Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety



 ,
,  ,
,  ,
, 

Abstract
1. Introduction
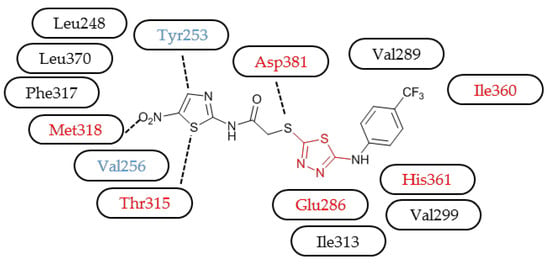
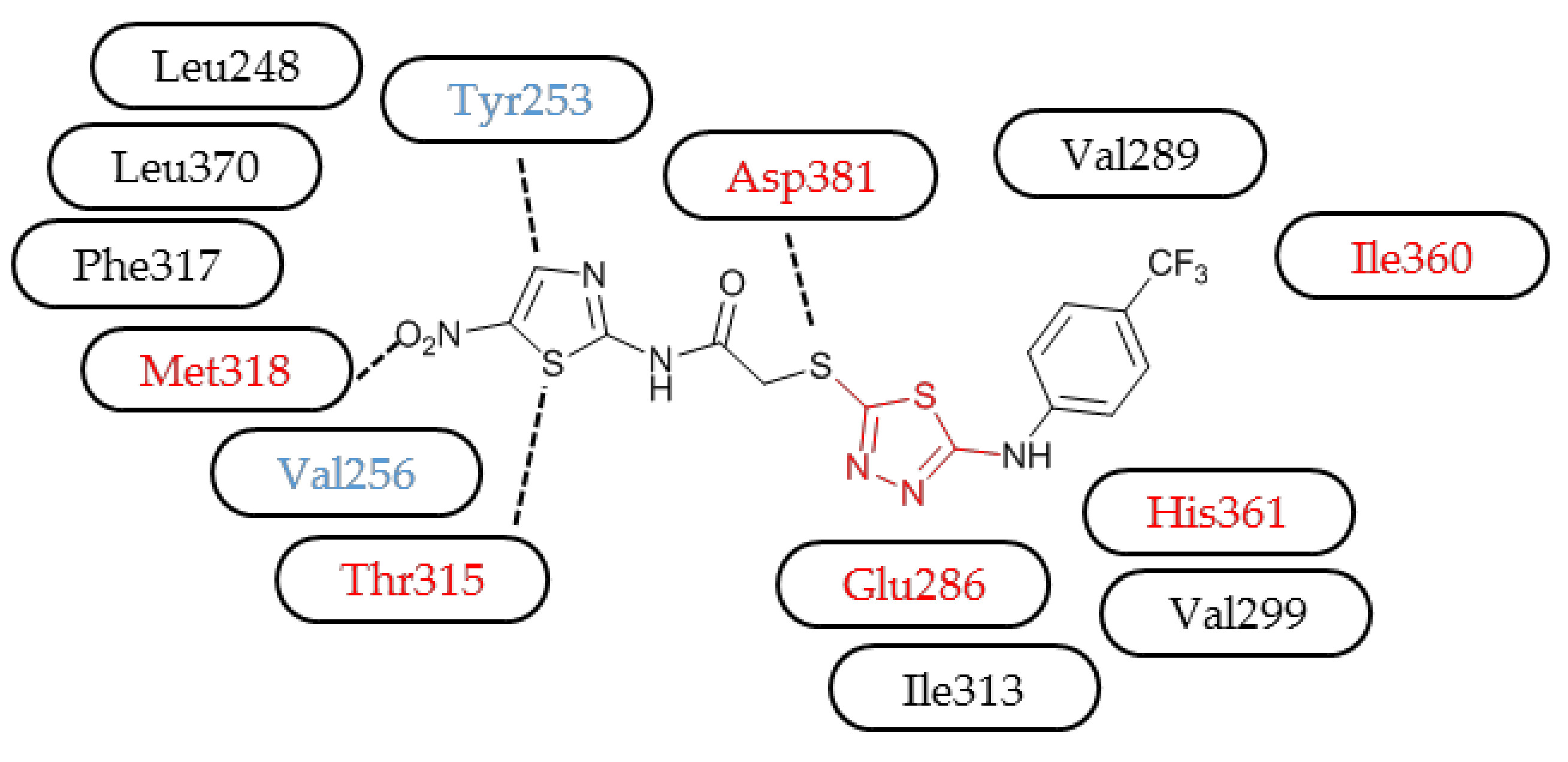
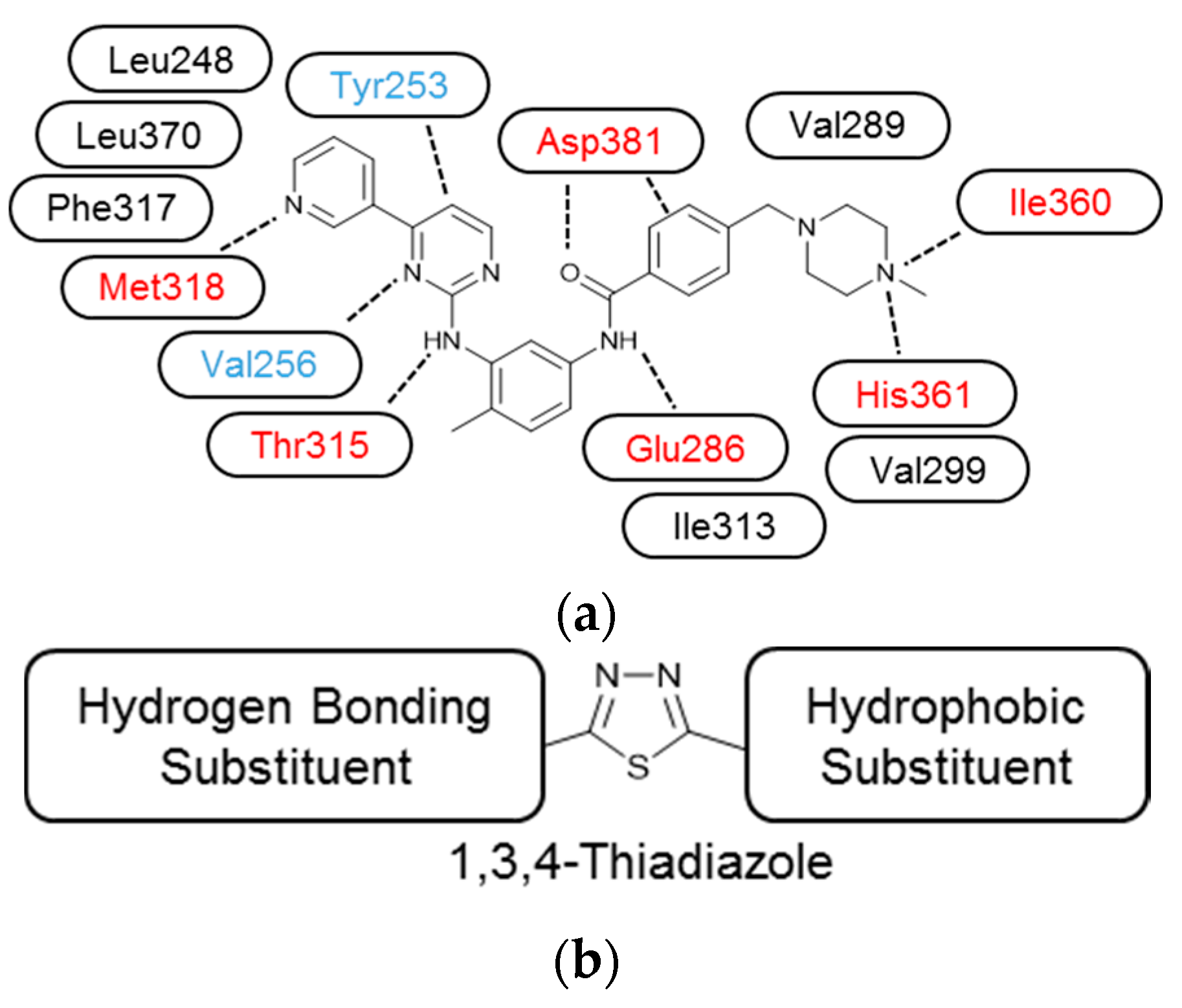
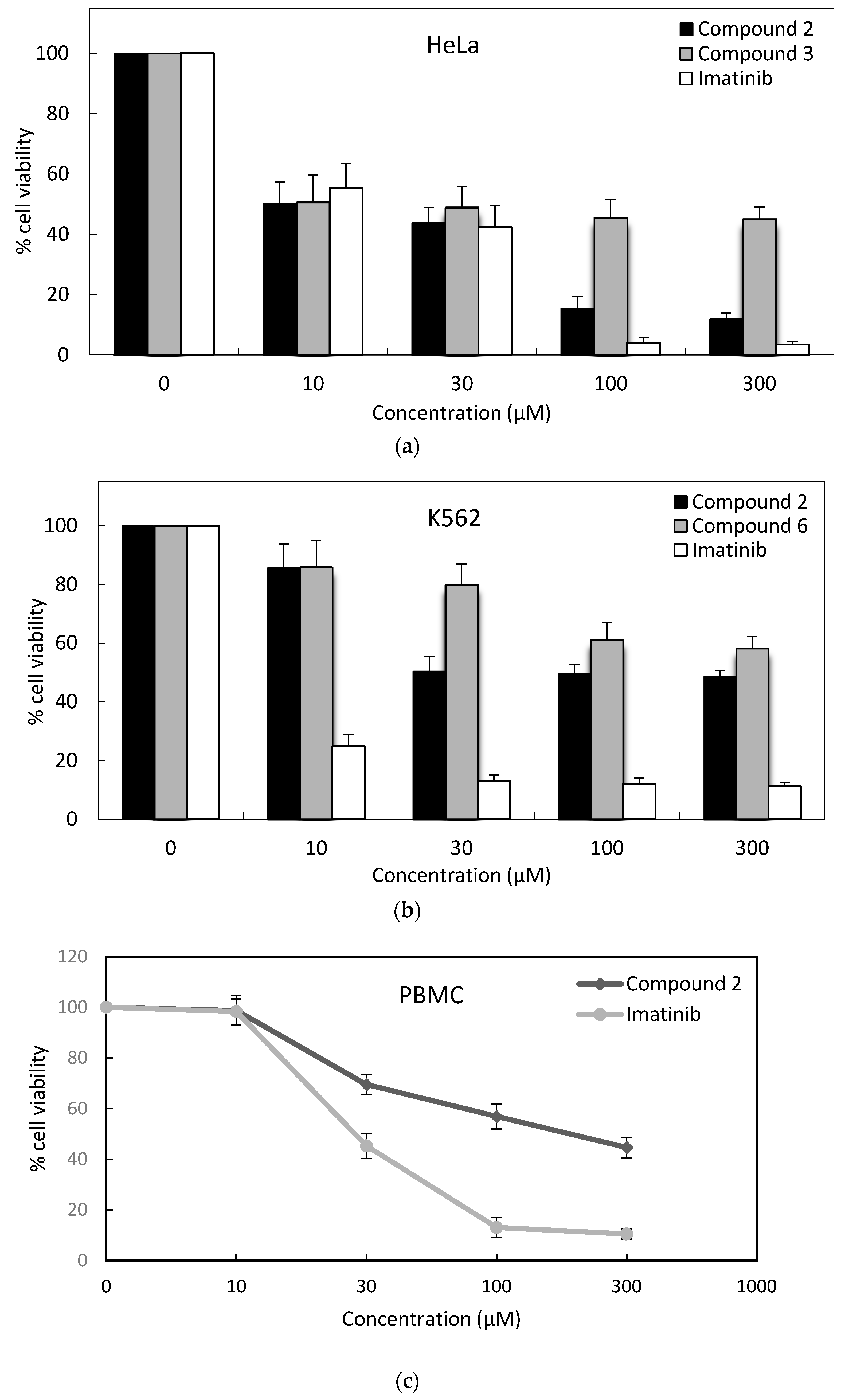
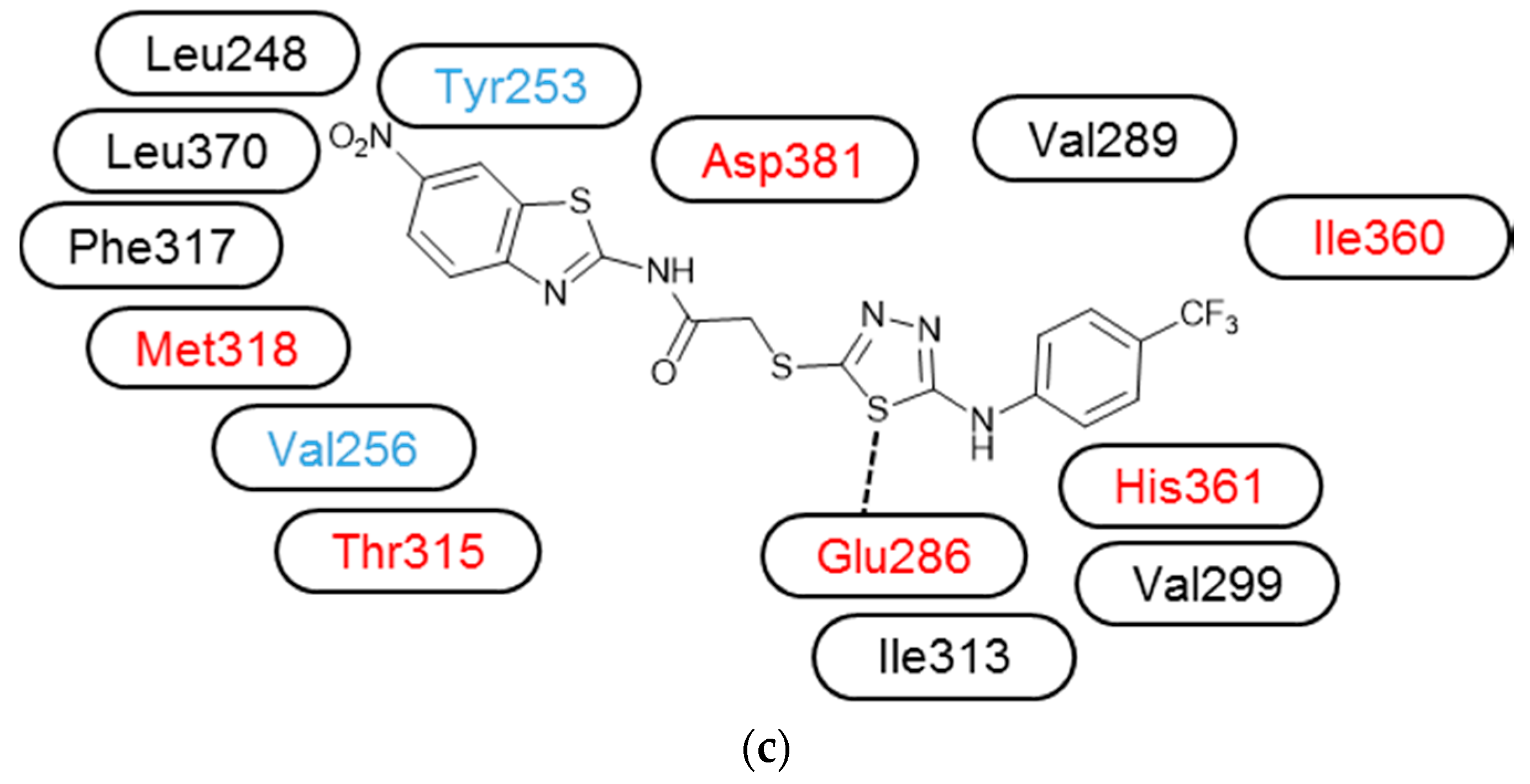
2. Results
3. Materials and Methods
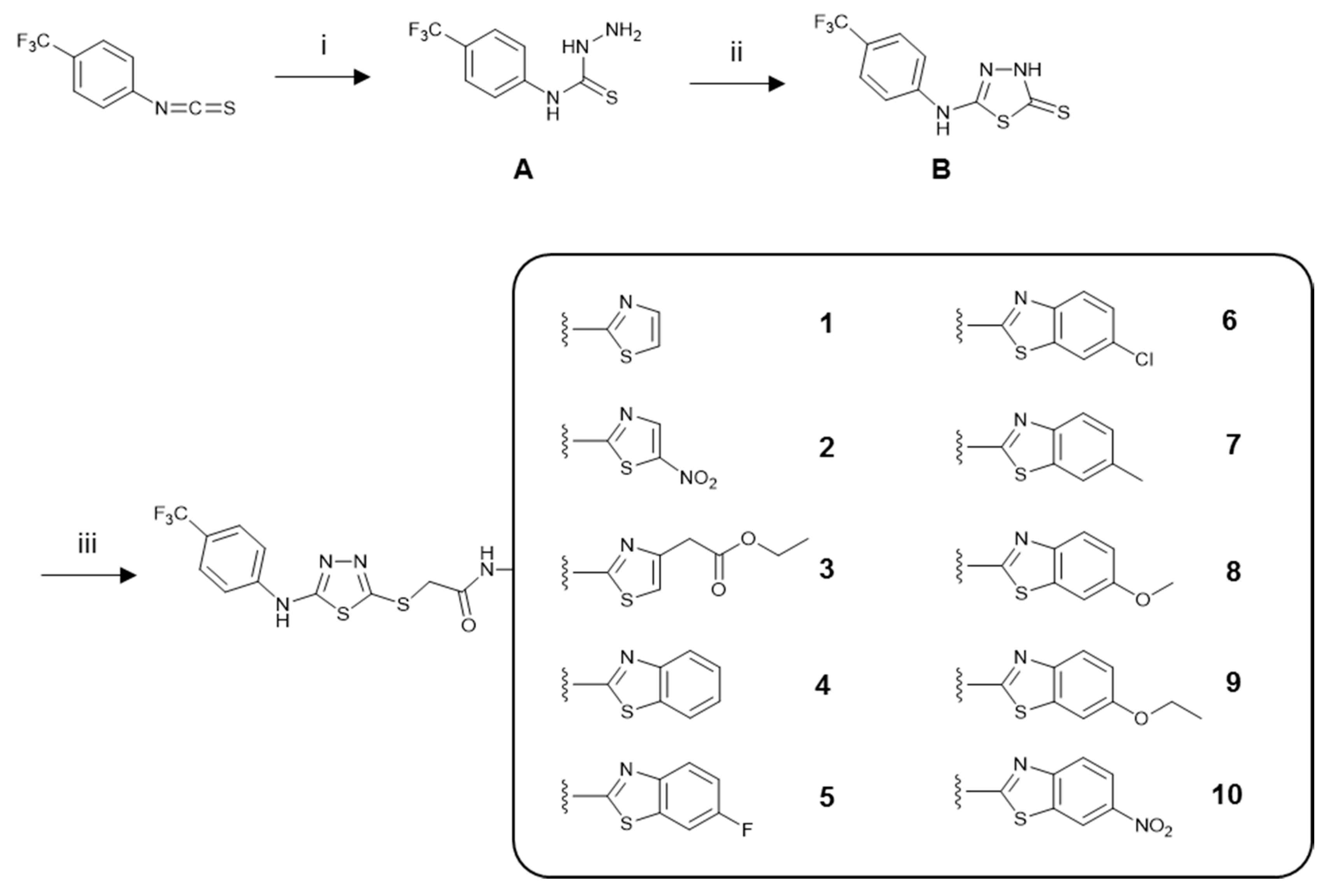
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of the Compounds
4-(4-(Trifluoromethyl)phenyl)thiosemicarbazide (A)
5-(4-(Trifluoromethyl)phenyl)amino-1,3,4-thiadiazole-2(3H)-thione (B)
N-(Aryl)-2-chloroacetamides
N-(Aryl)-2-[(5-((4-(trifluoromethyl)phenyl)amino)-1,3,4-thiadiazol-2-yl)thio]acetamide Derivatives (1–10)
3.2. Biochemistry
3.2.1. Cell Cultures
3.2.2. MTT Assay
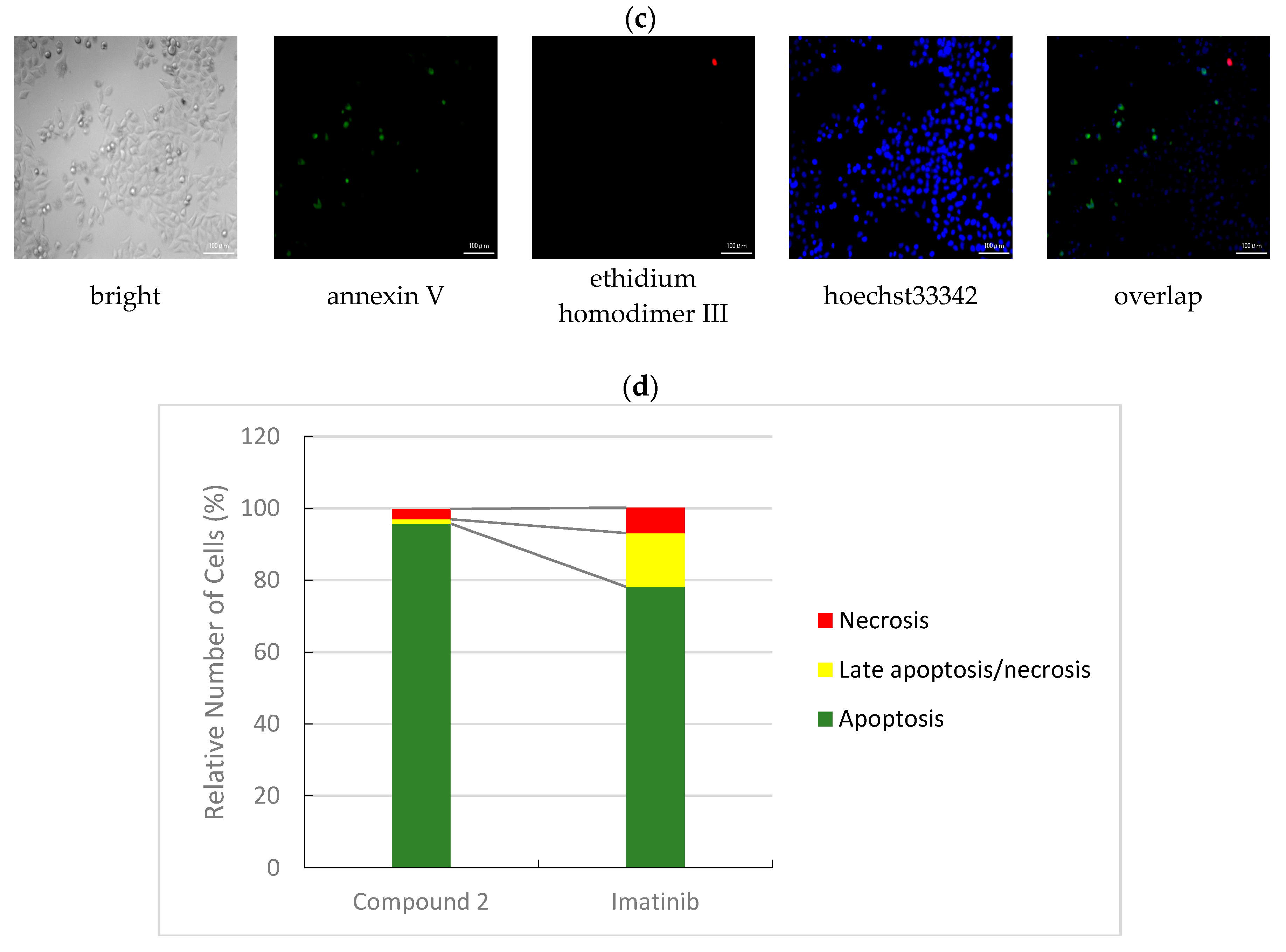
3.2.3. Apoptotic/Necrotic/Healthy Cells Detection Assay
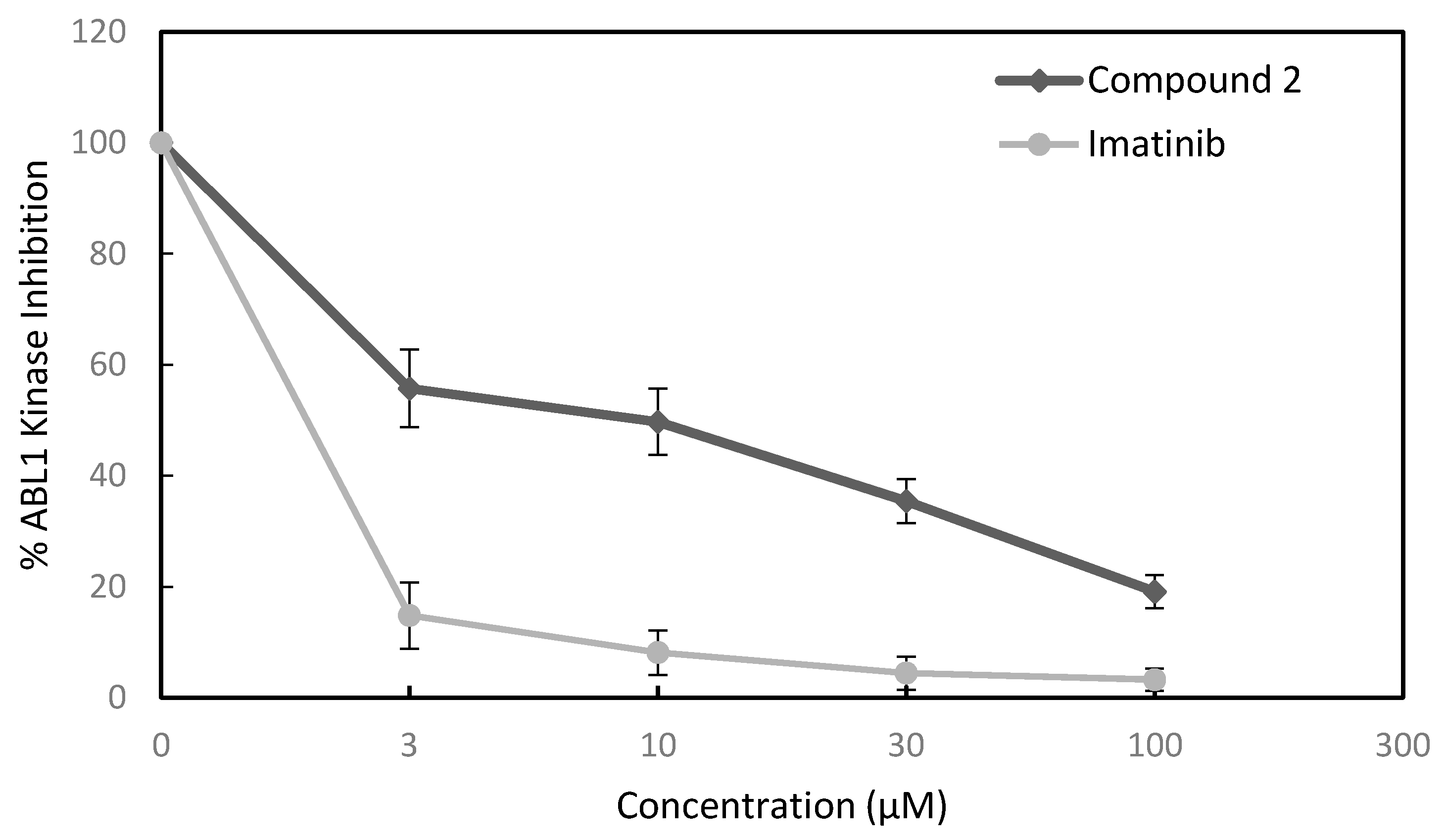
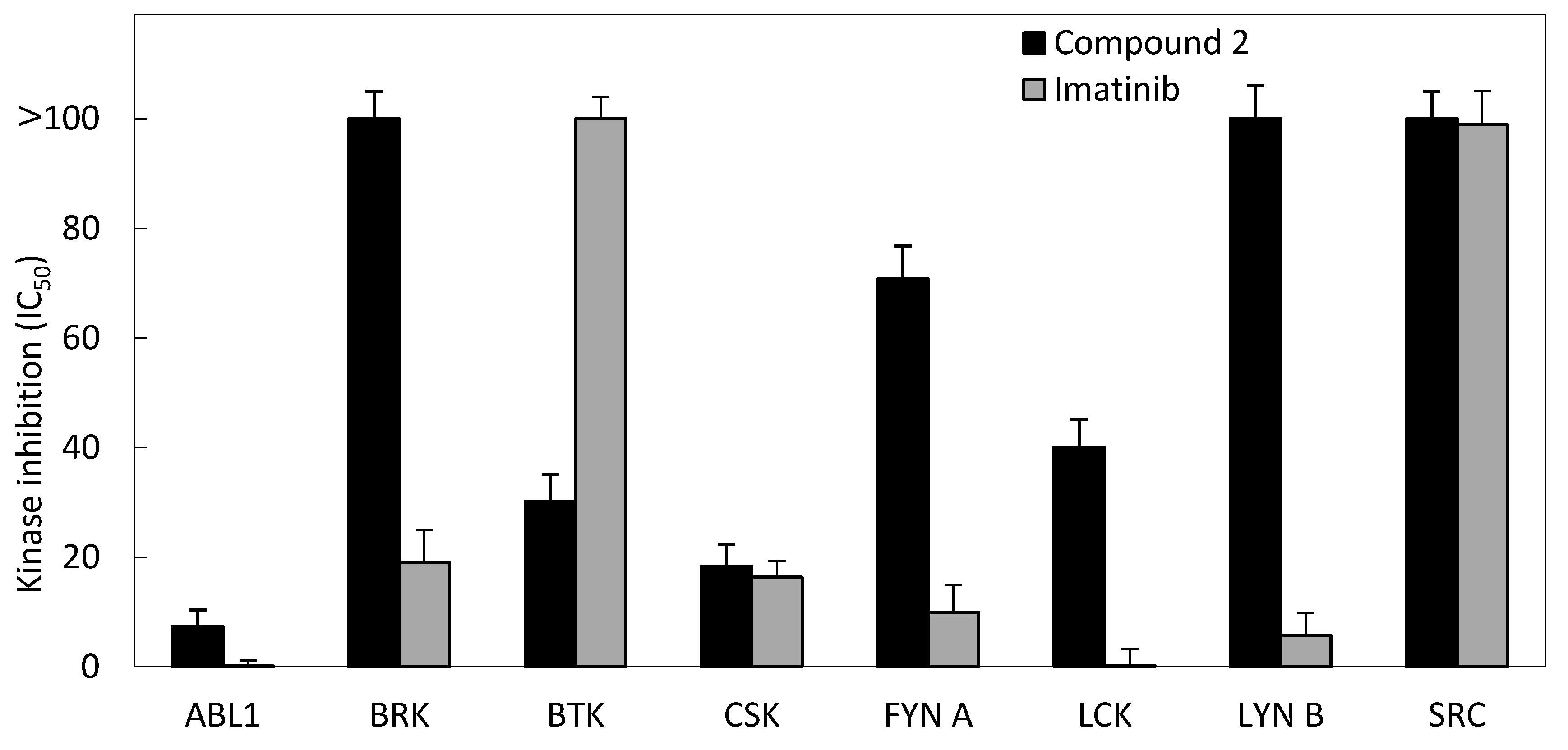
3.2.4. Kinase Inhibition Assay
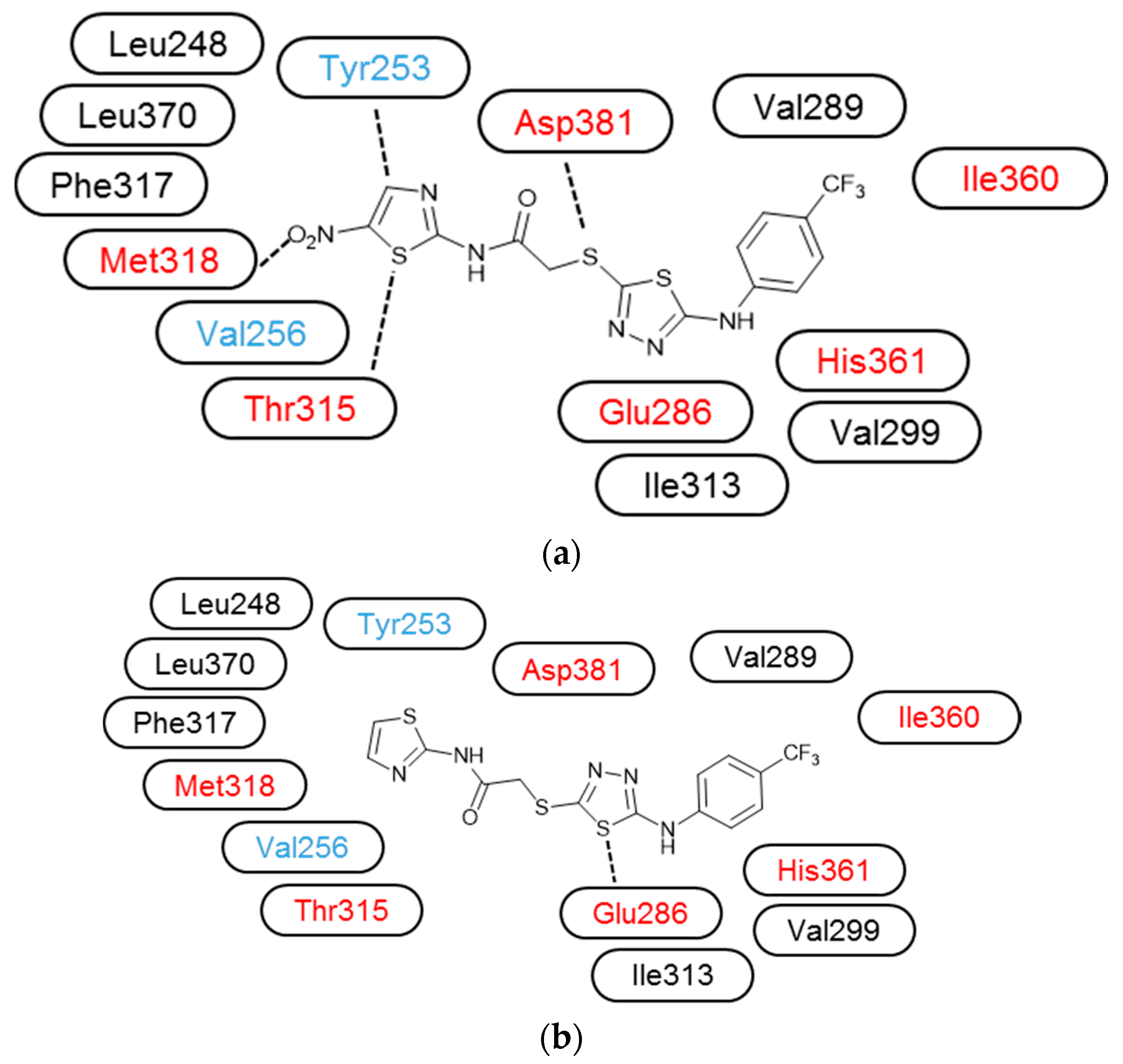
3.3. Molecular Modelling
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Popat, K.; McQueen, K.; Feeley, T.W. The Global Burden of Cancer. Best Pract. Res. Clin. Anaesthesiol. 2013, 27, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. (Ed.) Cancer Drug Resistance; Humana Press: Totowa, NJ, USA, 2006. [Google Scholar]
- Nussbaumer, S.; Bonnabry, P.; Veuthey, J.L.; Fleury-Souverain, S. Analysis of Anticancer Drugs: A Review. Talanta 2011, 85, 2265–2289. [Google Scholar] [CrossRef] [PubMed]
- Rebucci, M.; Michiels, C. Molecular Aspects of Cancer Cell Resistance to Chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Mazola, Y.; Rodríguez, R.; Mazola, Y. Protein Kinases as Targets for Drug Design. Biotecnol. Apl. 2008, 25, 7–15. [Google Scholar]
