Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
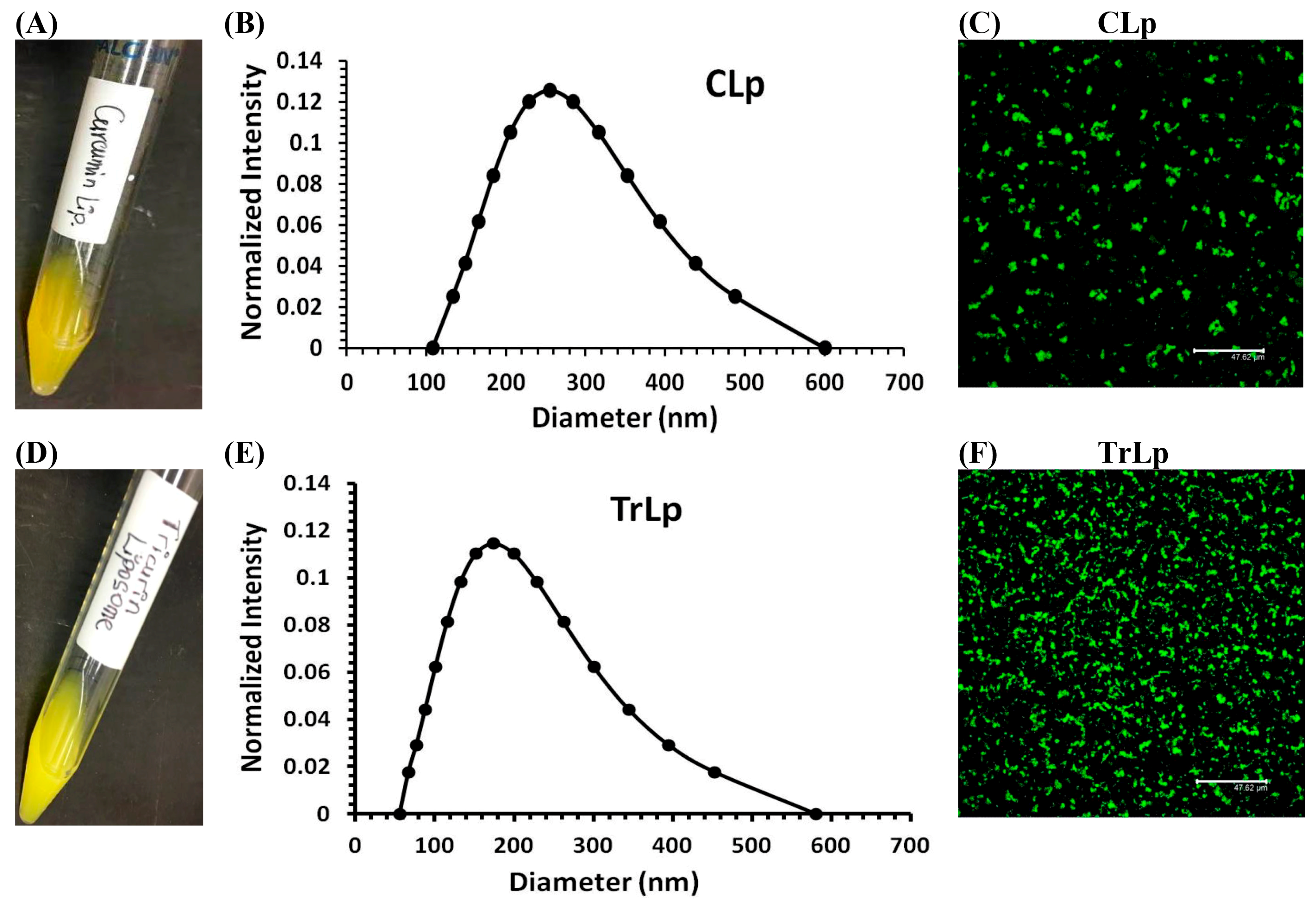
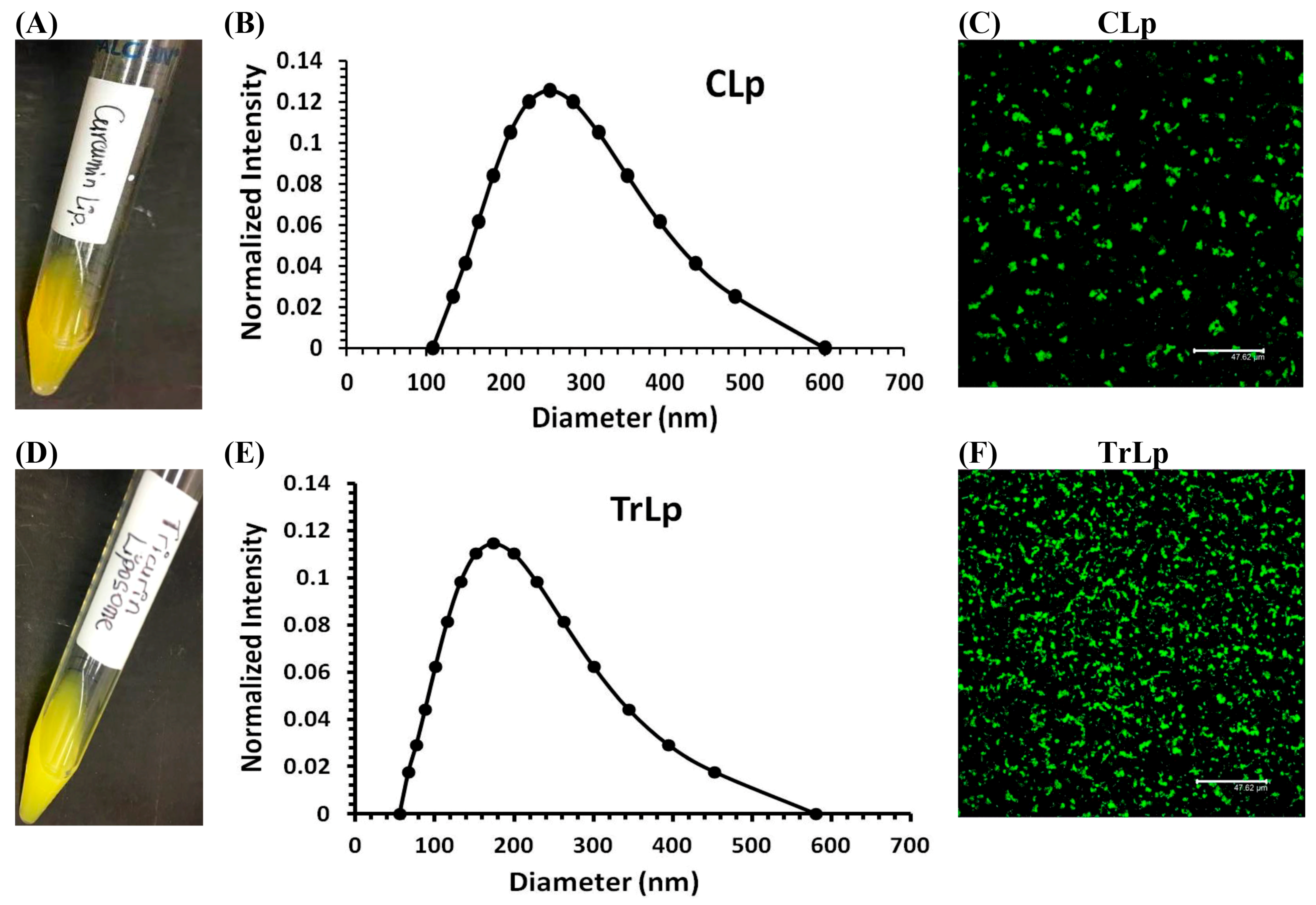
2.1. Physical Characterization of CLp and TrLp Particles
2.2. TrLp Is More Potent Than CLp in Eliminating GL261 Cells and Inhibiting Its Clonogenic Potential
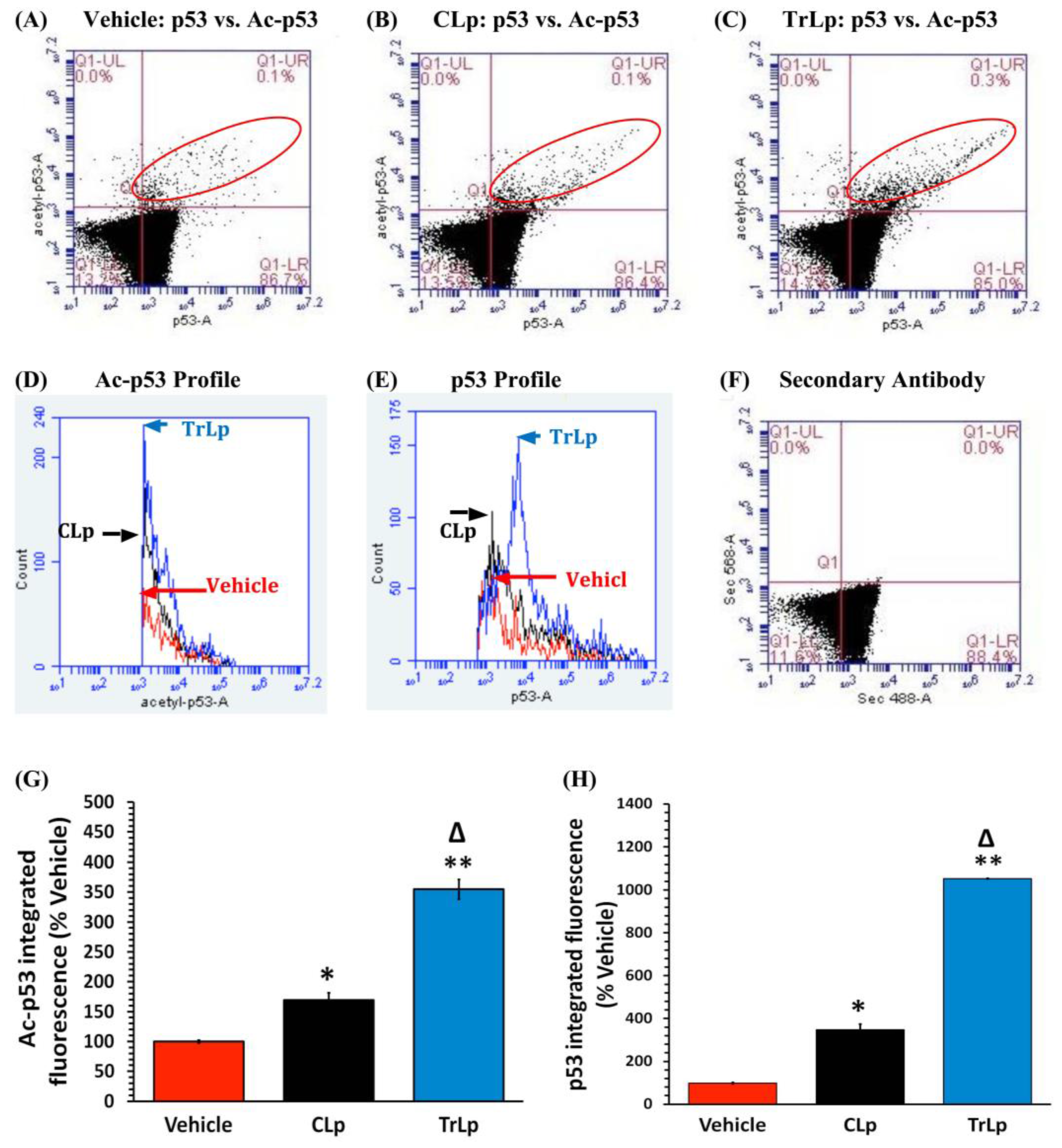
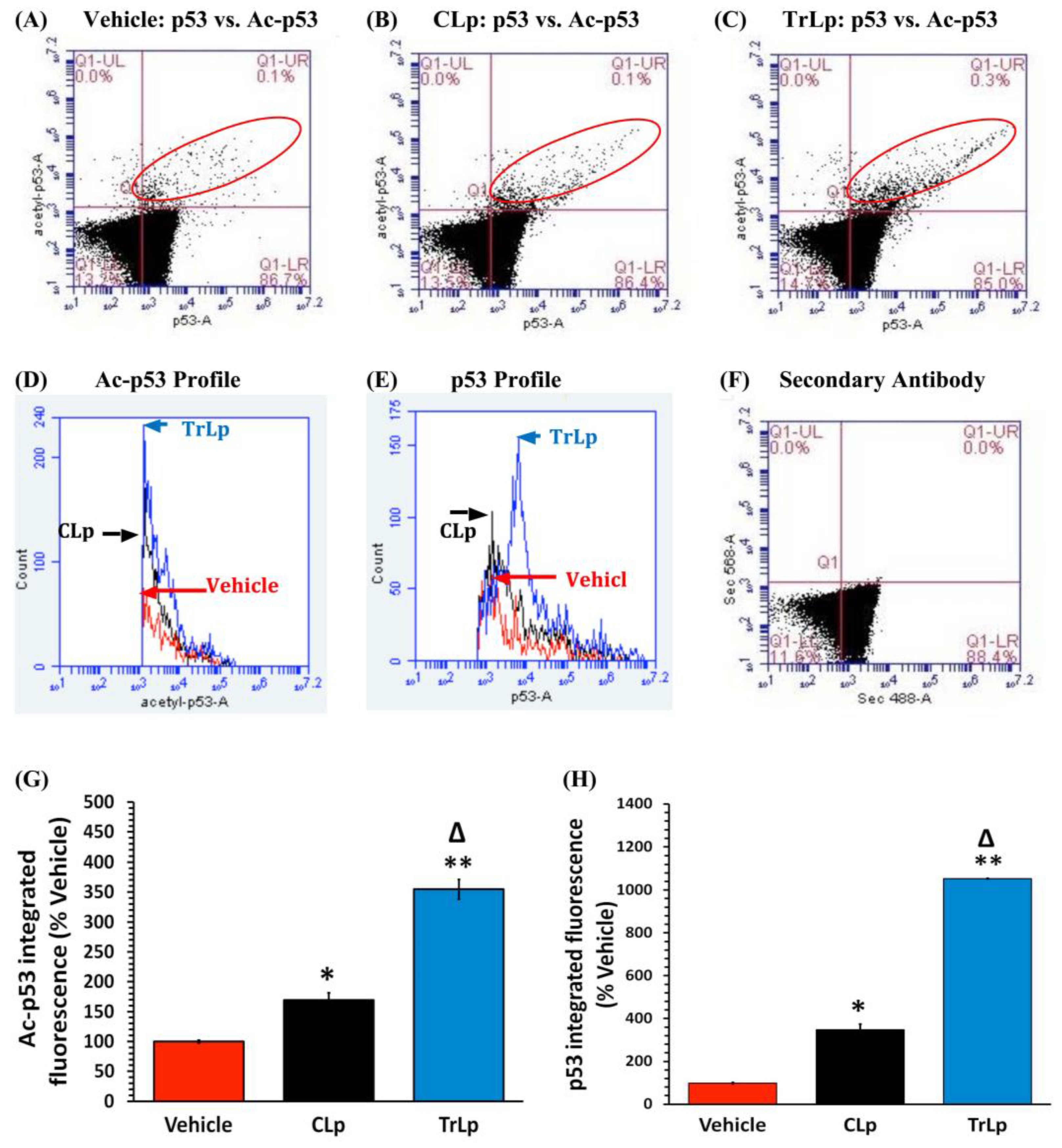
2.3. TrLp Potently Upregulates Activated p53 in Cultured GL261 Cells
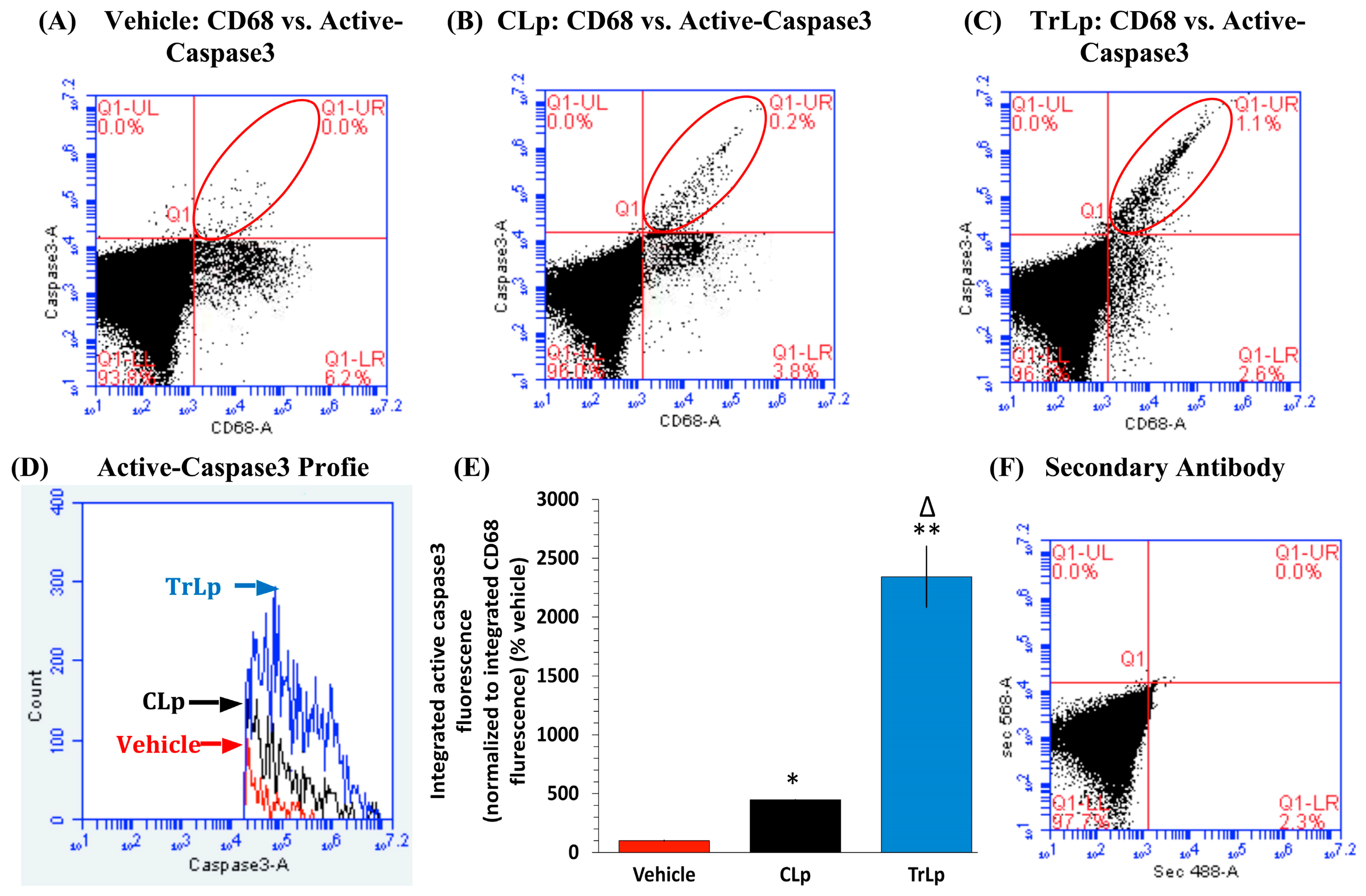
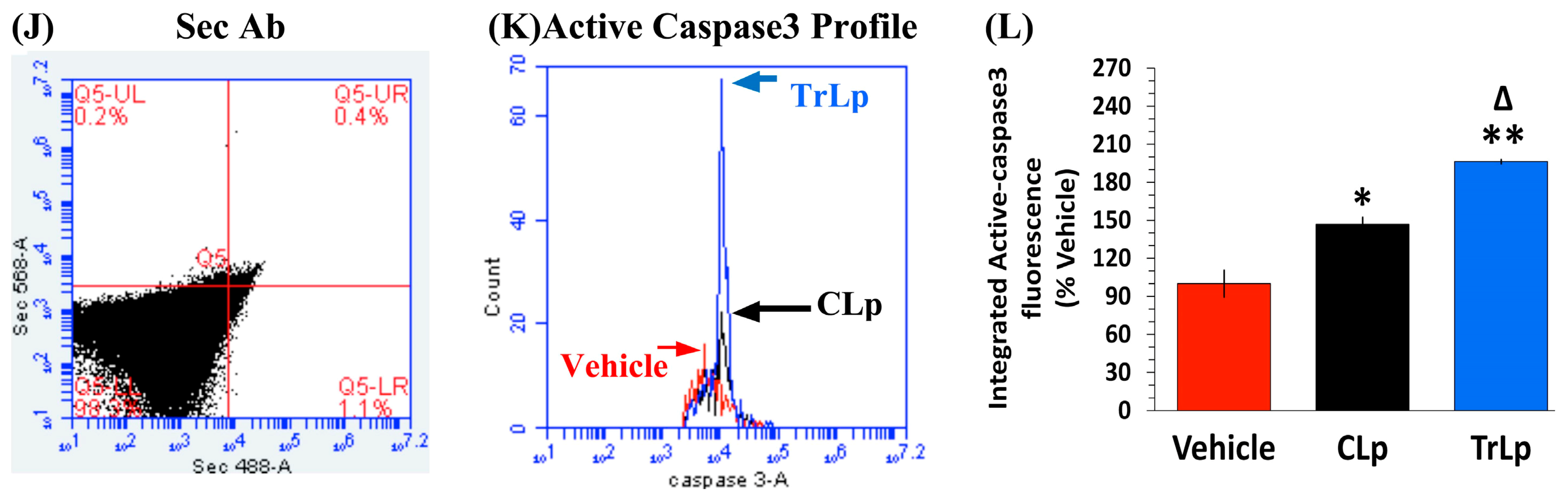
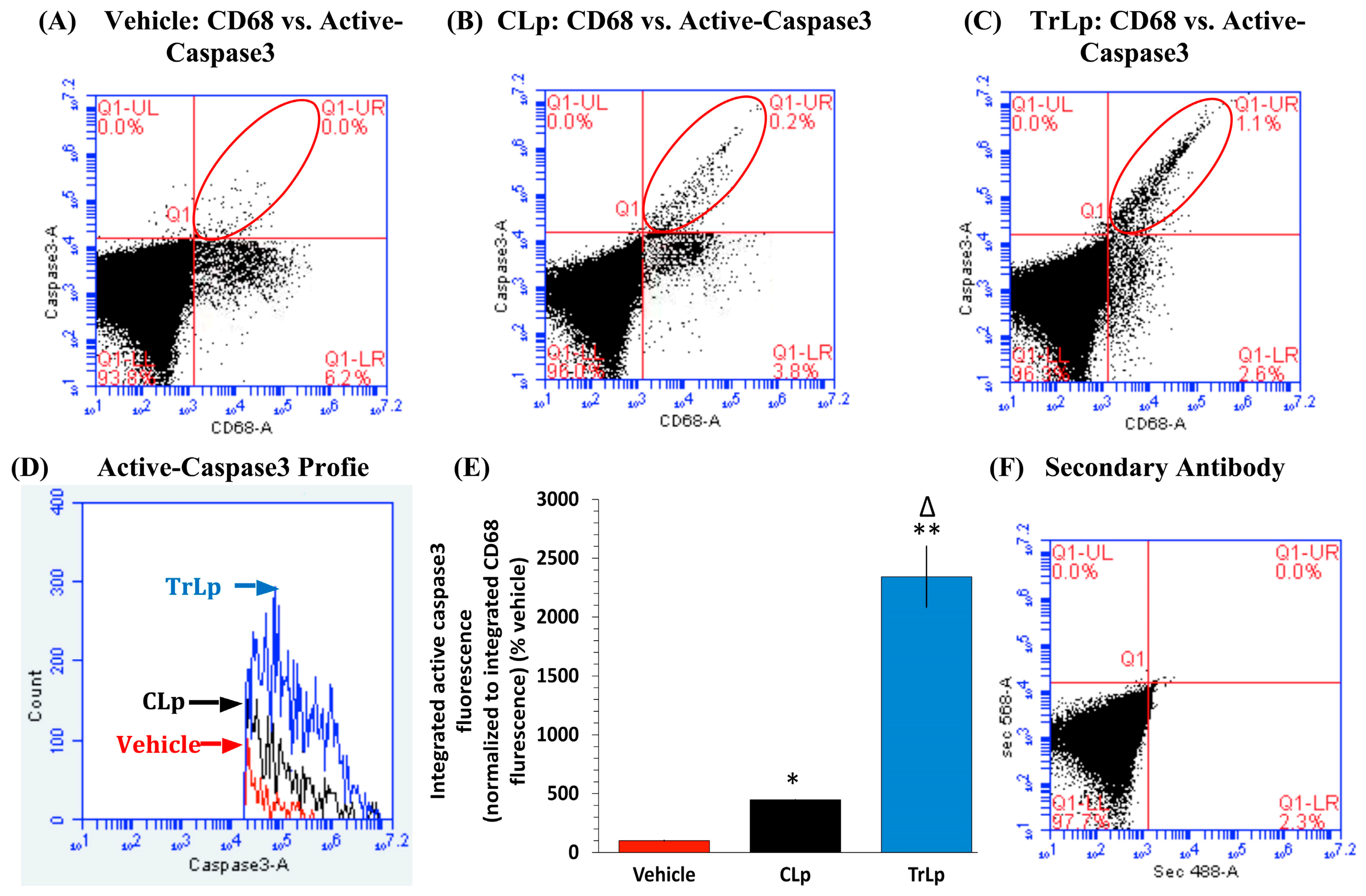
2.4. TrLp-Evoked Induction in Activated p53 Is Associated with caspase3 Activation in CD68(+) GL261 Cells
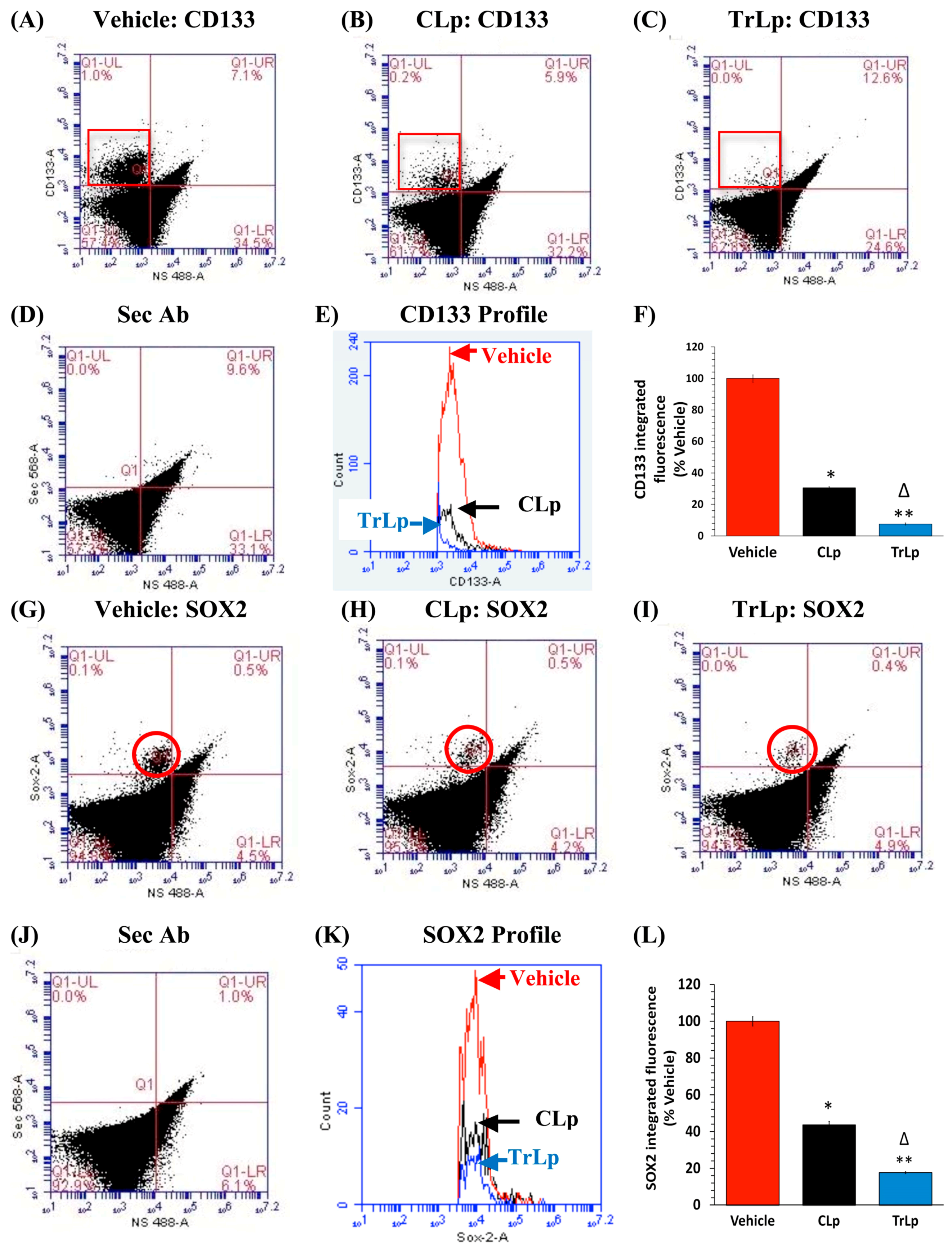
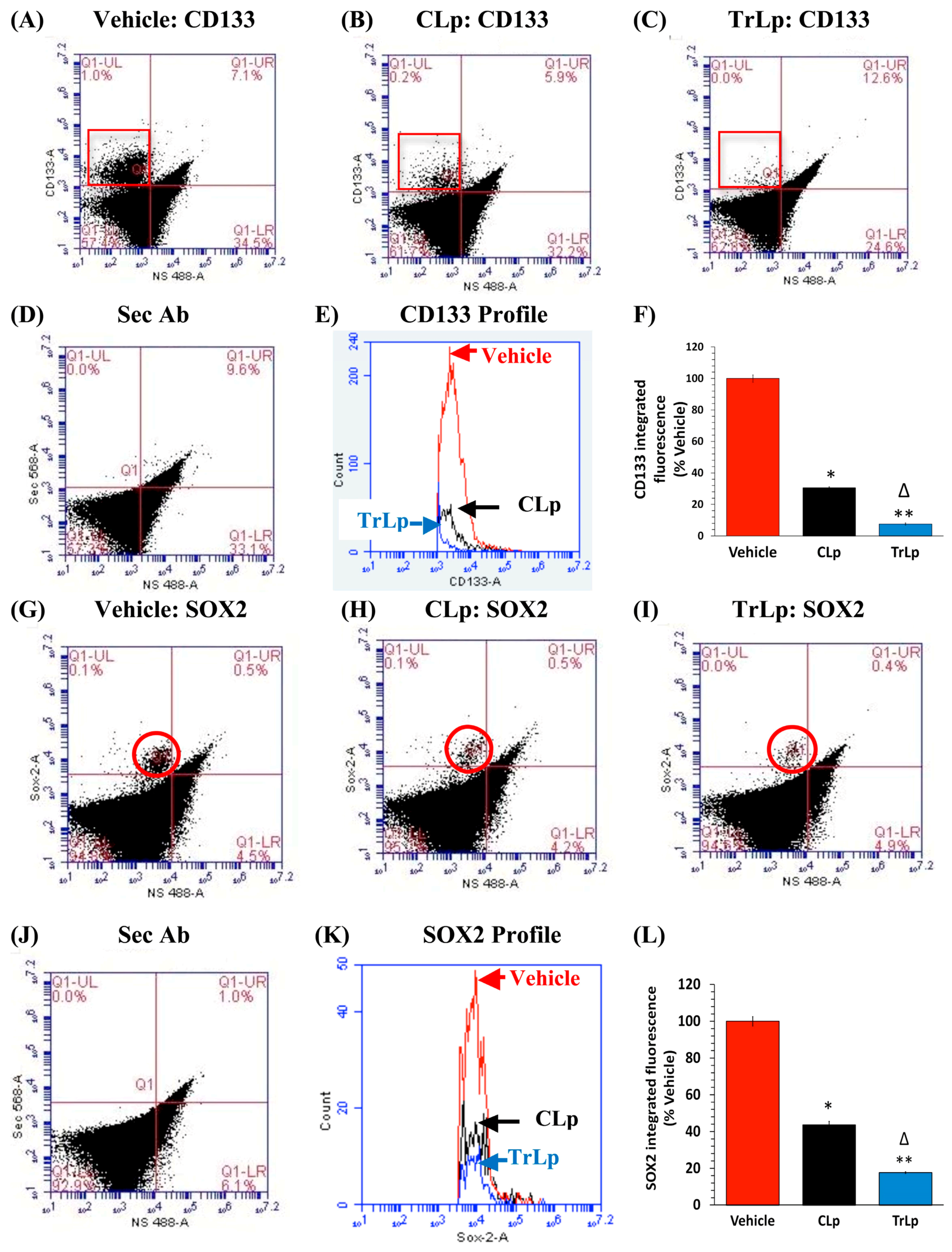
2.5. TrLp-Evoked Suppression of CD133(+) and SOX2(+) GL261 Stem Cells in Culture
2.6. TrLp Causes Increased Internalization of TrLp-Associated Curcumin in SOX2(+) GL261 Stem Cells in Culture
2.7. Pharmacokinetic Assessment of Curcuminoids (C) in the Plasma of TrLp-Injected Mice by HPLC
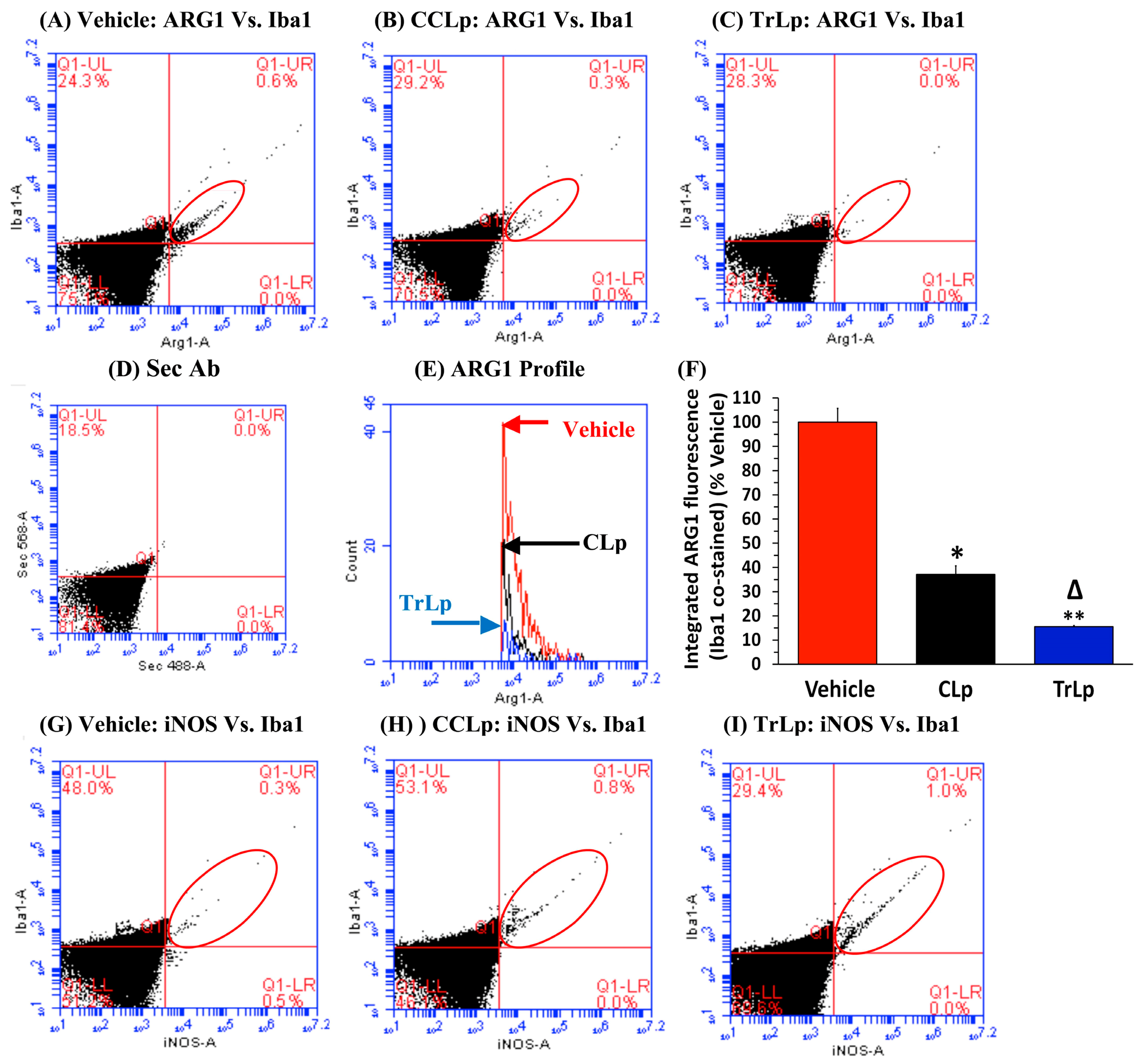
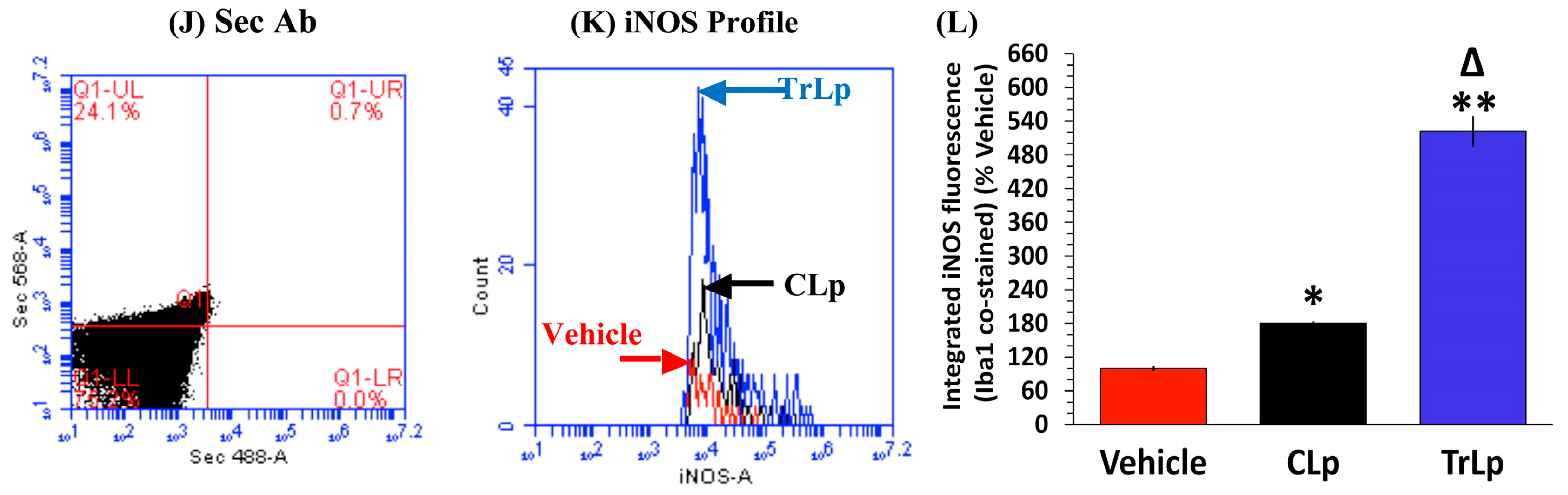
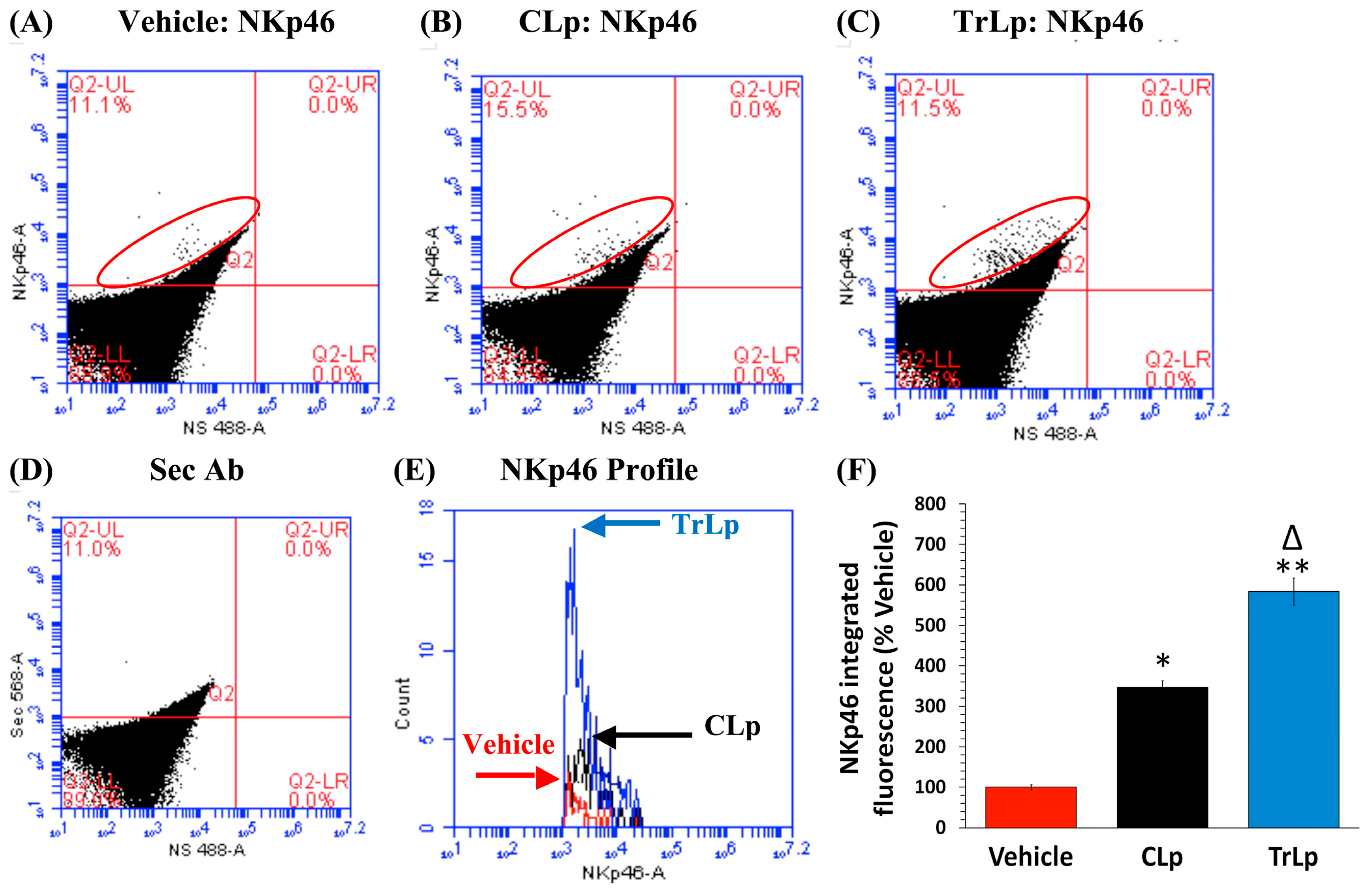
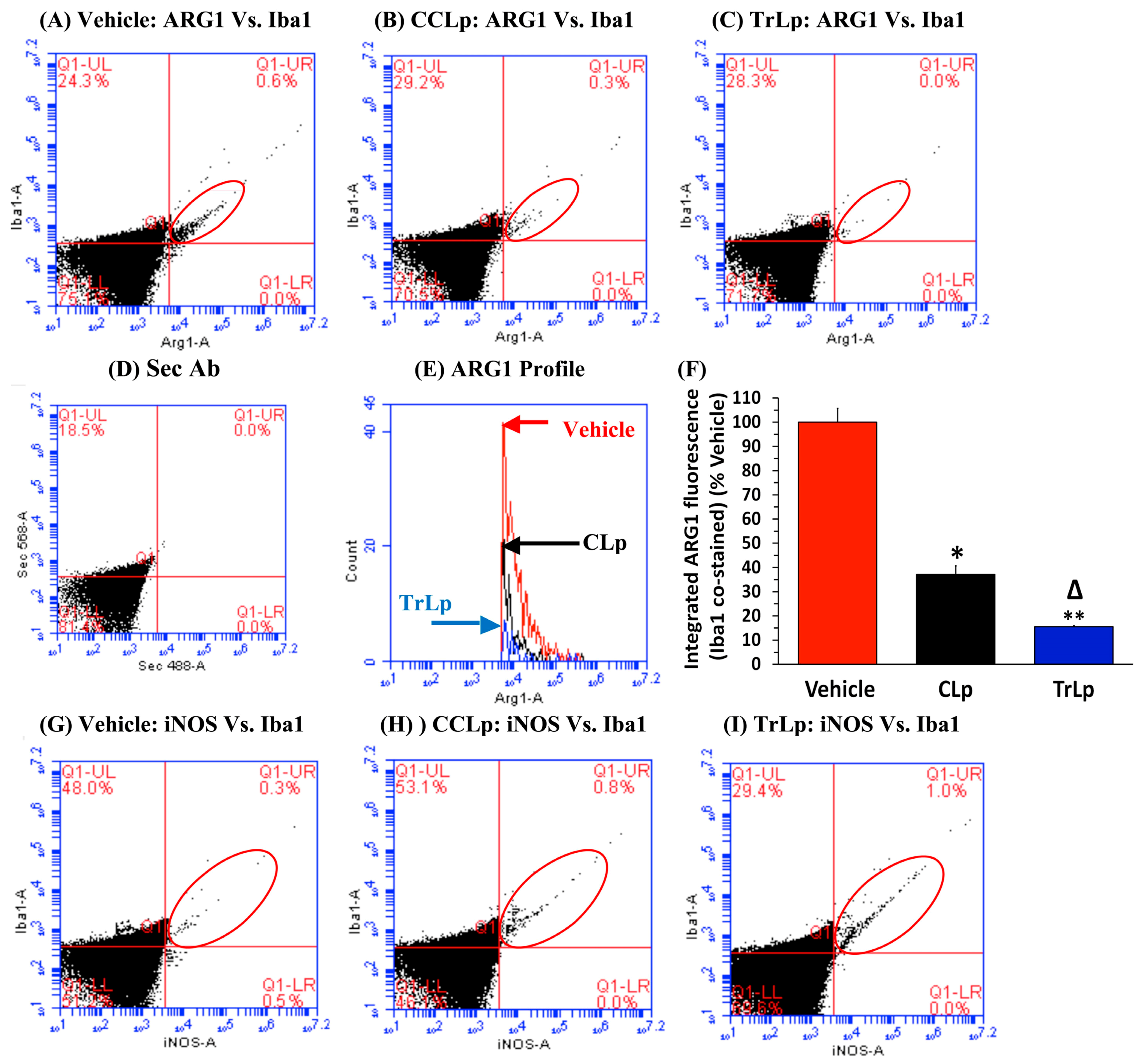
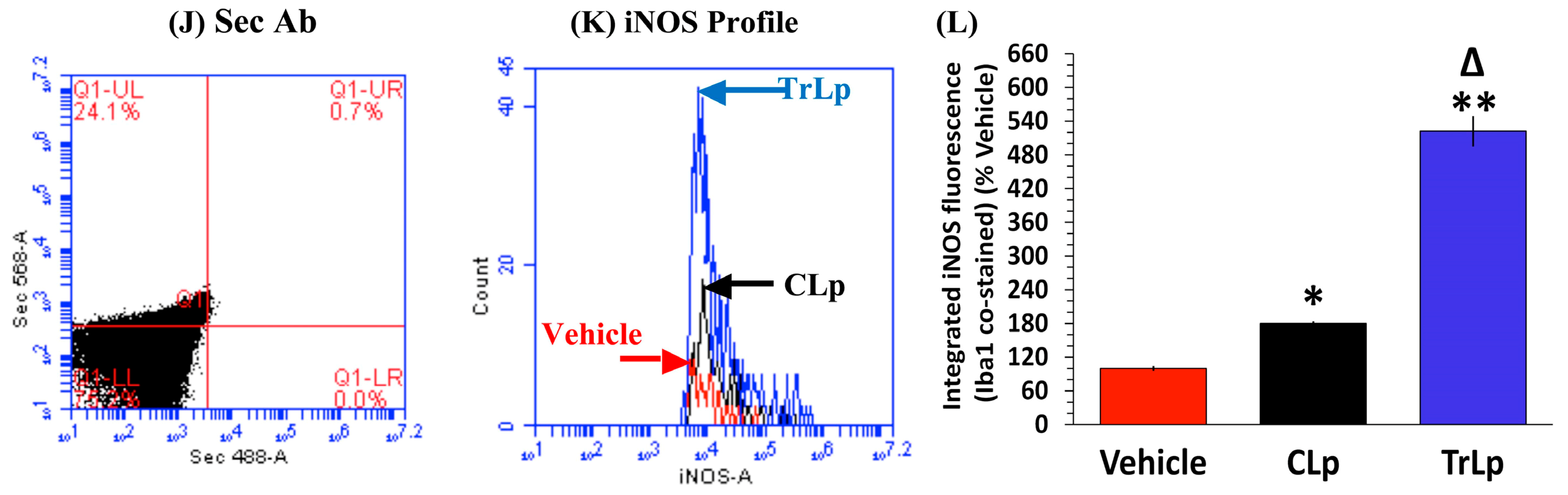
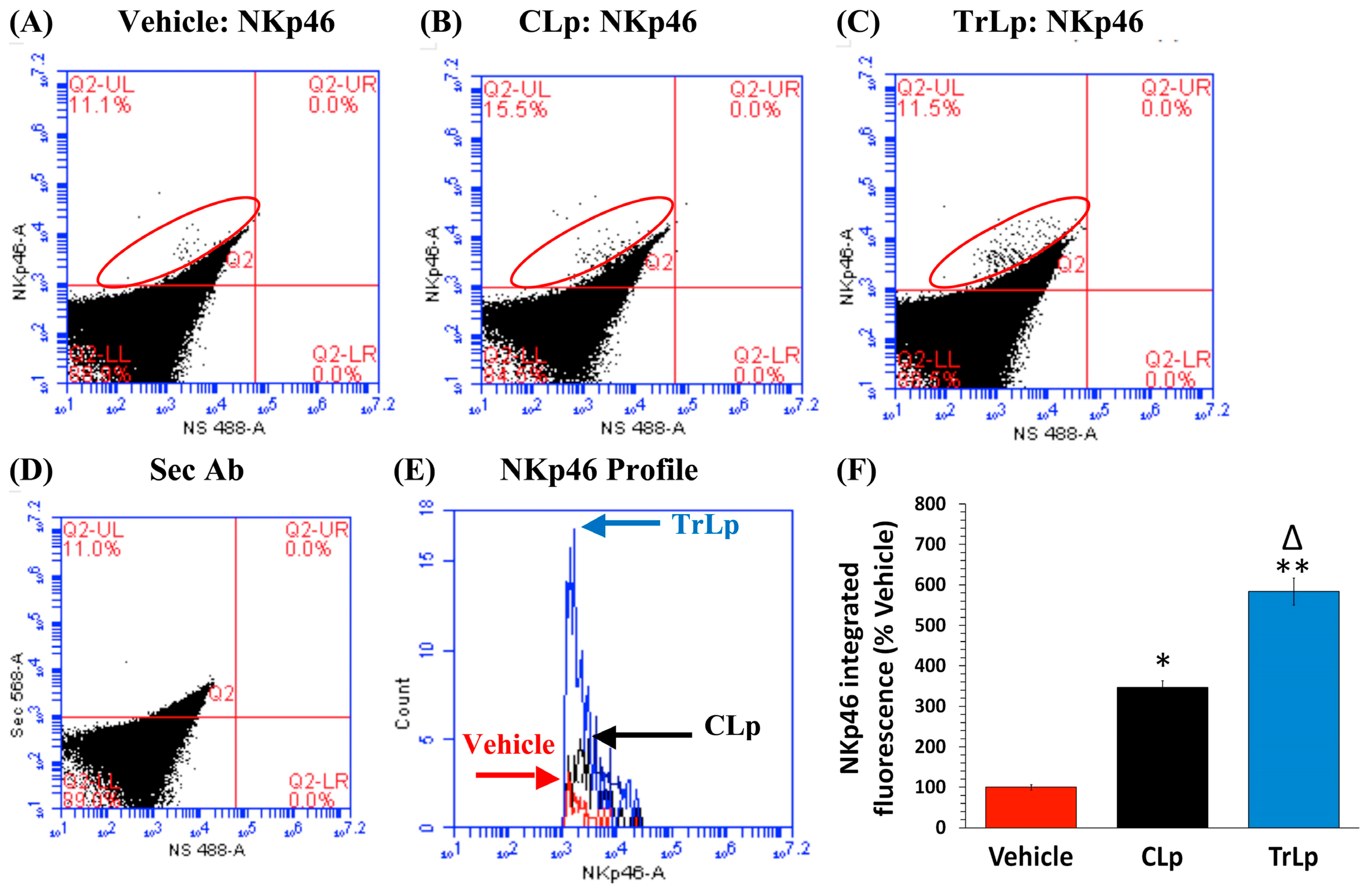
2.8. In a Short-Term (5-Day) Treatment Regimen, TrLp and CLp Cause Repolarization of iNOSlow Arg1high M2-Like Tumor-Associated Microglia/Macrophages (TAM) to iNOShigh Arg1low M1-Like TAM and Intra-Tumor Recruitment of NKp46(+) (Activated) Natural Killer (NK) Cells
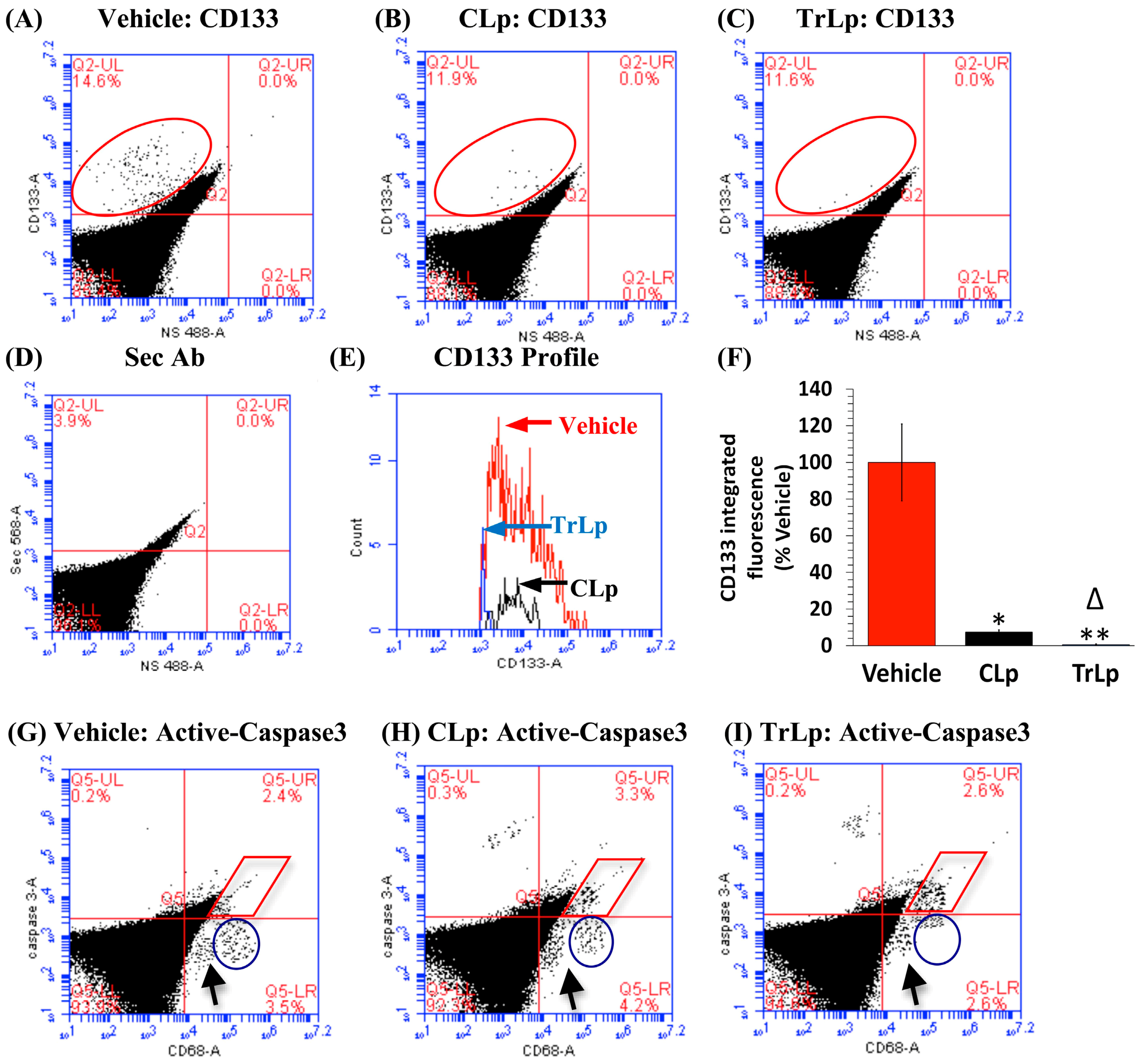
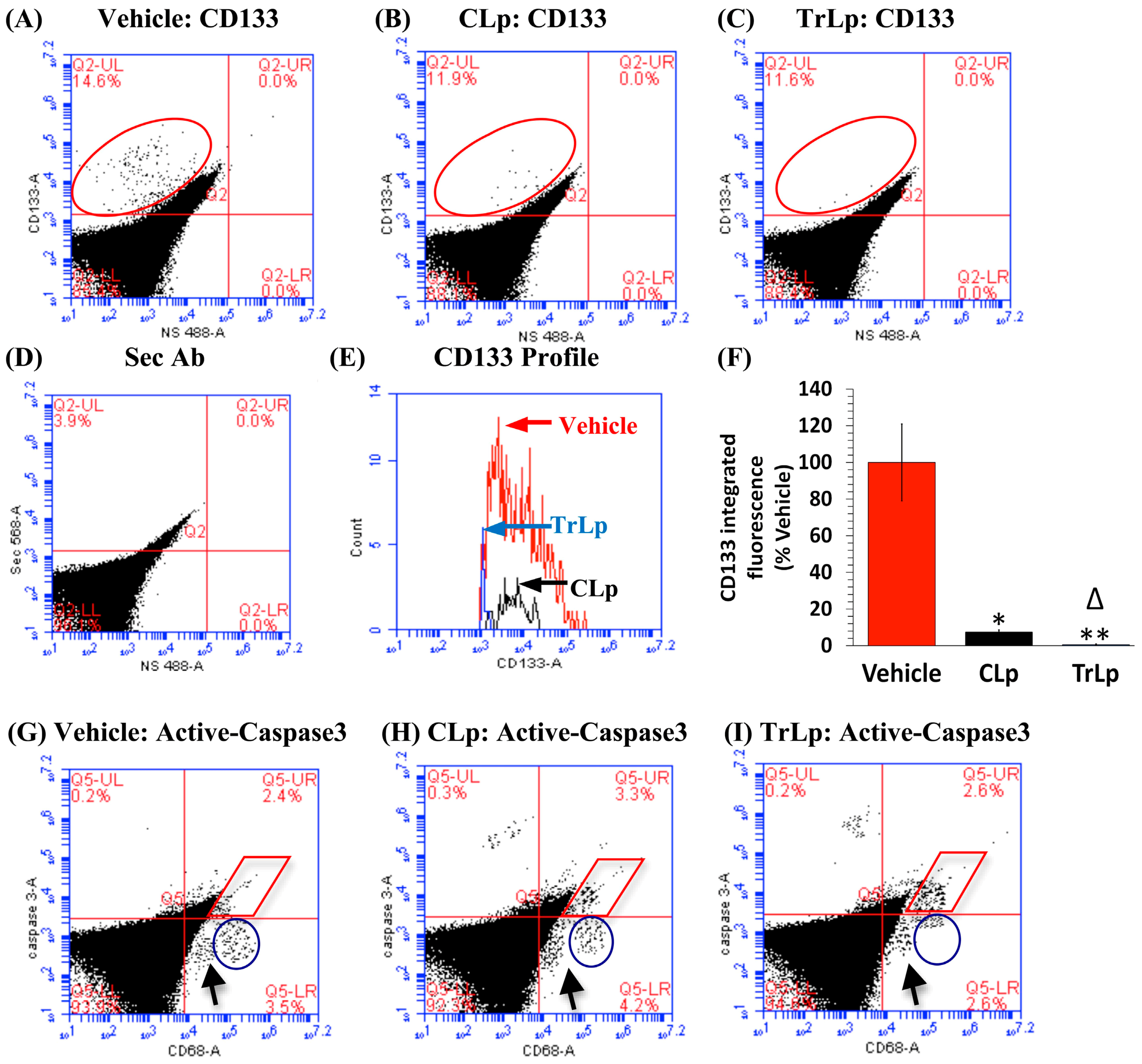
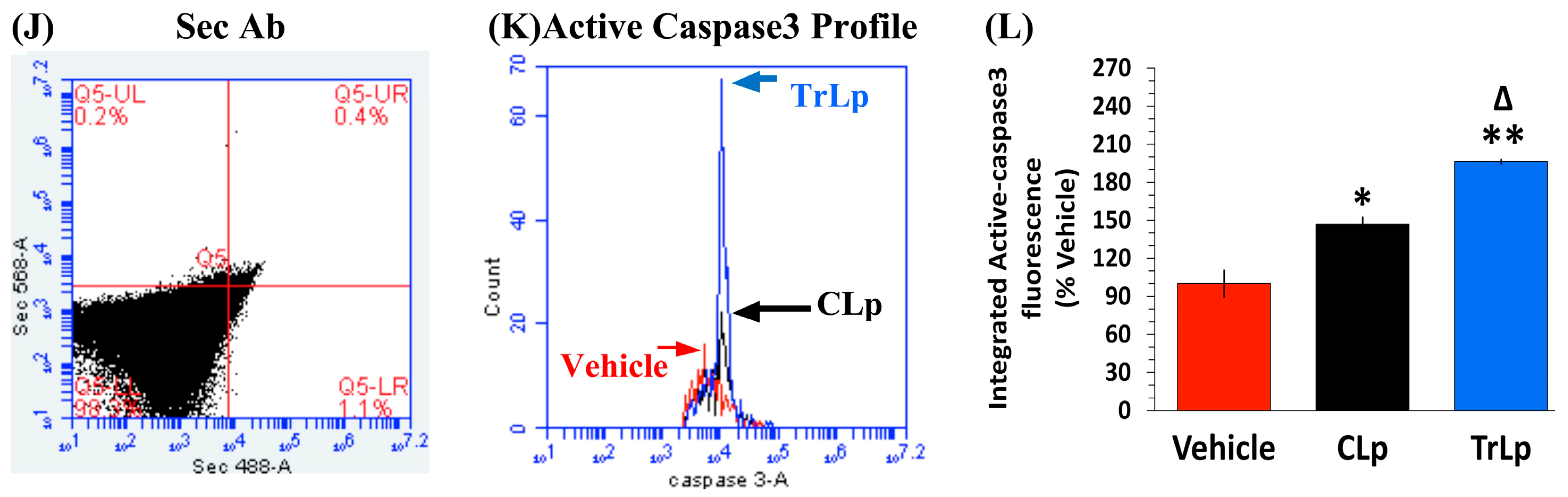
2.9. In the 5-Day Treatment of GBM Mice, TrLp and CLp Cause Suppression of CD133(+) GBM Stem Cells and Apoptosis of GL261-Evoked CD68high GBM Tumor Cells
2.10. Short-Term Treatment with TrLp and CLp Causes Tumor Shrinkage, but Long-Term Treatment Causes Rescue of Mice
3. Discussion
4. Methods and Materials
4.1. Animals
4.2. Curcumin & TriCurin
