Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.)

Abstract
1. Introduction
2. Results
2.1. Anthocyanin Composition and Content
2.2. mRNA Sequence
2.3. Sequence Alignment and Mapping to the Reference Genome
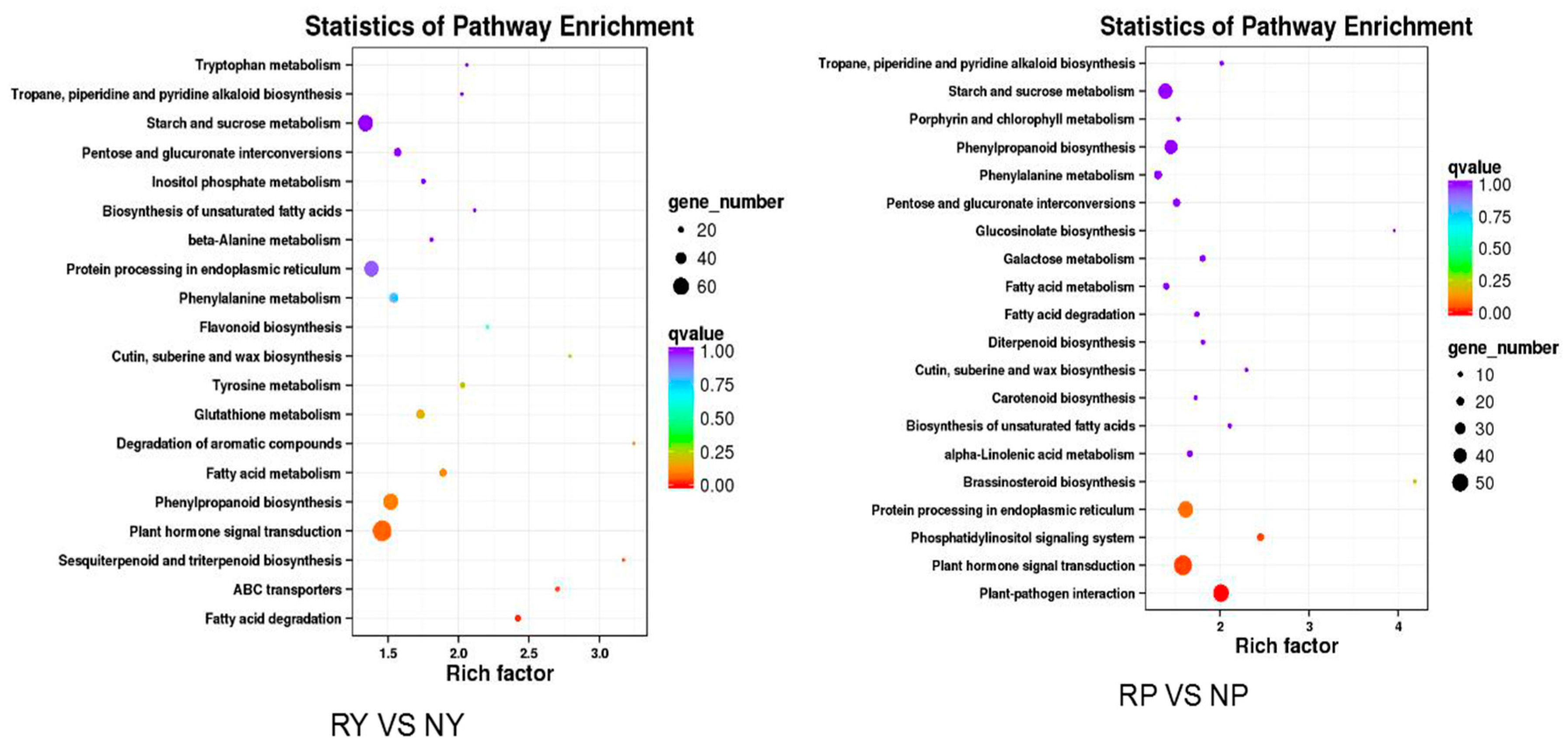
2.4. Analysis of Differentially Expressed Genes
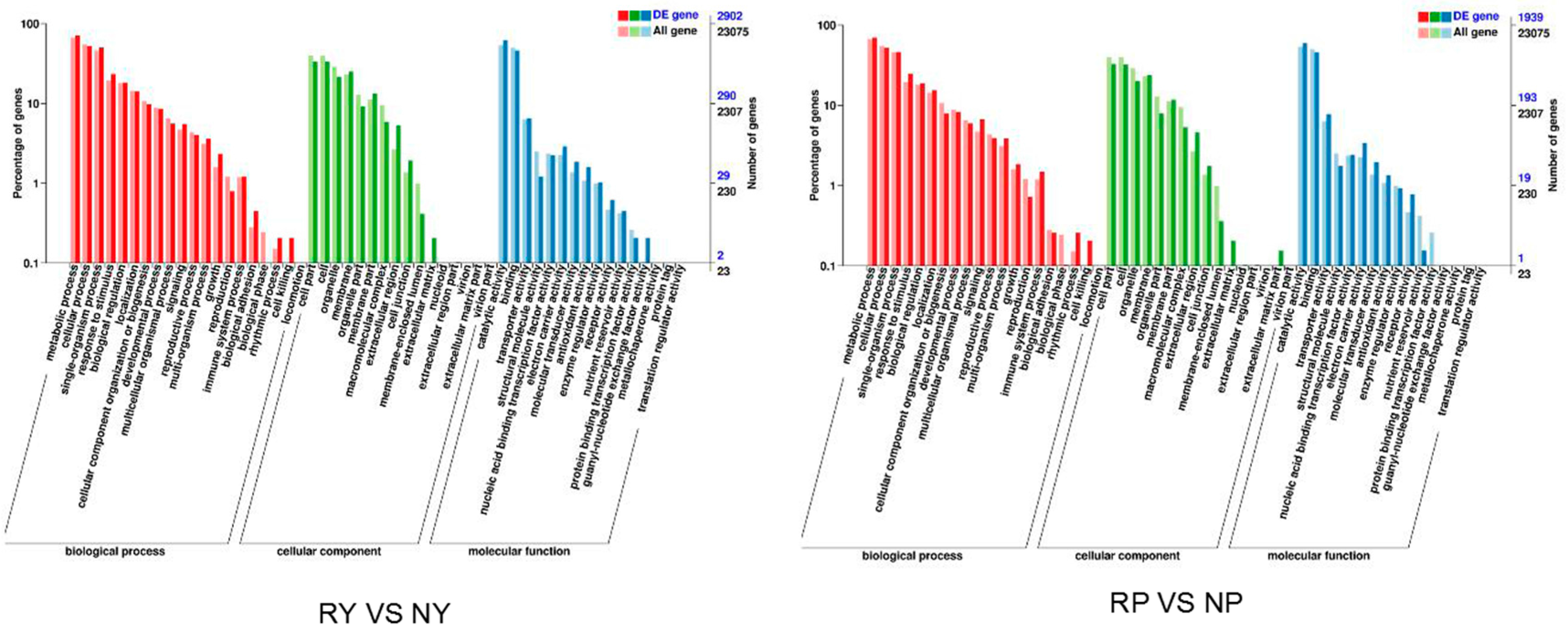
2.5. Functional Annotation and Classification
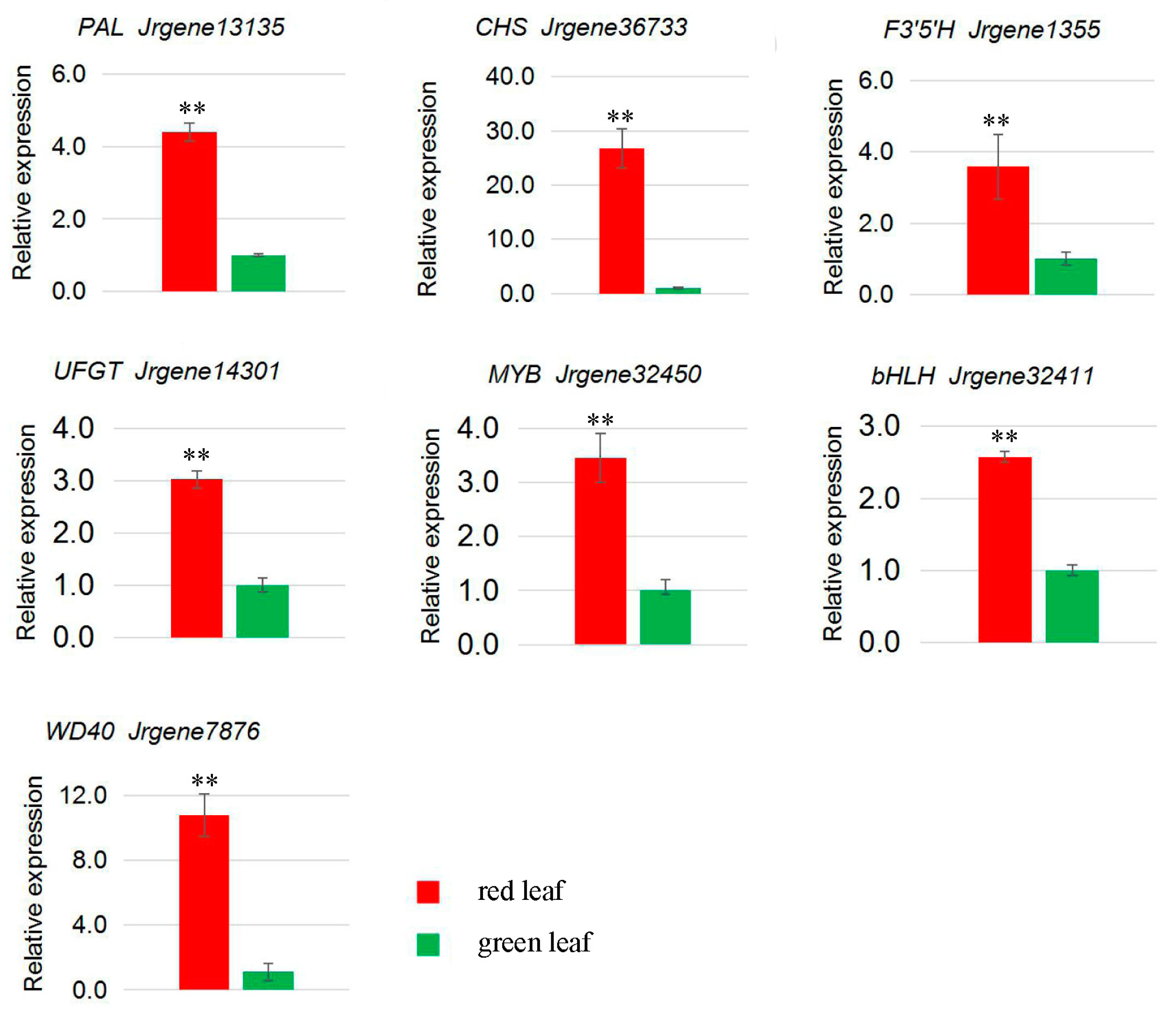
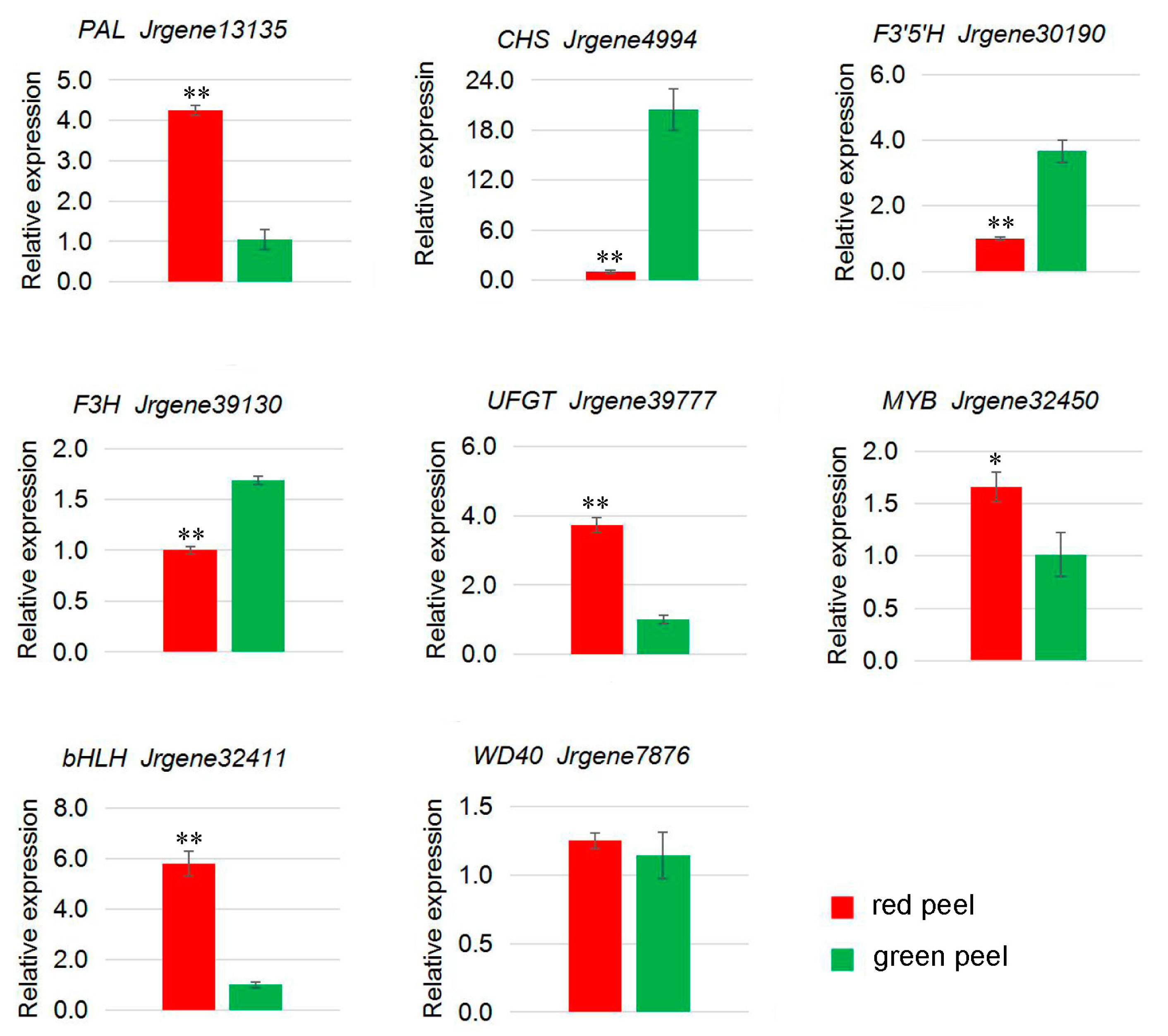
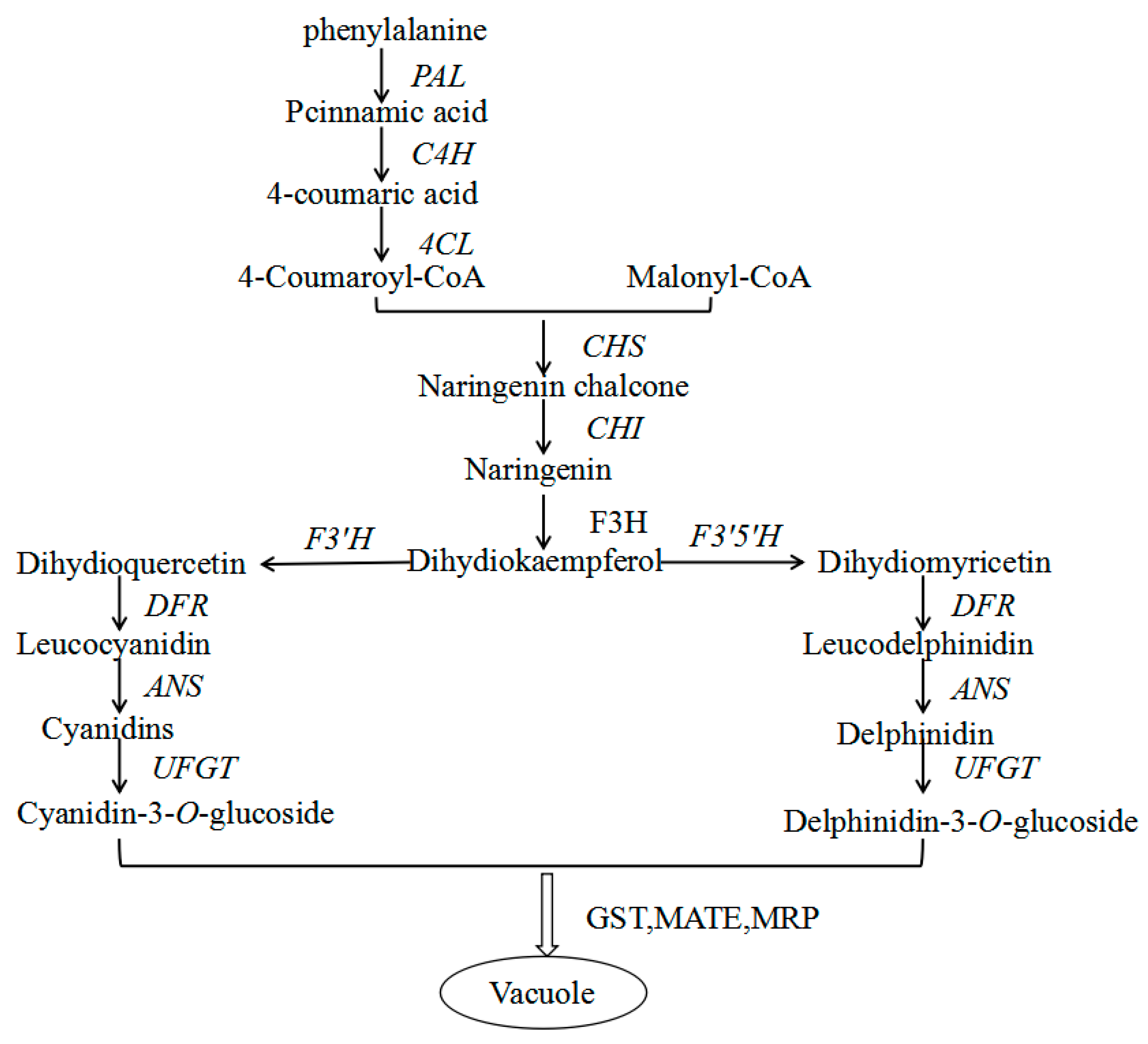
2.6. Genes from Walnut Transcriptome Involved in Anthocyanin Biosynthesis
3. Discussion
4. Materials and Methods
4.1. Anthocyanin Extraction
4.2. Total RNA Extraction
4.3. Library Preparation for Transcriptome Sequencing
4.4. Clustering and Sequencing
4.5. Transcriptome Assembly
4.6. Gene Functional Annotation
4.7. Sequence Alignment and Mapping to Reference Transcripts
4.8. Differential Gene Expression Analysis
4.9. qRT-PCR Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Aradhya, M.; Woeste, K.; Velasco, D. Genetic diversity, structure and differentiation in cultivated walnut (Juglans regia L.). Acta Hortic. 2010, 861, 127–132. [Google Scholar] [CrossRef]
- Xi, R.T.; Zhang, Y.P. Chinese Fruit Annals. Walnut, 1st ed.; China Forestry Publishing House: Beijing, China, 1996; pp. 1–9. ISBN 9787503814020. [Google Scholar]
- Mc Granahan, G.; Leslie, C. ‘Robert Livermore’, a Persian walnut cultivar with a red seedcoat. Hortic. Sci. 2004, 39, 1772. [Google Scholar]
- Hanbali, L.M.B.; Ghadieh, R.A.; Hasan, H.K.; Nakhal, Y.J.; Haddad, J. Measurement of antioxidant activity and antioxidant compounds under versatile extraction conditions: I. The immuno-biochemical antioxidant properties of sweet cherry (Prunus avium) extracts. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 173–187. [Google Scholar] [CrossRef]
- Kelebek, H.; Selli, S. Evaluation of chemical constituents and antioxidant activity of sweet cherry (Prunus avium L.) cultivars. Int. J. Food Sci. Technol. 2011, 46, 2530–2537. [Google Scholar] [CrossRef]
- Shi, M.Z.; Xie, D.Y. Biosynthesis and metabolic engineering of anthocyanins in Arabidopsis thaliana. Recent Pat. Biotechnol. 2014, 8, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-Y.; Seeram, N.P.; Nair, M.G.; Bourquin, L.D. Tart cherry anthocyanins inhibit tumor development in ApcMin mice and reduce proliferation of human colon cancer cells. Cancer Lett. 2003, 194, 13–19. [Google Scholar] [CrossRef]