- Krishnamurty, R.; Maly, D.J. Biochemical Mechanisms of Resistance to Small-Molecule Protein Kinase Inhibitors. ACS Chem. Biol. 2010, 5, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase Drug Discovery—What’s Next in the Field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting Cancer with Kinase Inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-Molecule Kinase Inhibitors: An Analysis of FDA-Approved Drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal Structures of the Kinase Domain of c-Abl in Complex with the Small Molecule Inhibitors PD173955 and Imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar] [PubMed]
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and Its Derivatives: A Review on Recent Progress in Biological Activities. Chem. Biol. Drug Des. 2013, 81, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-A Promising Structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.Y.; Wang, X.M.; Yang, Y.H.; Zhu, H.L. 1,3,4-Thiadiazole: Synthesis, Reactions, and Applications in Medicinal, Agricultural, and Materials Chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J. Biological and Pharmacological Activities of 1,3,4-Thiadiazole Based Compounds. Mini-Rev. Med. Chem. 2015, 15, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Alam, M.S.; Hamid, H. 1,3,4-Thiadiazoles: A Potent Multi Targeted Pharmacological Scaffold. Eur. J. Med. Chem. 2015, 92, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Dawood, K.M.; Farghaly, T.A. Thiadiazole Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2017, 27, 477–505. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, A. 1,3,4-Thiadiazole Based Anticancer Agents. Anti-Cancer Agents Med. Chem. 2016, 16, 1301–1314. [Google Scholar] [CrossRef]