4.3. Preparation of TriCurin Lipososme (TrLp) and and Curcumin Liposome (CLp)
4.4. Purification of Liposomes and Analysis of Liposomal Dimensions by Dynamic Light Scattering (DLS)
4.5. Confocal Imaging of Liposomes
4.6. Cell Culture
4.7. Determination of IC50 Using WST-1 Assay
4.8. Clonogenic Assay
4.9. Staining of GL261 Cells and GBM Tissue for Flow Cytometry
4.10. Immunocytochemical Staining of SOX2(+) GL261 Cells for Studying Curcumin Internalization and/or Stabilization
4.11. Extraction of Curcumin from the Plasma of Mice Treated with TriCurin Liposome and Quantification by HPLC Analysis
4.12. Implantation of GL261 Cells in Mice
4.13. Preparation of Dylight 800-CD68 Ab Adduct and Intranasal Treatment of GBM Mice
4.14. Treatment of Animals
4.15. Immunohistochemistry
4.16. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mahaley, M.S.; Mettlin, C.; Natarajan, N.; Laws, E.R.; Peace, B.B. Analysis of patterns of care of brain tumor patients in the united states: A study of the brain tumor section of the AANS and the CNS and the commission on cancer of the ACS. Clin. Neurosurg. 1990, 36, 347–352. [Google Scholar] [PubMed]
- Bleeker, F.E.; Molenaar, R.J.; Leenstra, S. Recent advances in the molecular understanding of glioblastoma. J. Neuro-Oncol. 2012, 108, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro Oncol. 2009, 11, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyns, B.; Chaskis, C.; Joosens, E.; Menten, J.; D’Hondt, L.; Branle, F.; Sadones, J.; Michotte, A. A multicenter cohort study of dose-dense temozolomide (21 of 28 days) for the treatment of recurrent anaplastic astrocytoma or oligoastrocytoma. Cancer Investig. 2008, 26, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Lin, J.H.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.B.; Sohn, S.; Krown, S.E.; Livington, P.O.; Wolchek, J.D.; Quinn, C.; Williams, L.; Foster, T.; Sepkowitz, K.A.; Chapman, P.B. Selective CD4 lymphopenia in melanoma patients treated with temozolomide: A toxicity with therapeutic implications. J. Clin. Oncol. 