- Knekt, P.; Jarvinen, R.; Seppanen, R.; Heliovaara, M.; Teppo, L.; Pukkala, E.; Aromaa, A. Dietary flavonoids and the risk of lung cancer and other malignant neoplasms. Am. J. Epidemiol. 1997, 146, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Cheng, F.; Wu, J.; Liu, B.; Zheng, S.; Liang, J.; Wang, X. Anthocyanin biosynthetic genes in Brassica rapa. BMC Genom. 2014, 15, 426. [Google Scholar] [CrossRef] [PubMed]
- Misyura, M.; Colasanti, J.; Rothstein, S.J. Physiological and genetic analysis of Arabidopsis thaliana anthocyanin biosynthesis mutants under chronic adverse environmental conditions. J. Exp. Bot. 2013, 64, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Tohge, T.; Zuther, E.; Fernie, A.R.; Hincha, D.K. Natural variation in flavonol and anthocyanin metabolism during cold acclimation in Arabidopsis thaliana accessions. Plant Cell Environ. 2015, 38, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Chia, L.S.; Goh, N.; Chia, T.; Brouillard, R. Analysis and biological activities of anthocyanins. Phytochemistry 2003, 64, 923–933. [Google Scholar] [CrossRef]
- Miller, R.; Owens, S.J.; Rorslett, B. Plants and colour-flowers and pollination. Opt. Laser Technol. 2011, 43, 282–294. [Google Scholar] [CrossRef]
- Pelletier, M.K.; Murrell, J.R.; Shirley, B.W. Characterization of flavonol synthase and leuco anthocyanidin dioxygenase genes in arabidopsis further evidence for differential regulation of “early” and “late” genes. Plant Physiol. 1997, 113, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Shibata, M.; Nishiyama, Y.; Springob, K.; Kitayama, M.; Shimada, N.; Aoki, T.; Ayabe, S.; Saito, K. Differential gene expression profiles of red and green forms of Perilla frutescens leading to comprehensive identification of anthocyanin biosynthetic genes. FEBS J. 2008, 275, 3494–3502. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kunihiro, A.; Tominaga-Wada, R. Arabidopsis CAPRICE (MYB) and GLABRA3 (bHLH) control tomato (Solanum lycopersicum) anthocyanin biosynthesis. PLoS ONE 2014, 9, e109093. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Li, M.; Ma, F.; Cheng, L. Phenylpropanoid metabolites and expression of key genes involved in anthocyanin biosynthesis in the shaded peel of apple fruit in response to sun exposure. Plant Physiol. Biochem. 2013, 69, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hichri, I.; Heppel, S.C.; Pillet, J.; Léon, C.; Czemmel, S.; Delrot, S.; Lauwergeat, V.; Bogs, J. The basic helix-loop-helix transcription factor MYC1 is involved in the regulation of the flavonoid biosynthesis pathway in grapevine. Mol. Plant 2010, 3, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Nicolè, S.; Barcaccia, G.; Erickson, D.L.; Kress, J.W.; Lucchin, M. The coding region of the UFGT gene is a source of diagnostic SNP markers that allow single-locus DNA genotyping for the assessment of cultivar identity and ancestry in grapevine (Vitis vinifera L.). BMC Res. Notes 2013, 6, 502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wei, G.