- Sun, J.; Yang, Y.S.; Li, W.; Zhang, Y.B.; Wang, X.L.; Tang, J.F.; Zhu, H.L. Synthesis, Biological Evaluation and Molecular Docking Studies of 1,3,4-Thiadiazole Derivatives Containing 1,4-Benzodioxan as Potential Antitumor Agents. Bioorg. Med. Chem. Lett. 2011, 21, 6116–6121. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, M.; Matysiak, J.; Szeliga, M.; Pożarowski, P.; Niewiadomy, A.; Albrecht, J.; Rzeski, W. 2-Amino-1,3,4-thiadiazole Derivative (FABT) Inhibits the Extracellular Signal-Regulated Kinase Pathway and Induces Cell Cycle Arrest in Human Non-Small Lung Carcinoma Cells. Bioorg. Med. Chem. Lett. 2012, 22, 5466–5469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, P.; Xuan, L.N.; Fu, X.Y.; Jing, F.; Li, S.; Liu, Y.M.; Chen, B.Q. Synthesis and Antitumor Activities of Novel Hybrid Molecules Containing 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Bearing Schiff Base Moiety. Bioorg. Med. Chem. Lett. 2014, 24, 5154–5156. [Google Scholar] [CrossRef] [PubMed]
- Yadagiri, B.; Gurrala, S.; Bantu, R.; Nagarapu, L.; Polepalli, S.; Srujana, G.; Jain, N. Synthesis and Evaluation of Benzosuberone Embedded with 1,3,4-Oxadiazole, 1,3,4-Thiadiazole and 1,2,4-Triazole Moieties as New Potential Anti Proliferative Agents. Bioorg. Med. Chem. Lett. 2015, 25, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumar, N.M.; Noel, B.; Shah, K. A Series of 2-Arylamino-5-(indolyl)-1,3,4-thiadiazoles as Potent Cytotoxic