Curcumin Analog DK1 Induces Apoptosis in Human Osteosarcoma Cells In Vitro through Mitochondria-Dependent Signaling Pathway


 ,
, 
 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
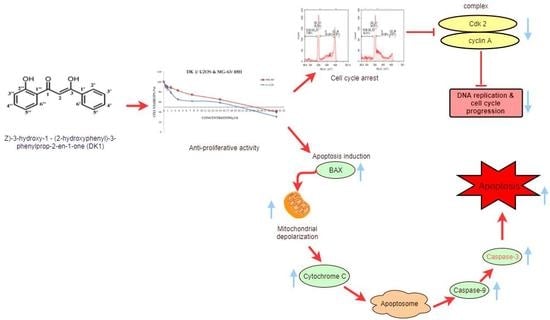
2.1. DK 1 Inhibits the Proliferation of U-2OS and MG-63
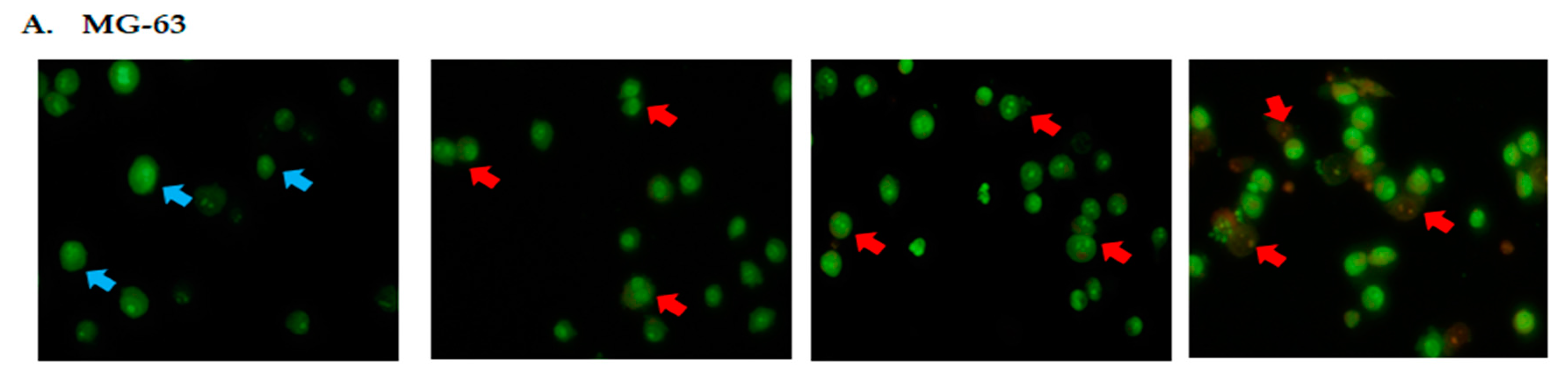
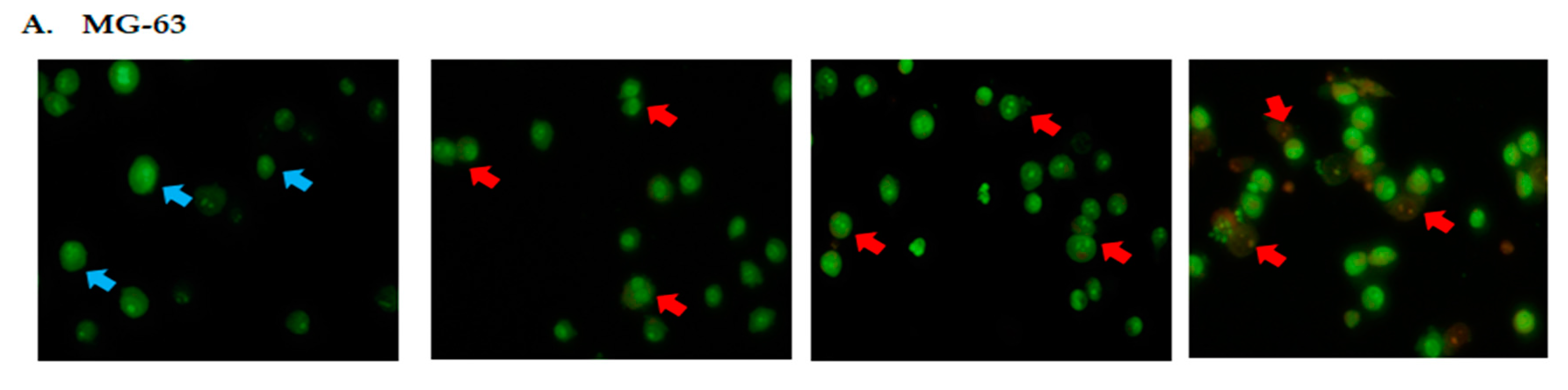
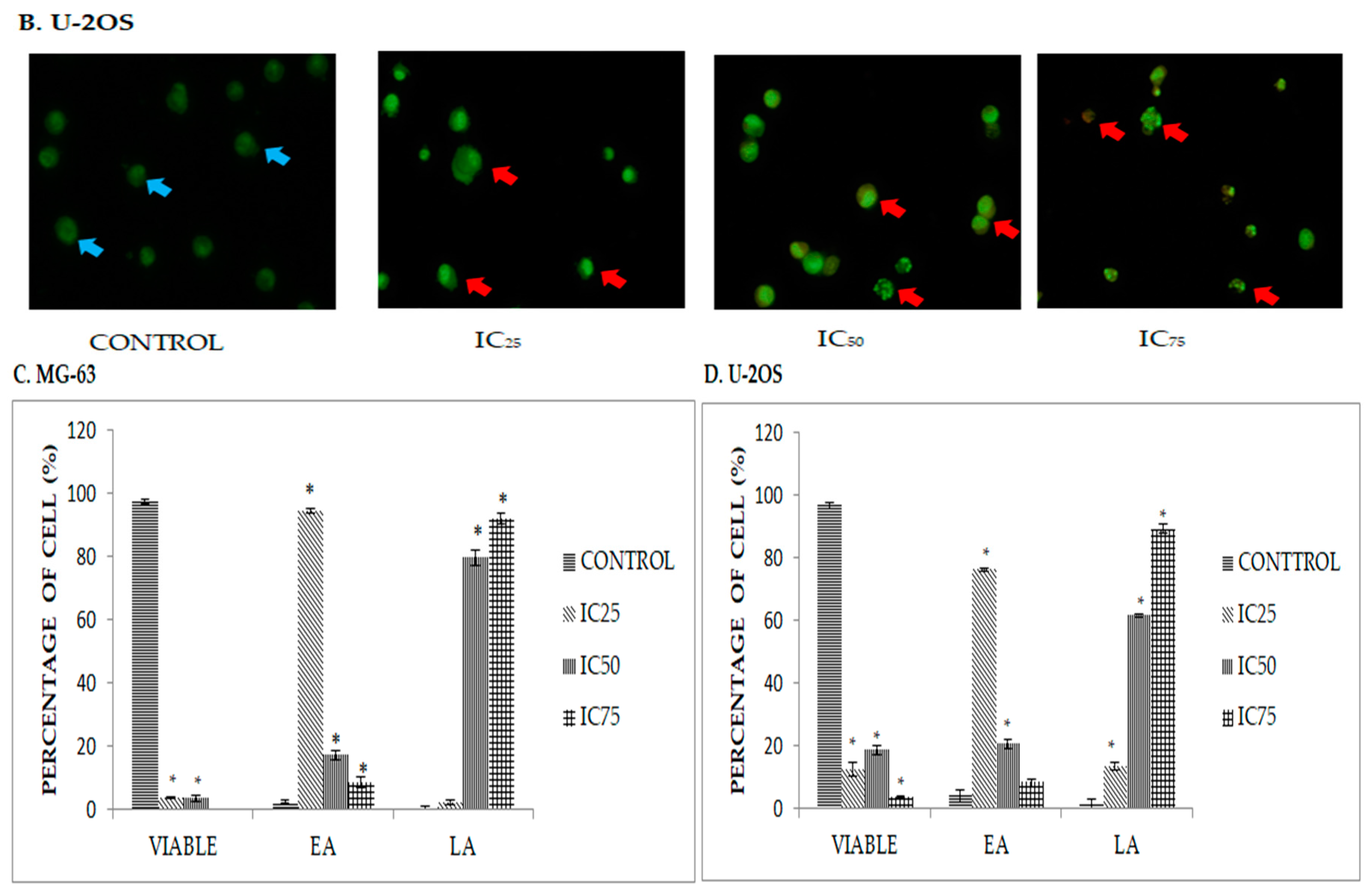
2.2. DK1 Altered the Morphological Appearances and Induces Apoptosis in U-2OS and MG-63
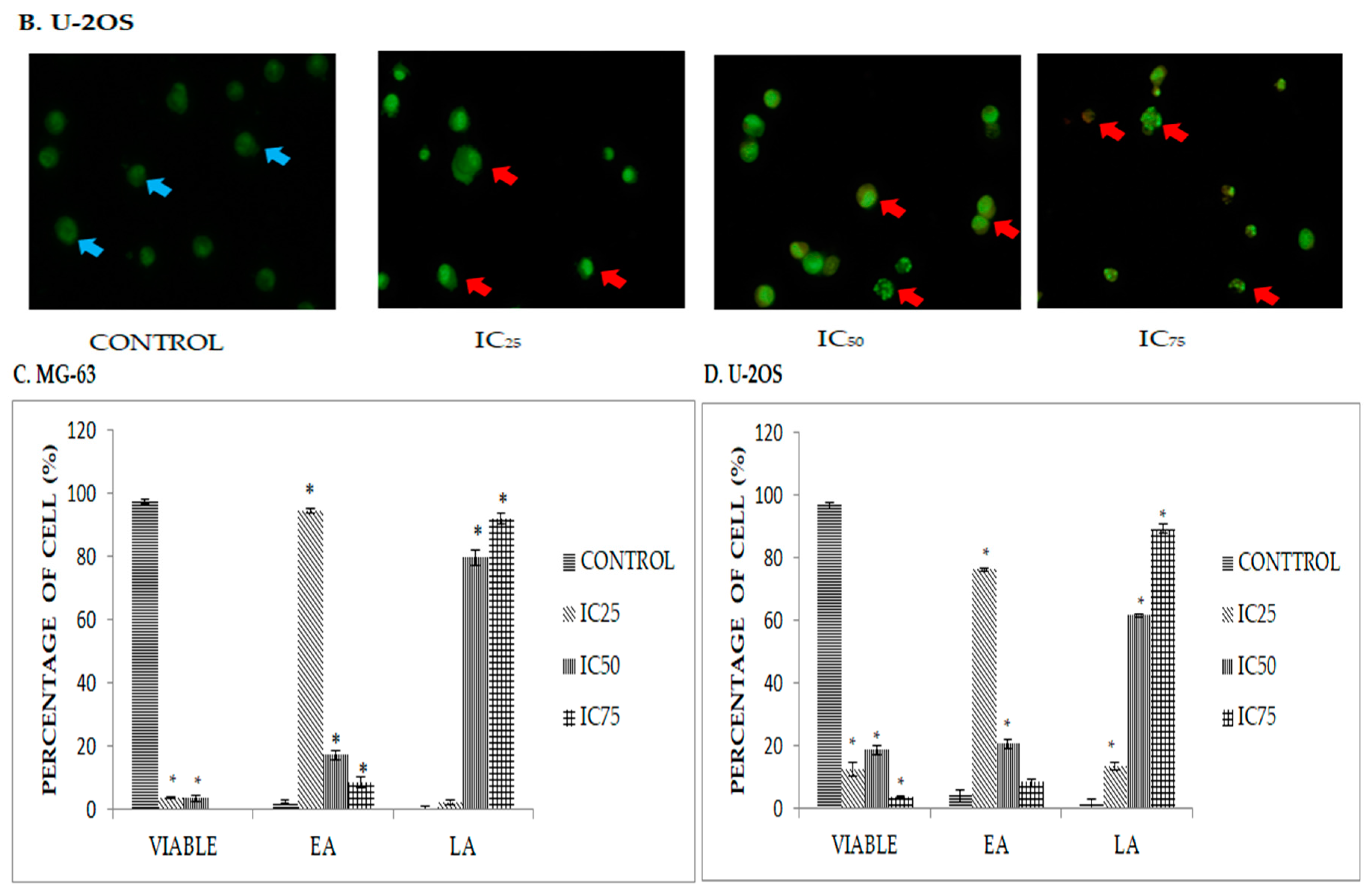
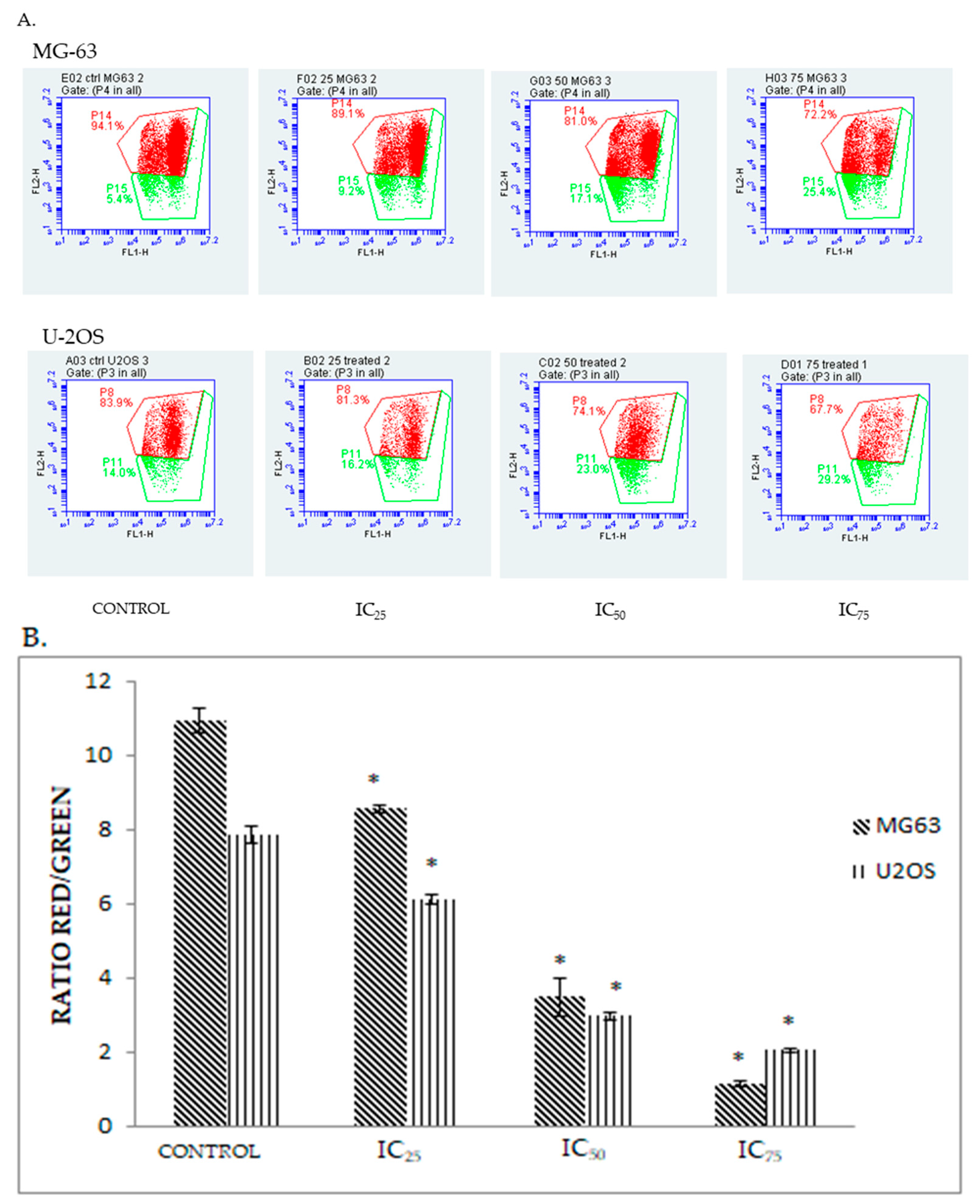
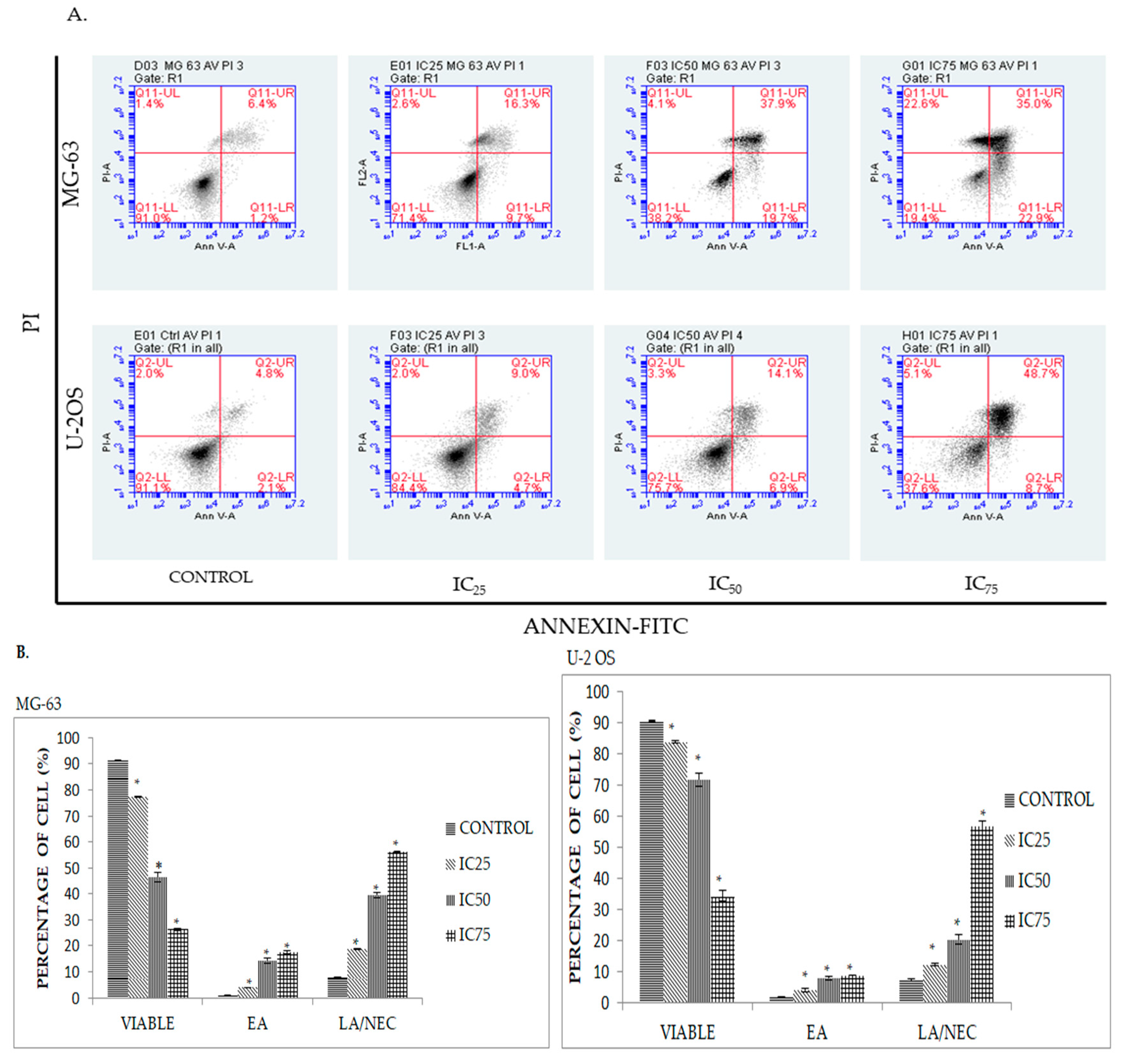
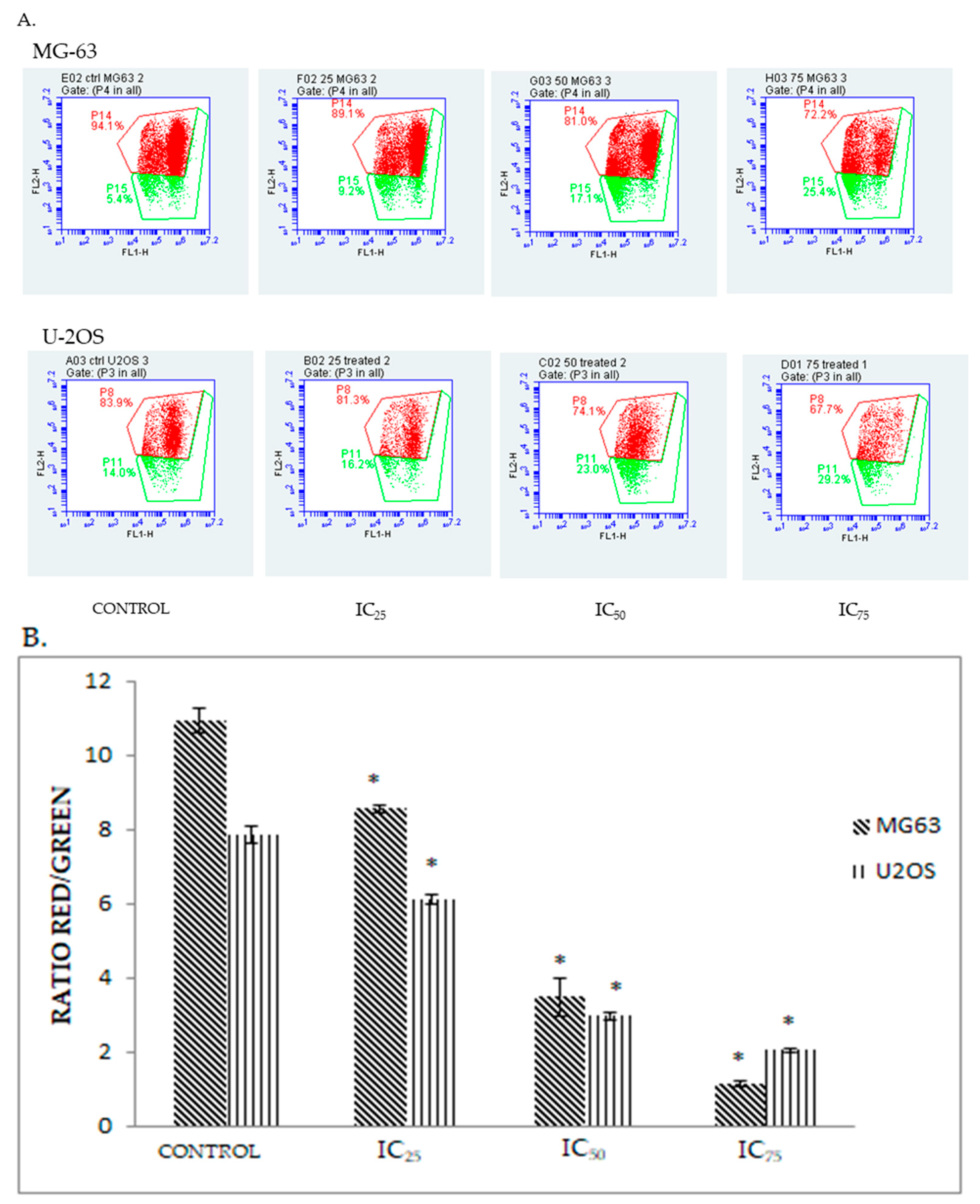
2.3. Quatification of Apoptotic Cell Death upon Exposure to DK1 via Annexin V/FITC Binding Assay
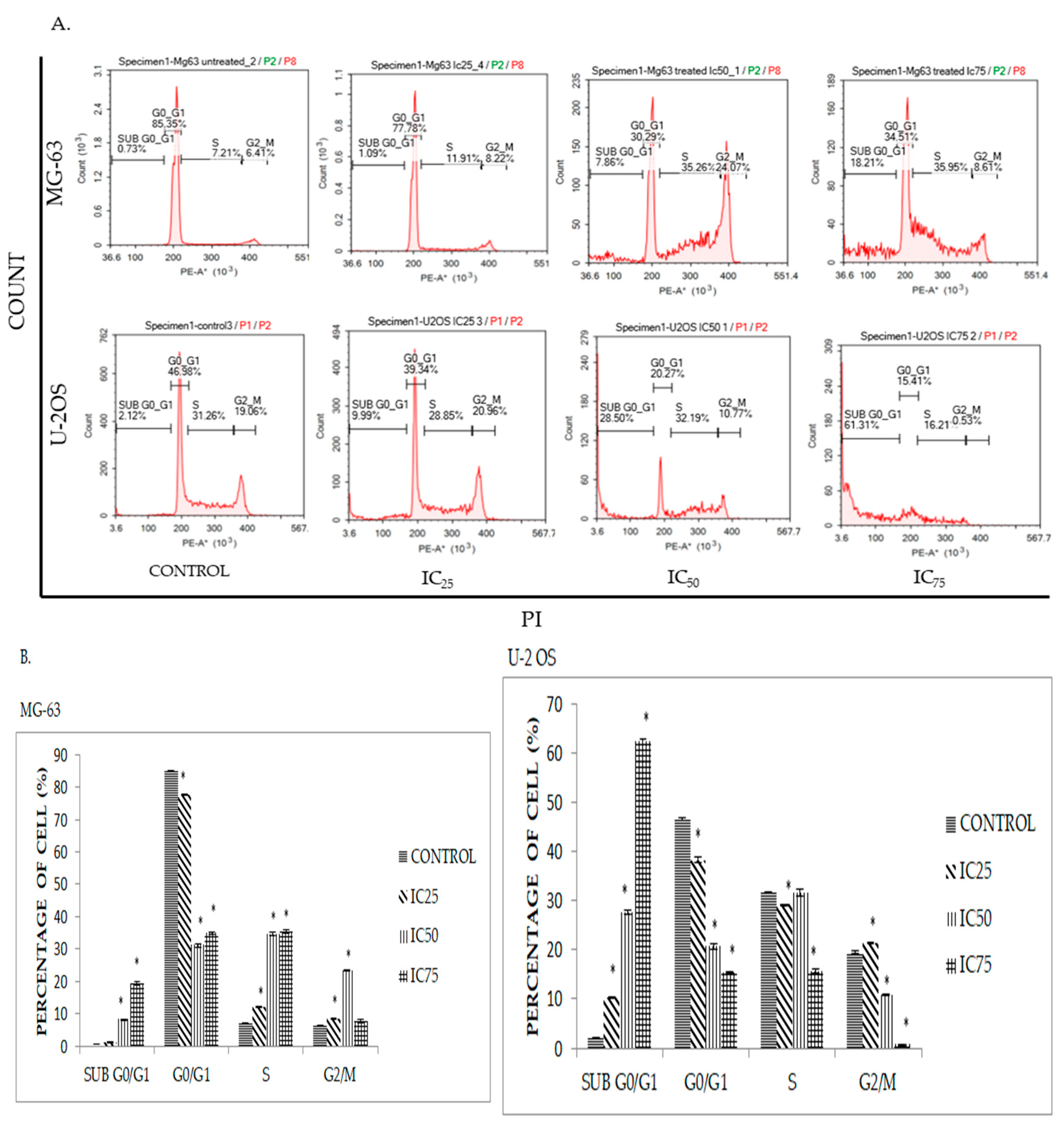
2.4. DK1 Induces Cell Cycle Accumulation at S Phase in MG-63 and U-2OS
2.5. DK 1 Induces Changes in the Mitochondrial Membrane Potential of U-2OS and MG-63
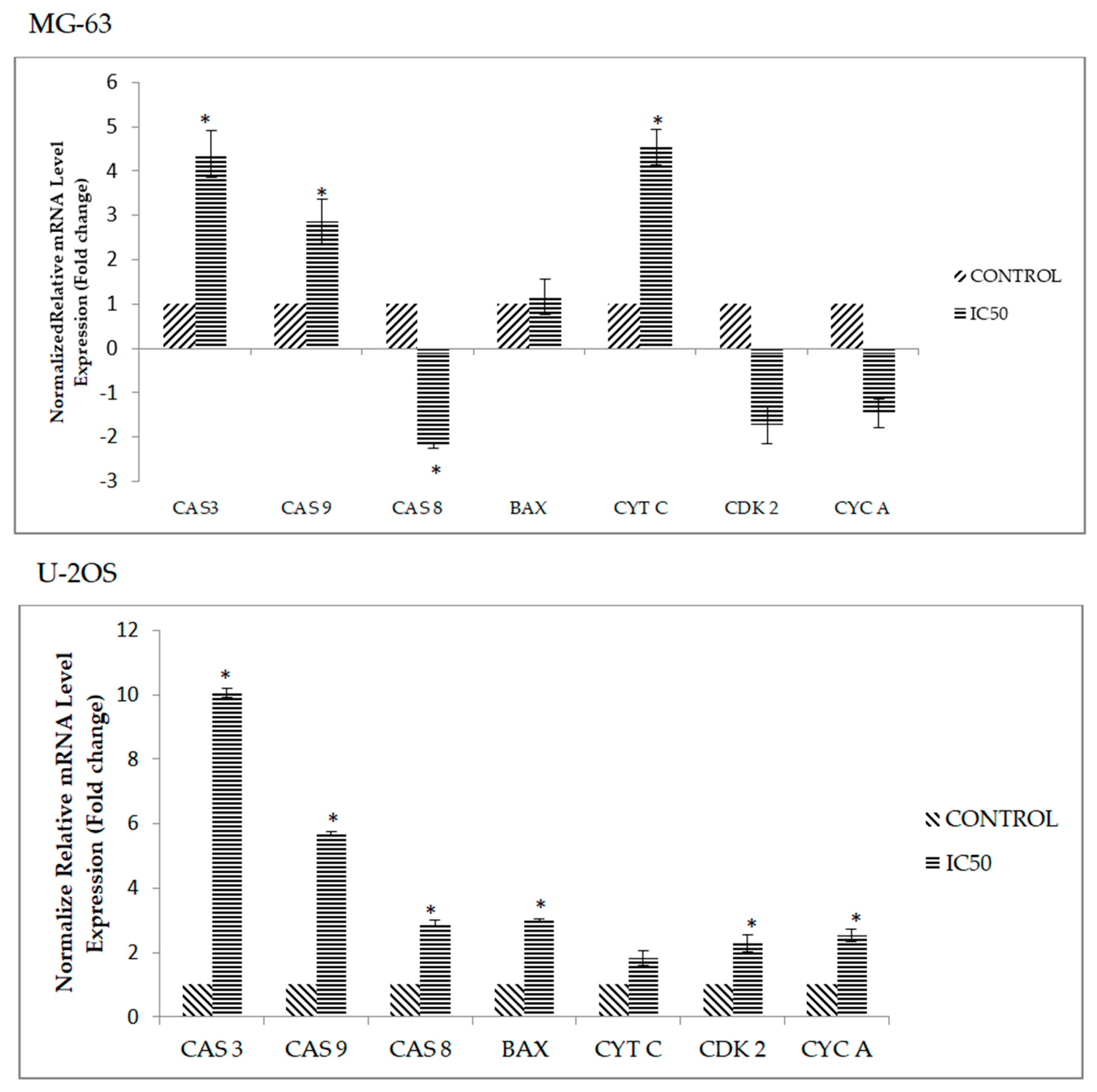
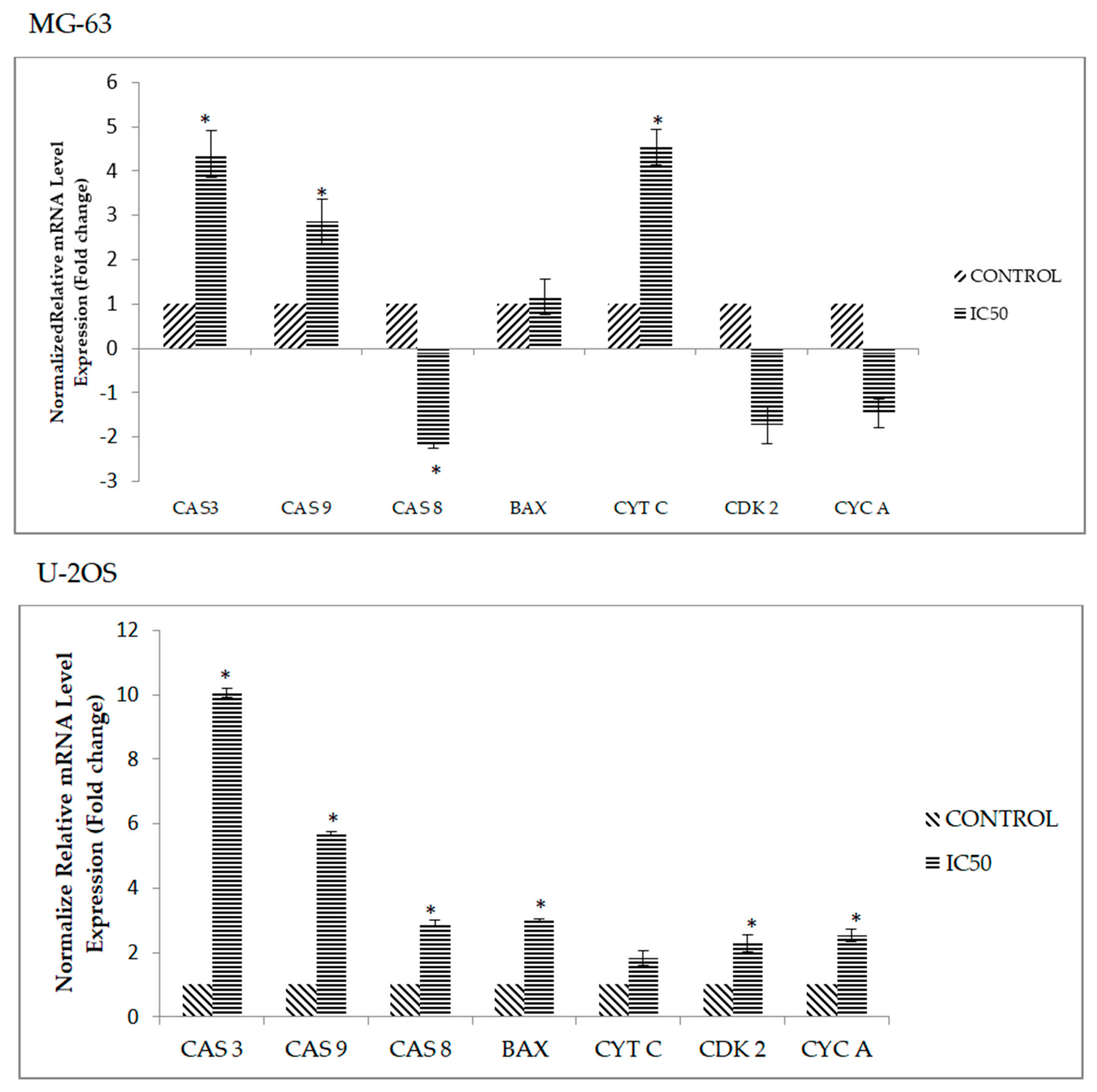
2.6. DK1 Regulates Several Apoptosis and Cell Cycle Related Genes and Protein
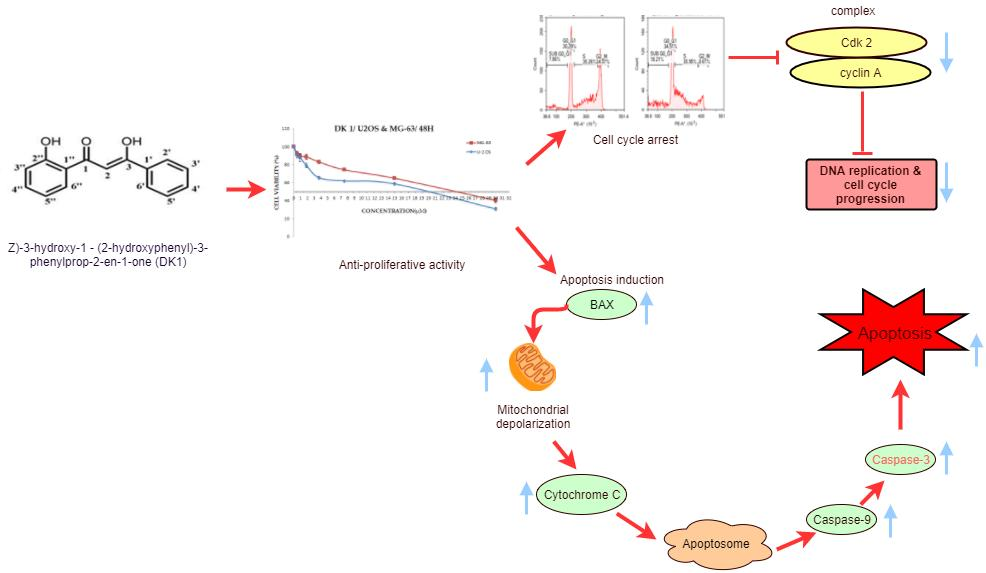
3. Discussion
4. Materials and Methods
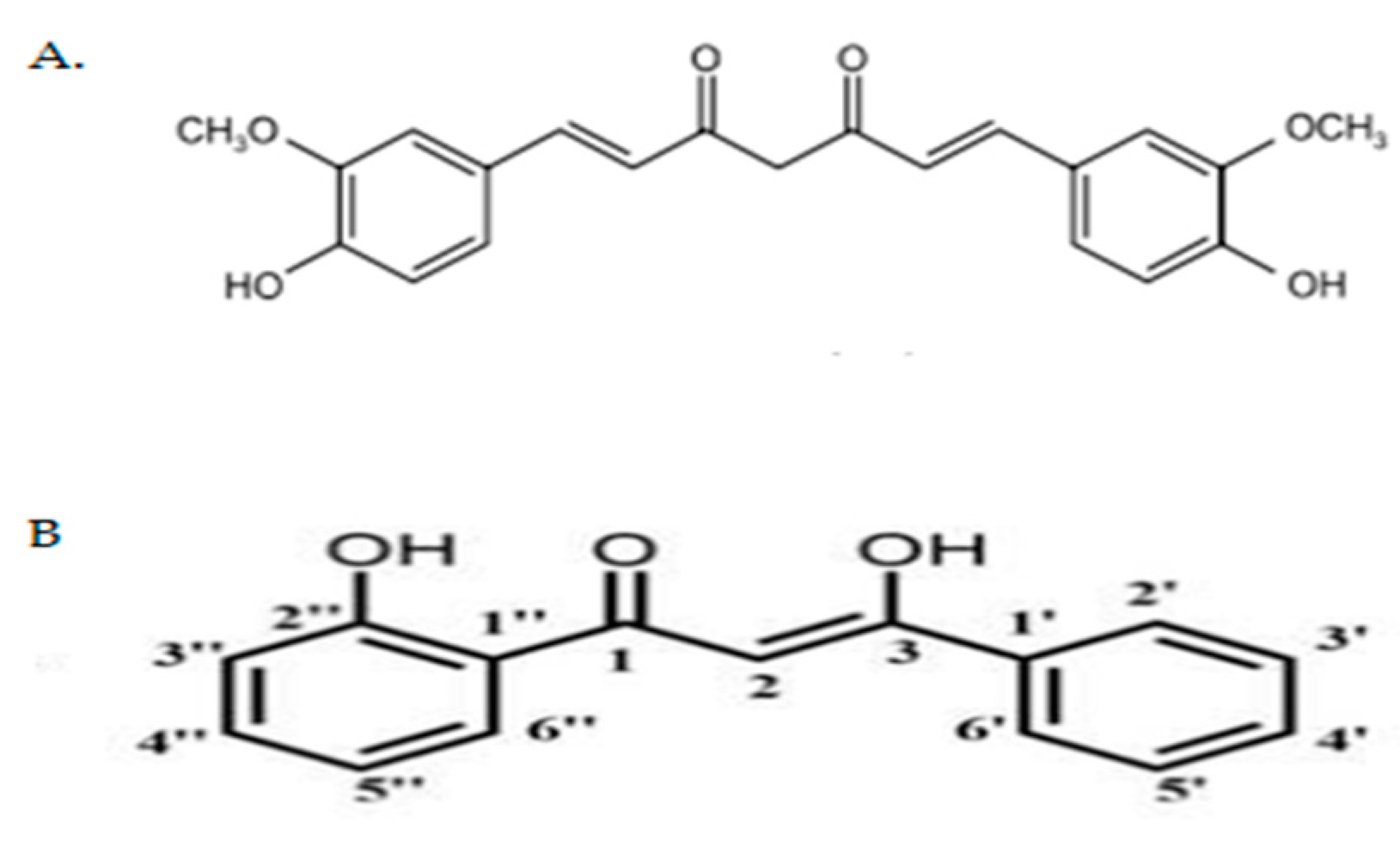
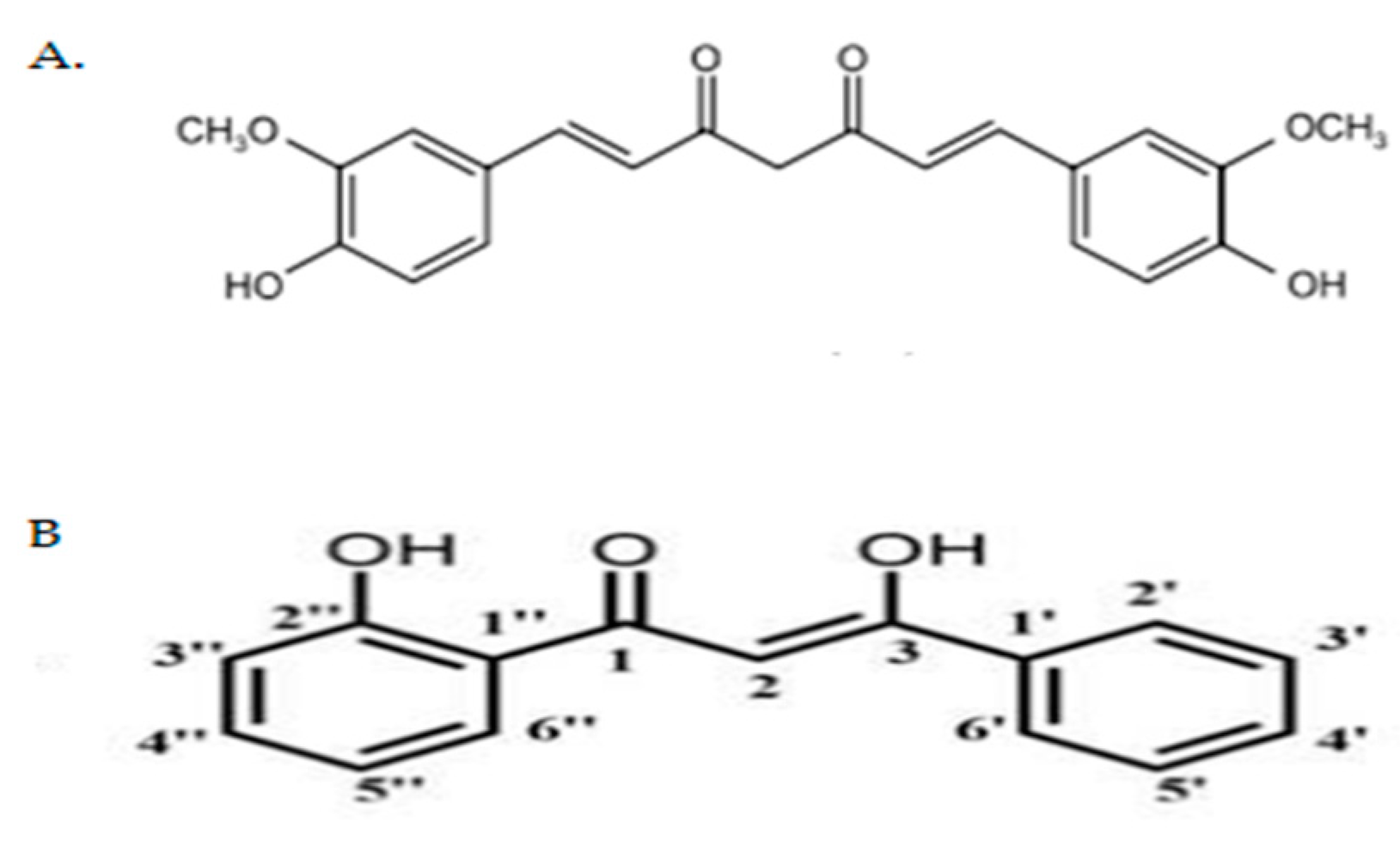
4.1. Preparation of Curcumin Analogue DK1
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Treatment
4.5. Florescence Detection Using Acridine Orange/Propidium Iodide (AO/PI) Double Staining Assay
4.6. Cell Cycle Analysis