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y. Unraveling themechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liao, L.; Zhou, H.; Gu, C.; Wang, L.; Han, Y. A small indel mutation in an anthocyanin transporter causes variegated colouration of peach flowers. J. Exp. Bot. 2015, 66, 7227–7239. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Huang, H.; Song, S.; Xie, D. Regulation of jasmonate-mediated stamen development and seed production by a bHLH-MYB complex in arabidopsis. Plant Cell 2015, 27, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shang, J.; Zhou, Y.; Wu, W.; Jie, B.; Wu, G. Determination of anthocyanins in the red walnut by UPLC-ESI-MS/MS. J. Food Sci. Rev. 2017. [Google Scholar]
- Martinez-Garcia, P.J.; Crepeau, M.W.; Puiu, D.; Gonzalez-Ibeas, D.; Whalen, J.; Stevens, K.A.; Paul, R.; Butterfield, T.S.; Britton, M.T.; Reagan, R.L.; et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 2016, 87, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Flavonoid transport mechanisms: How to go, and with whom. Trends Plant Sci. 2015, 20, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, X.; Zong, X.; Shu, H.; Gao, D.; Liu, Q. Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in the Red and Yellow Fruits of Sweet Cherry (Prunus avium L.). PLoS ONE 2015, 10, e0121164. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fan, X.; Zhang, Y.; Jiang, J.; Sun, H.; Liu, C. Transcriptome analysis of genes involved in anthocyanins biosynthesis and transport in berries of black and white spine grapes (Vitis davidii). Hereditas 2016, 153, 17. [Google Scholar] [CrossRef] [PubMed]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Winkel, B.S. Metabolic channeling in plants. Annu. Rev. Plant Biol. 2004, 55, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Brugliera, F. Flower colour. In Annual Plant Reviews, Volume 20: Flowering and Its Manipulation; Ainsworth, C., Ed.; Blackwell Publishing: Oxford, UK, 2007; pp. 201–239. [Google Scholar]
- Tanaka, Y. Flower colour and cytochromes p450. Phytochem. Rev. 2006, 5, 283–291. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Mizutani, M.; Fukui, Y.; Brugliera, F.; Holton, T. Engineering of the rose flavonoid biosynthetic pathway successfully generated blue-hued flowers accumulating delphinidin. Plant Cell Physiol. 