Agents. Eur. J. Med. Chem. 2012, 55, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Sun, F.; Hou, X.; Wang, F.; Yi, F.; Xu, W.; Fang, H. Design, Synthesis and Preliminary Bioactivity Studies of 1,3,4-Thiadiazole Hydroxamic Acid Derivatives as Novel Histone Deacetylase Inhibitors. Bioorg. Med. Chem. 2012, 20, 3865–3872. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Qin, Y.J.; Makawana, J.A.; Wang, Y.T.; Zhang, Y.Q.; Zhang, Y.L.; Yang, M.R.; Jiang, A.Q.; Zhu, H.L. Synthesis, Biological Evaluation and Molecular Modeling of 1,3,4-Thiadiazol-2-amide Derivatives as Novel Antitubulin Agents. Bioorg. Med. Chem. 2014, 22, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Wang, L.; Hou, X.; Wan, Y.; Xu, W.; Tang, W.; Fang, H. Improved Antiproliferative Activity of 1,3,4-Thiadiazole-Containing Histone Deacetylase (HDAC) Inhibitors by Introduction of the Heteroaromatic Surface Recognition Motif. Bioorg. Med. Chem. 2014, 22, 5766–5775. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Crespan, E.; Botta, G.; Falchi, F.; Maga, G.; Manetti, F.; Corradi, V.; Mancini, M.; Santucci, M.A.; Schenone, S.; et al. Discovery and SAR of 1,3,4-Thiadiazole Derivatives as Potent Abl Tyrosine Kinase Inhibitors and Cytodifferentiating Agents. Bioorg. Med. Chem. Lett. 2008, 18, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, L.; Khorand, A.; Aliabadi, A. Discovery of 2-Phenyl-N-(5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl)acetamide Derivatives as Apoptosis Inducers via the Caspase Pathway with Potential Anticancer Activity. Arch. Pharm. Chem. Life Sci. 2013, 346, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent Applications of 