Small Peptides Able to Suppress Prostaglandin E2 Generation in Renal Mesangial Cells





Abstract
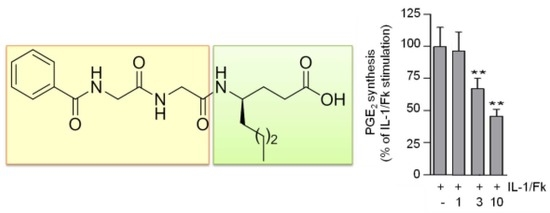
:
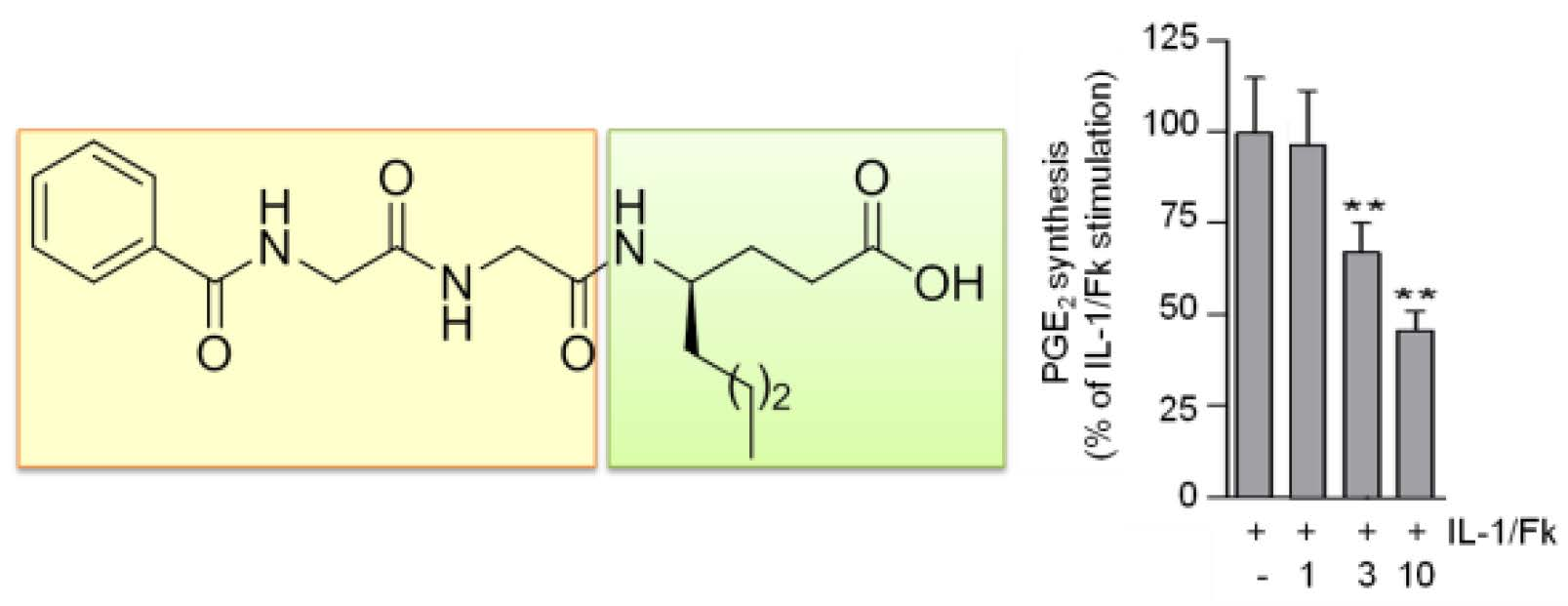{kind=link}