4.7. Annexin V/FITC Binding Assay
4.8. JC-1 MitoScreen Assay
4.9. Quantitive Real Time PCR Assay
4.10. Proteome Profiling of Apoptosis-Related Protein
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Broadhead, M.L.; Clark, J.C.M.; Myers, D.E.; Dass, C.R.; Choong, P.F.M. The molecular pathogenesis of osteosarcoma: A review. Sarcoma 2011, 2011, 959248. [Google Scholar] [CrossRef] [PubMed]
- Briccoli, A.; Rocca, M.; Salone, M.; Guzzardella, G.A.; Balladelli, A.; Bacci, G. High grade osteosarcoma of the extremities metastatic to the lung: Long-term results in 323 patients treated combining surgery and chemotherapy, 1985–2005. Surg. Oncol. 2010, 19, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, L.; Hong, Z.; Zheng, H.; Li, N.; Gao, H.; Chen, B.; Zhao, Y. Methanolic extract of pientze huang induces apoptosis signaling in human osteosarcoma MG63 cells via multiple pathways. Molecules 2016, 21, 283. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yuan, M.; Yuan, H.; Huang, X.; Sui, X.; Cui, X.; Tang, F.; Peng, J.; Lu, S.; Xu, W.; et al. Co-encapsulation of magnetic Fe3O4 nanoparticles and doxorubicin into biodegradable PLGA nanocarriers for intratumoral drug delivery. Int. J. Nanomed. 2012, 7, 1697–1708. [Google Scholar]
- Bao, M.; Cao, Z.; Yu, D.; Fu, S.; Zhang, G.; Yang, P.; Pan, Y.Y.; Yang, B.; Han, H.Y.; Zhou, Q. Columbamine suppresses the proliferation and neovascularization of metastatic osteosarcoma U2OS cells with low cytotoxicity. Toxicol. Lett. 