1,3-Thiazole Core Structure in the Identification of New Lead Compounds and Drug Discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Sikdar, P.; Bairagi, M. Recent Developments of 2-Aminothiazoles in Medicinal Chemistry. Eur. J. Med. Chem. 2016, 109, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent Patents on Thiazole Derivatives Endowed with Antitumor Activity. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Bioactive Thiazole and Benzothiazole Derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Noolvi, M.N.; Patel, H.M.; Kaur, M. Benzothiazoles: Search for Anticancer Agents. Eur. J. Med. Chem. 2012, 54, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S. A Comprehensive Review in Current Developments of Benzothiazole-Based Molecules in Medicinal Chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Pathak, D.P. Medicinal Significance of Benzothiazole Scaffold: An Insight View. J. Enzyme Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Singh, S.K. Benzothiazoles: How Relevant in Cancer Drug Design Strategy? Anticancer Agents Med. Chem. 2014, 14, 127–146. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, T.D.; Westwell, A.D. The Development of the Antitumour Benzothiazole Prodrug, Phortress, as a Clinical Candidate. Curr. Med. Chem. 2004, 11, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Turan-Zitouni, G.; Özkay, Y.; Özdemir, A.; Kaplancıklı, Z.A.; Altıntop, M.D. Synthesis of Some Benzothiazole Based Piperazinedithiocarbamate Derivatives and Evaluation of Their Anticancer Activities. Lett. Drug Des. Discov. 2011, 8, 830–837. [Google Scholar] [CrossRef]