2004, 22, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Happold, C.; Roth, P.; Wick, W.; Schmidt, N.; Florea, A.M.; Silginer, M.; Reifenberger, G.; Weller, M. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 2012, 122, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in immunotherapy for glioblastoma multiforme. J. Immunol. Res. 2017, 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Miao, L.; Wang, Y.; Xu, Z.; Zhao, Y.; Shen, Y.; Xiang, G.; Huang, L. Curcumin micelles remodel tumor microenvironment and enhance vaccine activity in an advanced melanoma model. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Baidoo, J.; Fried, A.; Atwi, D.; Dolai, S.; Boockvar, J.; Symons, M.; Ruggieri, R.; Raja, K.; Banerjee, P. Curcumin changes the polarity of tumor-associated microglia and eliminates glioblastoma. Int. J. Cancer 2016, 139, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Shiri, S.; Alizadeh, A.M.; Baradaran, B.; Farhanghi, B.; Shanehbandi, D.; Khodayari, S.; Khodayari, H.; Tavassoli, A. Dendrosomal curcumin suppresses metastatic breast cancer in mice by changing m1/m2 macrophage balance in the tumor microenvironment. Asian Pac. J. Cancer Prev. 2015, 16, 3917–3922. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Andreansky, S. Antitumor immunity and dietary compounds. Med. Sci. 2014, 2, 1–22. [Google Scholar] [CrossRef]
- Ye, X.-Z.; Xu, S.-L.; Xin, Y.-H.; Yu, S.-C.; Ping, Y.-F.; Chen, L.; Xiao, H.-L.; Wang, B.; Yi, L.; Wang, Q.-L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Neurology and Clinical Neuroscience E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Bhattacharyya, S.; Mandal, D.; Saha, B.; Sen, G.S.; Das, T.; Sa, G. Curcumin prevents tumor-induced T cell apoptosis through stat-5a-mediated bcl-2 induction. J. Biol. Chem. 2007, 282, 15954–15964. [Google Scholar] [CrossRef] [PubMed]