2012, 215, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.M.; Yeap, S.K.; Abu, N.; Lim, K.L.; Ky, H.; Pauzi, A.Z.M.; Ho, W.Y.; Tan, S.W.; Alan-Ong, H.K.; Zareen, S.; et al. Synthetic curcumin derivative DK1 possessed G2/M arrest and induced apoptosis through accumulation of intracellular ROS in MCF-7 breast cancer cells. Cancer Cell Int. 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xing, H.; Gao, Q.; Chen, G.; Lu, Y.; Wang, S.; Ma, D. Regulation of HtrA2/Omi by X-linked inhibitor of apoptosis protein in chemoresistance in human ovarian cancer cells. Gynecol. Oncol. 2005, 97, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Lozada-García, M.C.; Enríquez, R.G.; Ramírez-Apán, T.O.; Nieto-Camacho, A.; Palacios-Espinosa, J.F.; Custodio-Galván, Z.; Soria-Arteche, O.; Pérez-Villanueva, J. Synthesis of curcuminoids and evaluation of their cytotoxic and antioxidant properties. Molecules 2017, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg. Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Hong, H.M.; Kwon, D.D.; Pae, H.; Jeong, H.J. Dimethoxycurcumin, a structural analogue of curcumin, induces apoptosis in human renal carcinoma caki cells through the production of reactive oxygen species, the release of cytochrome c, and the activation of caspase-3. Korean J. Urol. 2010, 51, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Gerlier, D. Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