- Fusaki, N.; Iwamatsu, A.; Iwashima, M.; Fujisawa, J. Interaction between Sam68 and Src Family Tyrosine Kinases, Fyn and Lck, in T Cell Receptor Signaling. J. Biol. Chem. 1997, 272, 6214–6219. [Google Scholar] [CrossRef] [PubMed]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative Study of Several Algorithms for Flexible Ligand Docking. J. Comput. Aided Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Kaplancıklı, Z.A.; Altıntop, M.D.; Sever, B.; Cantürk, Z.; Özdemir, A. Synthesis and In Vitro Evaluation of New Thiosemicarbazone Derivatives as Potential Antimicrobial Agents. J. Chem. 2016, 2016, 1692540. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Can, Ö.D.; Demir Özkay, Ü.; Kaplancıklı, Z.A. Synthesis and Evaluation of New 1,3,4-Thiadiazole Derivatives as Antinociceptive Agents. Molecules 2016, 21, 1004. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Kaplancıklı, Z.A.; Turan-Zitouni, G.; Özdemir, A.; Demirci, F.; İşcan, G.; Revial, G. Synthesis of Some Novel Triazole Derivatives and Investigation of Their Antimicrobial Activities. Synth. Commun. 2011, 41, 2234–2250. [Google Scholar] [CrossRef]
- Karabacak, M.; Altıntop, M.D.; Çiftçi, H.İ.; Koga, R.; Otsuka, M.; Fujita, M.; Özdemir, A. Synthesis and Evaluation of New Pyrazoline Derivatives as Potential Anticancer Agents. Molecules 2015, 20, 19066–19084. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.F.S.; Iwamura, K.; Çiftçi, H.İ.; Koga, R.; Matsumoto, M.; Oba, Y.; Kurosaki, H.; Fujita, M.; Okamoto, Y.; Umezawa, K.; et al. Novel Metal Chelating Molecules with Anticancer Activity. Striking Effect of the Imidazole Substitution of the Histidine–Pyridine–Histidine System. Bioorg. Med. Chem. 2015, 23, 5476–5482. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; et al. A Clue to Unprecedented Strategy to HIV Eradication: “Lock-in and Apoptosis”. Sci. Rep. 2017, 7, 8957. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Sonoda, S.; Ejima, T.; Tanaka, A.; Koga, R.; Okamoto, Y.; Fujita, M.; Otsuka, M. Zinc-Mediated Binding of A Low-Molecular-Weight Stabilizer of the Host Anti-Viral Factor Apolipoprotein B mRNA-Editing Enzyme, Catalytic Polypeptide-Like 3G. Bioorg. Med. Chem. 2016, 24, 4398–4405. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, N.; Yildirim, H.; Tuyun, A.F.; Kara, E.M.; Celik, B.O.; Gupta, G.K.; Ciftci, H.I.; Fujita, M.; Otsuka, M.; Nasiri, H.R. Synthesis, Computational Study, and Evaluation of In Vitro Antimicrobial, Antibiofilm, and Anticancer Activities of New Sulfanyl Aminonaphthoquinone Derivatives. Lett. Drug Des. Discov. 2017, 14, 647–661. [Google Scholar] [CrossRef]
- Tanaka, A.; Radwan, M.O.; Hamasaki, A.; Ejima, A.; Obata, E.; Koga, R.; Tateishi, H.; Okamoto, Y.; Fujita, M.; Nakao, M.; et al. A novel inhibitor of farnesyltransferase with a zinc site recognition moiety and a farnesyl group. Bioorg. Med. Chem. Lett. 2017, 27, 3862–3866. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–10 are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) * | ||||
|---|---|---|---|---|---|
| K562 | MT-2 | Jurkat | HeLa | PBMC | |
| 1 | >300 | >300 | >300 | 135.1 ± 12 | - |
| 2 | 33.0 ± 4 | 166.8 ± 8 | 17.9 ± 4 | 12.4 ± 2 | 141.3 ± 16 |
| 3 | >300 | >300 | 129.4 ± 6 | 14.1 ± 5 | - |
| 4 | >300 | 267.3 ± 14 | >300 | 32.9 ± 2 | - |
| 5 | >300 | 78.5 ± 12 | 97.9 ± 10 | 17.3 ± 8 | - |
| 6 | >300 | 38.3 ± 2 | 54.4 ± 8 | 28.7 ± 10 | - |