- Debata, P.R.; Castellanos, M.R.; Fata, J.E.; Baggett, S.; Rajupet, S.; Szerszen, A.; Begum, S.; Mata, A.; Murty, V.V.; Opitz, L.M.; et al. A novel curcumin-based vaginal cream vacurin selectively eliminates apposed himan cervical cancer cells. Gynecol. Oncol. 2013, 129, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Langone, P.; Debata, P.R.; Dolai, S.; Curcio, G.M.; Inigo, J.D.; Raja, K.; Banerjee, P. Coupling to a cancer cell-specific antibody potentiates tumoricidal properties of curcumin. Int. J. Cancer 2012, 131, E569–E578. [Google Scholar] [CrossRef] [PubMed]
- Langone, P.; Debata, P.R.; Inigo, J.D.R.; Dolai, S.; Mukherjee, S.; Halat, P.; Mastroianni, K.; Curcio, G.M.; Castellanos, M.R.; Raja, K.; et al. Coupling to a glioblastoma-directed antibody potentiates anti-tumor activity of curcumin. Int. J. Cancer 2014, 135, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T. Targeting cancer stem cells by curcumin and clinical applications. Cancer Lett. 2014, 346, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.J.; Bisht, S.; Bar, E.E.; Maitra, A.; Eberhart, C.G. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol. Ther. 2011, 11, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Maher, D.M.; Bell, M.C.; O’Donnell, E.A.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Curcumin suppresses human papillomavirus oncoproteins, restores p53, Rb, and PTPN13 proteins and inhibits benzo[a]pyrene-induced upregulation of HPV E7. Mol. Carcinog. 2011, 50, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Debata, P.R.; Hussaini, R.; Chatterjee, K.; Baidoo, J.N.E.; Sampat, S.; Szerszen, A.; Navarra, J.P.; Fata, J.; Severinova, E.; et al. Unique synergistic formulation of curcumin, epicatechin gallate and resveratrol, tricurin, suppresses HPV E6, eliminates HPV+ cancer cells, and inhibits tumor progression. Oncotarget 2017, 8, 60904–60916. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Mukherjee, S.; Chang, Q.; Xie, X.; Li, H.; Castellanos, M.R.; Banerjee, P.; Iqbal, H.; Ivancic, R.; Wang, X.; et al. Tricurin, a novel formulation of curcumin, epicatechin gallate, and resveratrol, inhibits the tumorigenicity of human papillomavirus-positive head and neck squamous cell carcinoma. Oncotarget 2017, 8, 60025–60035. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.; Berliner, A.; Fernando, S.S.; Ranasinghe, B.; Ray, I.; Tariq, H.; Banerjee, P. Curcumin blocks brain tumor formation. Brain Res. 2009, 1266C, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhou, Y.; Wen, C.; Zhou, C.; Zhang, W.; Hu, X.; Wang, L.; You, C.; Shao, J. Curcumin sensitizes glioblastoma to temozolomide by simultaneously generating ROS and disrupting AKT/mTOR signaling. Oncol. Rep. 2014, 32, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Shakeri, A.; Rashidi, B.; Jalili, A.; Banikazemi, Z.; Sahebkar, A. Phytosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed. Pharmacother. 2017, 85, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Debata, P.R.; Curcio, G.M.; Mukherjee, S.; Banerjee, P. Causal factors for brain tumor and targeted strategies. In Universe of Scales: From Nanotechnology to Cosmology; Laughoff, S., Freund, F., Eds.; Springer: Cham, Switzerland, 2014; Volume 150, pp. 191–204. [Google Scholar]
- Li, L.; Braiteh, F.S.; Kurzrock, R. Liposome-encapsulated curcumin: In vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer 2005, 104, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Matloob, A.H.; Mourtas, S.; Klepetsanis, P.; Antimisiaris, S.G. Increasing the stability of curcumin in serum with liposomes or hybrid drug-in-cyclodextrin-in-liposome systems: A comparative study. Int. J. Pharm. 2014, 476, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.P.; Mukerjee, A.; Helson, L.; Gupta, R.; Vishwanatha, J.K. Efficacy of liposomal curcumin in a human pancreatic tumor xenograft model: Inhibition of tumor growth and angiogenesis. Anticancer Res. 2013, 33, 3603–3609. [Google Scholar] [PubMed]