2007, 48, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, Betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Seitz, C.; Eder, C.; Deiml, B.; Kellner, S.; Martens, S.; Forkmann, G. Cloning, functional identification and sequence analysis of flavonoid 3′-hydroxylase and flavonoid 3,5′-hydroxylase cDNA reveals independent evolution of flavonoid 3′,5′-hydroxylase in the Asteraceae family. Plant Mol. Biol. 2006, 61, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Ogata, J.; Kanno, Y.; Itoh, Y.; Tsugawa, H.; Suzuki, M. Plant biochemistry: Anthocyanin biosynthesis in roses. Nature 2005, 435, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Saitoh, M.; Hoshino, A.; Nitasaka, E.; Iida, S. Isolation of cDNAs for R2R3-Myb, bHLH and WDR transcriptional regulators and identification of c and ca mutations conferring white flowers in the Japanese morning glory. Plant Cell Physiol. 2006, 47, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, Y.; Wu, Q.; Luo, H.; Sun, Y.; Song, J. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genom. 2010, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Che, F.; Wang, L.; Meng, R.; Zhang, X.; Zhao, Z. Fruit coloration and anthocyanin biosynthesis after bag removal in non-red and red apples (Malus × domestica Borkh.). Molecules 2013, 18, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, Y.; Yang, S.; Xu, Y.; Chen, X. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta 2010, 232, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Soubeyrand, E.; Basteau, C.; Hilbert, G.; Leeuwen, C.v.; Delrot, S.; Gomès, E. Nitrogen supply affects anthocyanin biosynthetic and regulatory genes in grapevine cv. Cabernet-Sauvignon berries. Phytochemistry 2014, 103, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, M.; Lai, Z.; Fan, B.; Shi, K.; Zhou, Y.; Yu, J.; Chen, Z. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiol. 2010, 153, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Rosalesmendoza, S.; Zheng, D.; Lygin, A.V.; Korban, S.S. Ectopic expression of apple F3′H genes contributes to anthocyanin accumulation in the Arabidopsis tt7 mutant grown under nitrogen stress. Plant Physiol. 2010, 153, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Kakizaki, Y.; Nishihara, M. Isolation and characterization of GtMYBP3 and GtMYBP4, orthologues of R2R3-MYB transcription factors that regulate early flavonoid biosynthesis, in gentian flowers. J. Exp. Bot. 2012, 63, 6505–6517. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ishimaru, M.; Ding, C.K.; Yakushiji, H.; Goto, N. Comparison of UDP-glucose:flavonoid 3-O-glucosyltransferase (UFGT) gene sequences between white grapes (Vitis vinifera) and their sports with red skin. Plant Sci. 2001, 160, 543–550. [Google Scholar] [CrossRef]