- Aziz, M.; Abu, N.; Yeap, S.; Ho, W.; Omar, A.; Ismail, N.H.; Ahmad, S.; Pirozyan, M.R.; Akhtar, N.N.; Alitheen, N.B. Combinatorial cytotoxic effects of damnacanthal and doxorubicin against human breast cancer MCF-7 cells in vitro. Molecules 2016, 21, 1228. [Google Scholar] [CrossRef] [PubMed]
- Gerl, R.; Vaux, D.L. Apoptosis in the development and treatment of cancer. Carcinogenesis 2005, 26, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Koopman, G.; Reutlingsperger, C.P.M.; Kuijten, G.A.M. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1421. [Google Scholar] [PubMed]
- Ishaque, A.; Al-Rubeai, M. Use of intracellular pH and annexin-V flow cytometric assays to monitor apoptosis and its suppression by bcl-2 over-expression in hybridoma cell culture. J. Immunol. Methods 1998, 221, 43–57. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stewart, Z.A. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181–182, 475–481. [Google Scholar] [CrossRef]
- Etti, I.C.; Rasedee, A.; Hashim, N.M.; Abdul, A.B.; Kadir, A.; Keong, S.; Waziri, P.; Ibrahim, M.; Lim, K.L.; Etti, C.J. Artonin E induces p53-independent G1 cell cycle arrest and apoptosis through ROS-mediated mitochondrial pathway and livin suppression in MCF-7 cells. Drug Des. Dev. Ther. 2017, 11, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Abu, N.; Akhtar, M.N.; Yeap, S.K.; Lim, K.L.; Ho, W.Y.; Zulfadli, A.J.; Omar, A.R.; Sulaiman, M.R.; Abdullah, M.P.; Alitheen, N.B. Flavokawain A induces apoptosis in MCF-7 and MDA- MB231 and inhibits the metastatic