- Marczylo, T.H.; Verschoyle, R.D.; Cooke, D.N.; Morazzoni, P.; Steward, W.P.; Gescher, A.J. Comparison of systemic availability of curcumin with that of curcumin formulated with phosphatidylcholine. Cancer Chemother. Pharmacol. 2007, 60, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; Roth, J.A.; Tanaka, N. Overexpression of the wild-type p53 gene inhibits NF-kappaB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.S.; Mohanty, S.; Hossain, D.; Md, S.; Bhattacharyya, S.; Banerjee, S.; Chakraborty, J.; Saha, S.; Ray, P.; Bhattacharjee, P.; et al. Curcumin enhances the efficacy of chemotherapy by tailoring p65NFkB-p300 cross-talk in favor of p53–p300 in breast cancer. J. Biol. Chem. 2011, 286, 42232–42247. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Srivastava, R.K. Involvement of Bcl-2 family members, phosphatidylinositol 3’-kinase/AKT and mitochondrial p53 in curcumin (diferulolylmethane)-induced apoptosis in prostate cancer. Int. J. Oncol. 2007, 30, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Bassiouny, A.R.; Zaky, A. Synergistic effect of curcumin on temozolomide inhibition of cancer stem cell-like properties and reduced chemoresistance of glioblastoma C6. In Proceedings of the International Conference on Regenerative & Functional Medicine, Hilton San Antonio Airport, TX, USA, 12–14 November 2012; OMICS Group: Hilton San Antonio Airport, TX, USA, 2012; Volume 2. [Google Scholar]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Song, X.; Zhang, Y.; Chu, Y. Low-dose curcumin leads to the inhibition of tumor growth via enhancing CTL-mediated antitumor immunity. Int. Immunopharmacol. 2011, 11, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Doring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Brantley, E.C.; Guo, L.; Zhang, C.; Lin, Q.; Yokoi, K.; Langley, R.R.; Kruzel, E.; Maya, M.; Kim, S.W.; Kim, S.-J.; et al. Nitric oxide-mediated tumoricidal activity of murine microglial cells. Transl. Oncol. 