | 0.91, d (6.0) | 23.1, CH3 | 0.90, d (5.9) | 22.5, CH3 | 0.91, d (5.9) |
| Ac-CO | 170.9, C | |||||
| Ac-CH3 | 21.3, CH3 | 2.07, s | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zergainoh, N.; Ciavatta, M.L.; Carbone, M.; Bitam, F.; Aberkane, M.C.; Gavagnin, M. Exploring the Chemical Diversity of Algerian Plants: Three New Pentacyclic Triterpenoids from Launaea acanthoclada Roots. Molecules 2018, 23, 80. https://doi.org/10.3390/molecules23010080
Zergainoh N, Ciavatta ML, Carbone M, Bitam F, Aberkane MC, Gavagnin M. Exploring the Chemical Diversity of Algerian Plants: Three New Pentacyclic Triterpenoids from Launaea acanthoclada Roots. Molecules. 2018; 23(1):80. https://doi.org/10.3390/molecules23010080
Chicago/Turabian StyleZergainoh, Nabila, Maria Letizia Ciavatta, Marianna Carbone, Fatma Bitam, Mohamed Cherif Aberkane, and Margherita Gavagnin. 2018. "Exploring the Chemical Diversity of Algerian Plants: Three New Pentacyclic Triterpenoids from Launaea acanthoclada Roots" Molecules 23, no. 1: 80. https://doi.org/10.3390/molecules23010080
APA StyleZergainoh, N., Ciavatta, M. L., Carbone, M., Bitam, F., Aberkane, M. C., & Gavagnin, M. (2018). Exploring the Chemical Diversity of Algerian Plants: Three New Pentacyclic Triterpenoids from Launaea acanthoclada Roots. Molecules, 23(1), 80. https://doi.org/10.3390/molecules23010080