process in vitro. PLoS ONE 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lauvrak, S.U.; Munthe, E.; Kresse, S.H.; Stratford, E.W.; Namløs, H.M.; Meza-Zepeda, L.A.; Myklebost, O. Functional characterisation of osteosarcoma cell lines and identification of mRNAs and miRNAs associated with aggressive cancer phenotypes. Br. J. Cancer 2013, 109, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Ma, D.; Zhang, L.; Si, M.; Yin, H.; Li, J. Curcumin inhibits proliferation and invasion of osteosarcoma cells through in activation of Notch-1 signaling. FEBS J. 2012, 279, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Tao, J.G.; Wang, G.D.; Liu, S.P.; Zhao, H.X.; Liang, Q.D. In vitro antitumor activity of the ethyl acetate extract of Potentilla chinensis in osteosarcoma cancer cells. Mol. Med. Rep. 2016, 14, 3634–3640. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; He, J. Daphnoretin induces cell cycle arrest and apoptosis in human osteosarcoma (HOS) cells. Molecules 2012, 17, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “Sub-G1” peaks on DNA content histograms. Cytometry 2007, 71A, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, M.C.; Crosariol, M.; Loro, E.; Li, Z.; Pestell, R.G. Cyclins and cell cycle control in cancer and disease. Genes Cancer 2012, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Lee, M.K.; Kim, J.H. Curcumin induces cell cycle arrest and apoptosis in human osteosarcoma (HOS) cells. Anticancer Res. 2009, 29, 5039–5044. [Google Scholar] [PubMed]