2010, 3, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Caligiuri, M.A. Human natural killer cell development and biology. Blood Rev. 2006, 20, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Leon, L.; Jeannin, J.F.; Bettaieb, A. Post-translational modifications induced by nitric oxide (no): Implication in cancer cells apoptosis. Nitric Oxide Biol. Chem. 2008, 19, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rahat, M.A.; Hemmerlein, B. Macrophage-tumor cell interactions regulate the function of nitric oxide. Front. Physiol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.; Safta, T.B.; Ziani, L.; Chouaib, S. Mechanisms of cytotoxic lymphocyte-mediated apoptosis and relationship with the tumor suppressor p53. Crit. Rev. Immunol. 2015, 35, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Gertsch, J.; Guttinger, M.; Heilmann, J.; Sticher, O. Curcumin differentially modulates mRNA profiles in Jurkat T and human peripheral blood mononuclear cells. Bioorg. Med. Chem. 2003, 11, 1057–1063. [Google Scholar] [CrossRef]
- Kang, T.H.; Lee, J.H.; Song, C.K.; Han, H.D.; Shin, B.C.; Pai, S.I.; Hung, C.F.; Trimble, C.; Lim, J.S.; Kim, T.W.; et al. Epigallocatechin-3-gallate enhances CD8+ T cell-mediated antitumor immunity induced by DNA vaccination. Cancer Res. 2007, 67, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, G.X.; Wang, W.; Li, T.; Yuan, Y.K. Resveratrol induces apoptosis, influences IL-6 and exerts immunomodulatory effect on mouse lymphocytic leukemia both in vitro and in vivo. Int. Immunopharmacol. 2007, 7, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.K.; Nargi, D.; Randolph, C.; Narayanan, B.A. Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in pten knockout mice. Int. J. Cancer 2009, 125, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, R.; Beulaja, M.; Arulvasu, C.; Sellamuthu, S.; Dinesh, D.; Prabhu, D.; Babu, G.; Vaseeharan, B.; Prabhu, N.M. Synergistic anticancer activity of curcumin and catechin: An in vitro study using human cancer cell lines. Microscopy Res. Tech. 2012, 75, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.P.; Banerjee, S.; Nautiyal, J.; Patel, B.B.; Patel, V.; Du, J.; Yu, Y.; Elliott, A.A.; Levi, E.; Sarkar, F.H. Curcumin synergizes with resveratrol to inhibit colon cancer. Nutr. Cancer 2009, 61, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lund, K.C.; Pantuso, T. Combination effects of quercetin, resveratrol and curcumin on in vitro intestinal absorption. J. Restor. Med. 2014, 3, 112–120. [Google Scholar] [CrossRef]