| 7 | >300 | 197.9 ± 11 | >300 | 30.2 ± 4 | - |
| 8 | >300 | >300 | >300 | 30.9 ± 4 | - |
| 9 | >300 | >300 | 96.1 ± 7 | 69.4 ± 7 | - |
| 10 | >300 | >300 | >300 | 75.3 ± 9 | - |
| Imatinib | 5.0 ± 2 | 9.7 ± 3 | 6.7 ± 2 | 15.2 ± 6 | 28.3 ± 6 |
| Kinase | IC50 (μM) | |
|---|---|---|
| Compound 2 | Imatinib | |
| ABL1 | 7.4 | 0.2 |
| BRK | >100 | 19 |
| BTK | 30.2 | >100 |
| CSK | 18.4 | 16.4 |
| FYN A | 70.8 | 10 |
| LCK | 40.1 | 0.3 |
| LYN B | >100 | 5.8 |
| SRC | >100 | 99 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altıntop, M.D.; Ciftci, H.I.; Radwan, M.O.; Sever, B.; Kaplancıklı, Z.A.; Ali, T.F.S.; Koga, R.; Fujita, M.; Otsuka, M.; Özdemir, A. Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety. Molecules 2018, 23, 59. https://doi.org/10.3390/molecules23010059
Altıntop MD, Ciftci HI, Radwan MO, Sever B, Kaplancıklı ZA, Ali TFS, Koga R, Fujita M, Otsuka M, Özdemir A. Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety. Molecules. 2018; 23(1):59. https://doi.org/10.3390/molecules23010059
Chicago/Turabian StyleAltıntop, Mehlika Dilek, Halil Ibrahim Ciftci, Mohamed O. Radwan, Belgin Sever, Zafer Asım Kaplancıklı, Taha F. S. Ali, Ryoko Koga, Mikako Fujita, Masami Otsuka, and Ahmet Özdemir. 2018. "Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety" Molecules 23, no. 1: 59. https://doi.org/10.3390/molecules23010059
APA StyleAltıntop, M. D., Ciftci, H. I., Radwan, M. O., Sever, B., Kaplancıklı, Z. A., Ali, T. F. S., Koga, R., Fujita, M., Otsuka, M., & Özdemir, A. (2018). Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety. Molecules, 23(1), 59. https://doi.org/10.3390/molecules23010059