- Forbes-hernández, T.Y.; Giampieri, F.; Gasparrini, M.; Mazzoni, L.; Quiles, J.L.; Alvarez-suarez, J.M.; Battino, M. The effects of bioactive compounds from plant foods on mitochondrial function: A focus on apoptotic mechanisms. Food Chem. Toxicol. 2014, 68, 154–182. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 1471–2121. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Eskes, R.; Antonsson, B.; Osen-sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+. J. Cell Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Shih, Y.-L.; Lee, M.-H.; Au, M.-K.; Chen, Y.-L.; Lu, H.-F.; Chung, J.-G. Bufalin induces apoptosis of human osteosarcoma U-2 OS cells through endoplasmic reticulum stress, caspase- and mitochondria-dependent signaling pathways. Molecules 2017, 22, 437. [Google Scholar] [CrossRef] [PubMed]
- Martinez-ruiz, G.; Maldonado, V.; Ceballos-cancino, G.; Grajeda, J.P.R.; Melendez-zajgla, J. Role of Smac/DIABLO in cancer progression. J. Exp. Clin. Cancer Res. 2008, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Akagi, T.; Hashikawa, T. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004, 11, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Chau, L. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds curcumin analog DK1 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | IC50 (µM) |
|---|---|
| MG-63 | 23.8 ± 0.8 |
| U-2OS | 19.6 ± 0.3 |
| 3T3 | >30 |
| Cell Lines | Proteins | Relative Intensity (Fold Change) | Regulation |