- Chen, M.H. Cardiac dysfunction induced by novel targeted anticancer therapy: An emerging issue. Curr. Cardiol. Rep. 2009, 11, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, M.; Bondy, M.L.; Wiencke, J.; Yost, M. Environmental risk factors for primary malignant brain tumors: A review. J. Neuro-Oncol. 1993, 17, 47–64. [Google Scholar] [CrossRef]
- Davis, F.G.; Malinski, N.; Haenszel, W.; Chang, J.; Flannery, J.; Gershman, S.; Dibble, R.; Bigner, D.D. Primary brain tumor incidence rates in four united states regions, 1985–1989: A pilot study. Neuroepidemiology 1996, 15, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Ernstoff, M.S.; Fadul, C.E. Immunotherapy for the treatment of glioblastoma. Cancer J. (Sudbury, Mass.) 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Baidoo, J.N.E.; Sampat, S.; Mancuso, A.; David, L.; Cohen, L.S.; Zhou, S.; Banerjee, P. Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells. Molecules 2018, 23, 201. https://doi.org/10.3390/molecules23010201
Mukherjee S, Baidoo JNE, Sampat S, Mancuso A, David L, Cohen LS, Zhou S, Banerjee P. Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells. Molecules. 2018; 23(1):201. https://doi.org/10.3390/molecules23010201
Chicago/Turabian StyleMukherjee, Sumit, Juliet N. E. Baidoo, Samay Sampat, Andrew Mancuso, Lovena David, Leah S. Cohen, Shuiqin Zhou, and Probal Banerjee. 2018. "Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells" Molecules 23, no. 1: 201. https://doi.org/10.3390/molecules23010201
APA StyleMukherjee, S., Baidoo, J. N. E., Sampat, S., Mancuso, A., David, L., Cohen, L. S., Zhou, S., & Banerjee, P. (2018). Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells. Molecules, 23(1), 201. https://doi.org/10.3390/molecules23010201