- Griesser, M.; Hoffmann, T.; Bellido, M.L.; Rosati, C.; Fink, B.; Kurtzer, R.; Aharoni, A.; Munozblanco, J.; Schwab, W. Redirection of flavonoid biosynthesis through the down-regulation of an anthocyanidin glucosyltransferase in ripening strawberry fruit. Plant Physiol. 2008, 146, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Aguadé, M. Nucleotide sequence variation at two genes of the phenylpropanoid pathway, the FAH1 and F3H genes, in Arabidopsis thaliana. Mol. Biol. Evol. 2001, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Hori, W.; Hosokawa, M.; Tatsuzawa, F.; Doi, M. Post-transcriptional silencing of chalcone synthase is involved in phenotypic lability in petals and leaves of bicolor dahlia (Dahlia variabilis) ‘Yuino’. Planta 2017, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.G.; Jiang, W.; Mantri, N.; Bao, X.Q.; Chen, S.L.; Tao, Z.M. Transcriptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alata L.) tubers. BMC Genom. 2015, 16, 346. [Google Scholar] [CrossRef] [PubMed]
- Matus, J.T.; Poupin, M.J.; Cañón, P.; Bordeu, E.; Alcalde, J.A.; Arce-Johnson, P. Isolation of WDR and bHLH genes related to flavonoid synthesis in grapevine (Vitis vinifera L.). Plant Mol. Biol. 2010, 72, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.W.; Davies, K.M.; Lewis, D.H.; Zhang, H.; Montefiori, M.; Brendolise, C.; Boase, M.R.; Ngo, H.; Jameson, P.E.; Schwinn, K.E. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in Eudicots. Plant Cell 2014, 26, 962–980. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Tan, H.; Ma, Z.; Huang, J. DELLA Proteins Promote Anthocyanin Biosynthesis via Sequestering MYBL2 and JAZ Suppressors of the MYB/bHLH/WD40 Complex in Arabidopsis thaliana. Mol. Plant. 2016, 9, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gao, L.; Wang, H.; Chen, Y.; Yang, H.; Wei, C.; Wan, X.; Xia, T. The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct. Integr. Genom. 2013, 13, 75–98. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Tian, Y.; Chen, K.; Wang, X.; Hao, Y. The apple WD40 protein MdTTG1 interacts with bHLH but not MYB proteins to regulate anthocyanin accumulation. J. Plant Physiol. 2012, 169, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Schaart, J.G.; Dubos, C.; Romero De La Fuente, I.; Van Houwelingen, A.; De Vos, R.C.; Jonker, H.H.; Xu, W.; Routaboul, J.M.; Lepiniec, L.; Bovy, A.G. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria × ananassa) fruits. New Phytol. 2013, 197, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Takos, A.M.; Jaffe, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Honda, C.; Hatsuyama, Y.; Igarashi, M.; Bessho, H.; Moriguchi, T. Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol. 2007, 48, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Li, Y.Y.; Zhang, Q.Y.; Li, M.; Sun, C.H.; Yu, J.Q.; Hao, Y.J. The R2R3-MYB transcription factor MdMYB73 is involved in malate accumulation and vacuolar acidification in apple. Plant J. 2017, 91, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef] [PubMed]