|---|---|---|---|
| U-2 OS | Pro-caspase 3 | 2.0 * ± 0.15 | Up |
| Cleaved caspase 3 | 4.6 * ± 0.07 | Up | |
| Bax | 4.5 * ±0.38 | Up | |
| Cytochrome c | 3.0 * ± 0.35 | Up | |
| Fas | 2.2 * ± 0.05 | Up | |
| HO-1/HMOX1/HSP32 | −0.6 ± 0.05 | Down | |
| HTRA2/Omi | 3.3 * ± 0.13 | Up | |
| SMAC/Diablo | 2.9 * ± 0.04 | Up | |
| MG-63 | Pro-caspase 3 | −0.6 * ± 0.03 | Down |
| Cleaved caspase 3 | 1.4 * ± 0.12 | Up | |
| Bax | −0.8 ± 0.02 | Down | |
| Cytochrome c | −0.8 ± 0.08 | Down | |
| Fas | −0.4 * ± 0.01 | Down | |
| HO-1/HMOX1/HSP32 | −0.4 * ± 0.13 | Down | |
| HTRA2/Omi | −0.6 * ± 0.12 | Down | |
| SMAC/Diablo | −0.8 *± 0.01 | Down |
| Genes | Accession Number | Forward Primers | Reverse Primers | Amplicon Size (bp) |
|---|---|---|---|---|
| Caspase 3 | NM_004346 | AGAACTGGACTGTGGCATTGAG | GCTTGTCGGCATACTGTTTCAG | 191 |
| Caspase 9 | NM_001229 | TGTCCTACTCTACTTTCCCAGGTTTT | GTGAGCCCACTGCTCAAAGAT | 101 |
| Caspase 8 | NM_001228 | CATCCAGTCACTTTGCCAGA | GCATCTGTTTCCCCATGTTT | 128 |
| BAX | BC014175 | CAAGAAGCTGAGCGAGTGT | CAGTTGAAGTTGCCGTCAGA | 153 |
| Cytochrome C | NM_018947.5 | GGGCCAAATCTCCATGGTCT | GGCAGTGGCCAATTATTACTC | 246 |
| Cyclin A | NM_001237 212 | GATGCTGACCCATACCTCAAG | GGTGAAGGTCCATGAGACAAG | 160 |
| CDK 2 | NM_052827 | GAAGATGGACGGAGCTTGTT | TGGAGGAGAGGGTGAGATTAG | 173 |
| ACTB | NM_001101.3 | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG | 184 |
| 18srRNA | X03205 | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | 151 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, M.N.M.; Hussin, Y.; Che Rahim, N.F.; Nordin, N.; Mohamad, N.E.; Yeap, S.K.; Yong, C.Y.; Masarudin, M.J.; Cheah, Y.K.; Abu, N.; et al. Curcumin Analog DK1 Induces Apoptosis in Human Osteosarcoma Cells In Vitro through Mitochondria-Dependent Signaling Pathway. Molecules 2018, 23, 75. https://doi.org/10.3390/molecules23010075
Aziz MNM, Hussin Y, Che Rahim NF, Nordin N, Mohamad NE, Yeap SK, Yong CY, Masarudin MJ, Cheah YK, Abu N, et al. Curcumin Analog DK1 Induces Apoptosis in Human Osteosarcoma Cells In Vitro through Mitochondria-Dependent Signaling Pathway. Molecules. 2018; 23(1):75. https://doi.org/10.3390/molecules23010075
Chicago/Turabian StyleAziz, Muhammad Nazirul Mubin, Yazmin Hussin, Nurul Fattin Che Rahim, Noraini Nordin, Nurul Elyani Mohamad, Swee Keong Yeap, Chean Yeah Yong, Mas Jaffri Masarudin, Yoke Kqueen Cheah, Nadiah Abu, and et al. 2018. "Curcumin Analog DK1 Induces Apoptosis in Human Osteosarcoma Cells In Vitro through Mitochondria-Dependent Signaling Pathway" Molecules 23, no. 1: 75. https://doi.org/10.3390/molecules23010075
APA StyleAziz, M. N. M., Hussin, Y., Che Rahim, N. F., Nordin, N., Mohamad, N. E., Yeap, S. K., Yong, C. Y., Masarudin, M. J., Cheah, Y. K., Abu, N., Akhtar, M. N., & Alitheen, N. B. (2018). Curcumin Analog DK1 Induces Apoptosis in Human Osteosarcoma Cells In Vitro through Mitochondria-Dependent Signaling Pathway. Molecules, 23(1), 75. https://doi.org/10.3390/molecules23010075