- Medina-Puche, L.; Cumplido-Laso, G.; Amil-Ruiz, F.; Hoffmann, T.; Ring, L.; Rodríguez-Franco, A.; Caballero, J.L.; Schwab, W.; Muñoz-Blanco, J.; Blanco-Portales, R. MYB10 plays a major role in the regulation of flavonoid/phenylpropanoid metabolism during ripening of Fragaria ananassa fruits. J. Exp. Bot. 2014, 65, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; De Vos, C.H.; Wein, M.; Sun, Z.; Greco, R.; Kroon, A.; Mol, J.N.; O’Connell, A.P. The strawberry FaMYB1 transcription factor suppresses anthocyanin and flavonol accumulation in transgenic tobacco. Plant J. 2001, 28, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Kadomura-Ishikawa, Y.; Miyawaki, K.; Takahashi, A.; Noji, S. RNAi-mediated silencing and over expression of the FaMYB1 gene and its effect on anthocyanin accumulation in strawberry fruit. Biol. Plant. 2015, 59, 677–685. [Google Scholar] [CrossRef]
- Suzuki, S.; Miyata, K.; Hara, M.; Tsukaya, K.N.H.; Takase, M.; Hayama, R.; Mizoguchi, T. A loss-of-function mutation in the DWARF4/PETANKO5 gene enhances the late-flowering and semi-dwarf phenotypes of the Arabidopsis clock mutant lhy-12;cca1-101 under continuous light without affecting FLC expression. Plant Biotechnol. 2016, 33, 315–321. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Baudry, A.; Caboche, M.; Lepiniec, L. TT8 controls its own expression in a feedback regulation involving TTG1 and homologous MYB and bHLH factors, allowing a strong and cell-specific accumulation of flavonoidsin Arabidopsis thaliana. Plant J. 2006, 46, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Määttä, K.R.; Kamal-Eldin, A.; Törrönen, A.R. High-performance liquid chromatography (HPLC) analysis of phenolic compounds in berries with diode array and electrospray ionization mass spectrometric (MS) detection: Ribes species. Agric. Food Chem. 2003, 51, 6736–6744. [Google Scholar]
- Lin, L.Z.; Harnly, J.; Zhang, R.W.; Fan, X.E.; Chen, H.J. Quantitation of the hydroxycinnamic acid derivatives and the glycosides of flavonols and flavones by UV absorbance after identification by LC-MS. Agric. Food Chem. 2012, 60, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Wang, W.; Wang, J.; Wei, H.; Yan, X.; Liu, Q. The application of SYBR Green I real-time quantitative RT-PCR in quantitative analysis of sweet cherry viruses in different tissues. Acta Phytophylacica Sin. 2012, 39, 497–502. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I. Trinity: Reconstructing a fulllength transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-through put functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2007, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, W.A.; Dhir, R.; Kelly, L.A.; Petrosko, P.; Krill-Burger, J.M.; Sciulli, C.M.; Lyons-Weiler, M.A.; Chandran, U.R.; Lomakin, A.; Masterson, R.V.; et al. Serum protein profiles predict coronary artery disease in symptomatic patients referred for coronary angiography. BMC Med. 2012, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Period and Position | Anthocyanins (mg kg−1) | |||
|---|---|---|---|---|
| Delphinidin-3-O-Galactoside | Delphinidin-3-O-Glucoside | Cyanidin-3-O-Galactoside | Cyanidin-3-O-Arabinoside | |
| Red leaf (RY) | 0.080 | 0.293 | 0.676 | 0.123 |
| Green leaf (NY, CK) | ND | ND | ND | ND |
| Red fruit peel (RP) | ND | 0.283 | ND | ND |
| Green fruit peel (NP, CK) | ND | ND | ND | ND |
| Samples Name | Raw Reads | Clean Reads | Clean Bases | GC Content (%) | Q30 (%) |
|---|---|---|---|---|---|
| RY-1 | 23,306,215 | 22,779,186 | 6,676,568,814 | 45.68 | 94.05 |
| RY-2 | 24,247,568 | 23,769,824 | 6,973,950,086 | 45.83 | 94.14 |
| RY-3 | 21,691,848 | 21,343,231 | 6,272,412,100 | 46.01 | 93.78 |
| NY-1 | 23,258,402 | 22,787,786 | 6,645,288,228 | 46.01 | 93.80 |
| NY-2 | 26,601,923 | 26,078,697 | 7,635,444,098 | 45.96 | 93.83 |
| NY-3 | 24,983,554 | 24,403,548 | 7,115,739,742 | 46.11 | 93.95 |
| RP-1 | 24,568,131 | 24,232,274 | 7,126,004,890 | 46.58 | 93.94 |
| RP-2 | 33,080,603 | 32,451,498 | 9,482,999,428 | 46.44 | 94.35 |
| RP-3 | 28,654,652 | 28,000,182 | 8,248,082,522 | 46.32 | 93.90 |
| NP-1 | 22,070,960 | 21,796,810 | 6,436,963,652 | 46.40 | 93.98 |
| NP-2 | 21,233,811 | 20,845,557 | 6,190,372,176 | 46.26 | 93.66 |
| NP-3 | 22,560,037 | 21,829,578 | 6,392,561,574 | 46.26 | 94.26 |
| Samples Name | Total Reads | Mapped Reads | Uniq Mapped Reads | Multiple Map Reads | Reads Map to ‘+’ | Reads Map to ‘−’ |
|---|---|---|---|---|---|---|
| RY-1 | 45,558,372 | 37,795,360 (82.96%) | 34,764,686 (76.31%) | 3,030,674 (6.65%) | 18,136,253 (39.81%) | 18,093,059 (39.71%) |
| RY-2 | 47,539,648 | 39,824,315 (83.77%) | 36,730,552 (77.26%) | 3,093,763 (6.51%) | 19,145,848 (40.27%) | 19,100,686 (40.18%) |
| RY-3 | 42,686,462 | 35,339,915 (82.79%) | 32,530,754 (76.21%) | 2,809,161 (6.58%) | 16,959,449 (39.73%) | 16,919,275 (39.64%) |
| NY-1 | 45,575,572 | 38,308,495 (84.05%) | 35,308,726 (77.47%) | 2,999,769 (6.58%) | 18,402,601 (40.38%) | 18,390,534 (40.35%) |
| NY-2 | 52,157,394 | 43,816,115 (84.01%) | 40,369,542 (77.40%) | 3,446,573 (6.61%) | 21,050,148 (40.36%) | 21,040,603 (40.34%) |
| NY-3 | 48,807,096 | 41,183,630 (84.38%) | 37,772,027 (77.39%) | 3,411,603 (6.99%) | 19,718,031 (40.40%) | 19,707,282 (40.38%) |
| RP-1 | 48,464,548 | 41,247,795 (85.11%) | 37,712,174 (77.81%) | 3,535,621 (7.30%) | 19,584,181 (40.41%) | 19,515,493 (40.27%) |
| RP-2 | 64,902,996 | 55,737,684 (85.88%) | 51,160,661 (78.83%) | 4,577,023 (7.05%) | 26,528,679 (40.87%) | 26,456,969 (40.76%) |
| RP-3 | 56,000,364 | 48,249,805 (86.16%) | 44,167,278 (78.87%) | 4,082,527 (7.29%) | 22,926,566 (40.94%) | 22,845,563 (40.80%) |
| NP-1 | 43,593,620 | 36,841,808 (84.51%) | 33,972,850 (77.93%) | 2,868,958 (6.58%) | 17,632,332 (40.45%) | 17,616,465 (40.41%) |
| NP-2 | 41,691,114 | 35,541,476 (85.25%) | 32,900,607 (78.92%) | 2,640,869 (6.33%) | 17,063,308 (40.93%) | 17,047,411 (40.89%) |
| NP-3 | 43,659,156 | 37,161,184 (85.12%) | 34,291,341 (78.54%) | 2,869,843 (6.57%) | 17,795,242 (40.76%) | 17,780,682 (40.73%) |
| Gene_ID | FPKM Value | Annotation | |
|---|---|---|---|
| RY | NY | ||
| Jrgene13135 | 155.58 | 25.96 | PAL |
| Jrgene36733 | 65.03 | 1.98 | CHS |
| Jrgene1355 | 26.26 | 4.93 | F3′5′H |
| Jrgene14301 | 25.70 | 5.12 | UFGT |
| Jrgene32450 | 294.37 | 156.67 | MYB |
| Jrgene32411 | 1.92 | 0.52 | bHLH |
| Jrgene7876 | 81.21 | 18.15 | WD40 |
| Gene_ID | FPKM Value | Annotation | |
|---|---|---|---|
| RP | NP | ||
| Jrgene13135 | 286.55 | 21.74 | PAL |
| Jrgene4994 | 0.47 | 4.70 | CHS |
| Jrgene30190 | 0.14 | 6.09 | F3′5′H |
| Jrgene39130 | 0.22 | 9.03 | F3H |
| Jrgene39777 | 52.47 | 11.28 | UFGT |
| Jrgene32450 | 30.76 | 5.21 | MYB |
| Jrgene32411 | 9.50 | 0.56 | bHLH |
| Jrgene7876 | 47.44 | 31.35 | WD40 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Luo, X.; Wu, C.; Cao, S.; Zhou, Y.; Jie, B.; Cao, Y.; Meng, H.; Wu, G. Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.). Molecules 2018, 23, 25. https://doi.org/10.3390/molecules23010025
Li Y, Luo X, Wu C, Cao S, Zhou Y, Jie B, Cao Y, Meng H, Wu G. Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.). Molecules. 2018; 23(1):25. https://doi.org/10.3390/molecules23010025
Chicago/Turabian StyleLi, Yongzhou, Xiang Luo, Cuiyun Wu, Shangyin Cao, Yifei Zhou, Bo Jie, Yalong Cao, Haijun Meng, and Guoliang Wu. 2018. "Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.)" Molecules 23, no. 1: 25. https://doi.org/10.3390/molecules23010025
APA StyleLi, Y., Luo, X., Wu, C., Cao, S., Zhou, Y., Jie, B., Cao, Y., Meng, H., & Wu, G. (2018). Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.). Molecules, 23(1), 25. https://doi.org/10.3390/molecules23010025