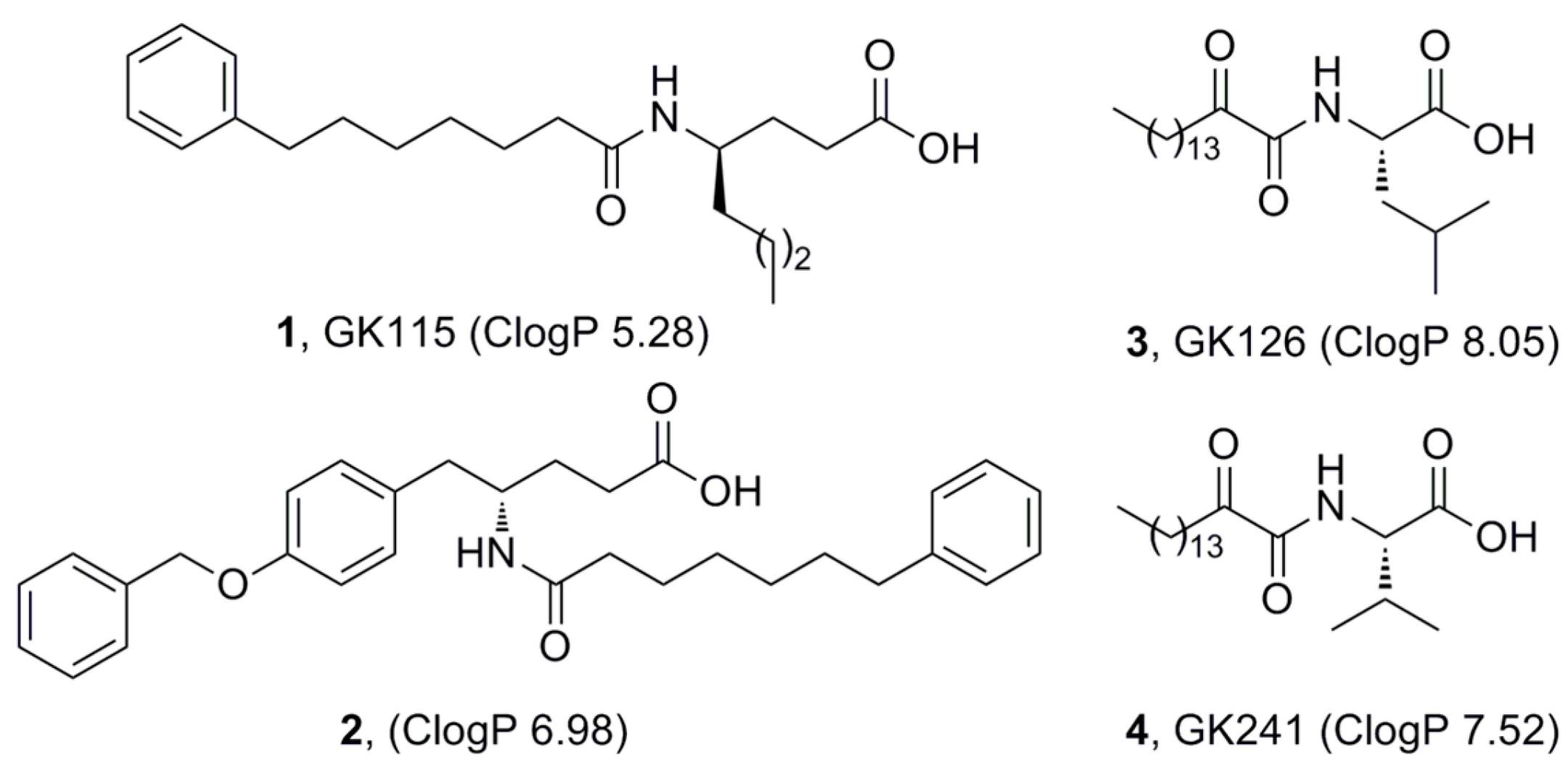{kind=link}
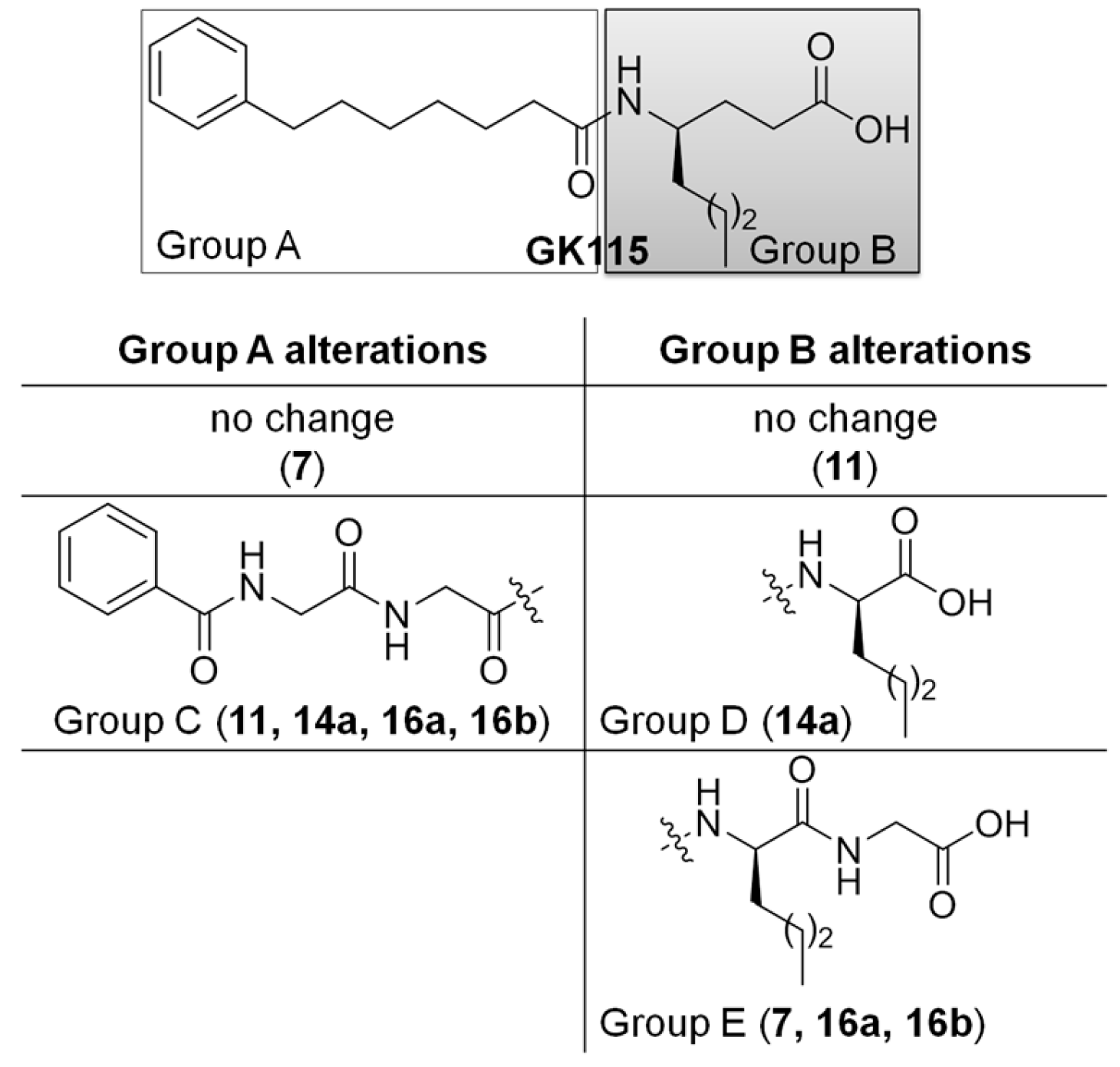{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
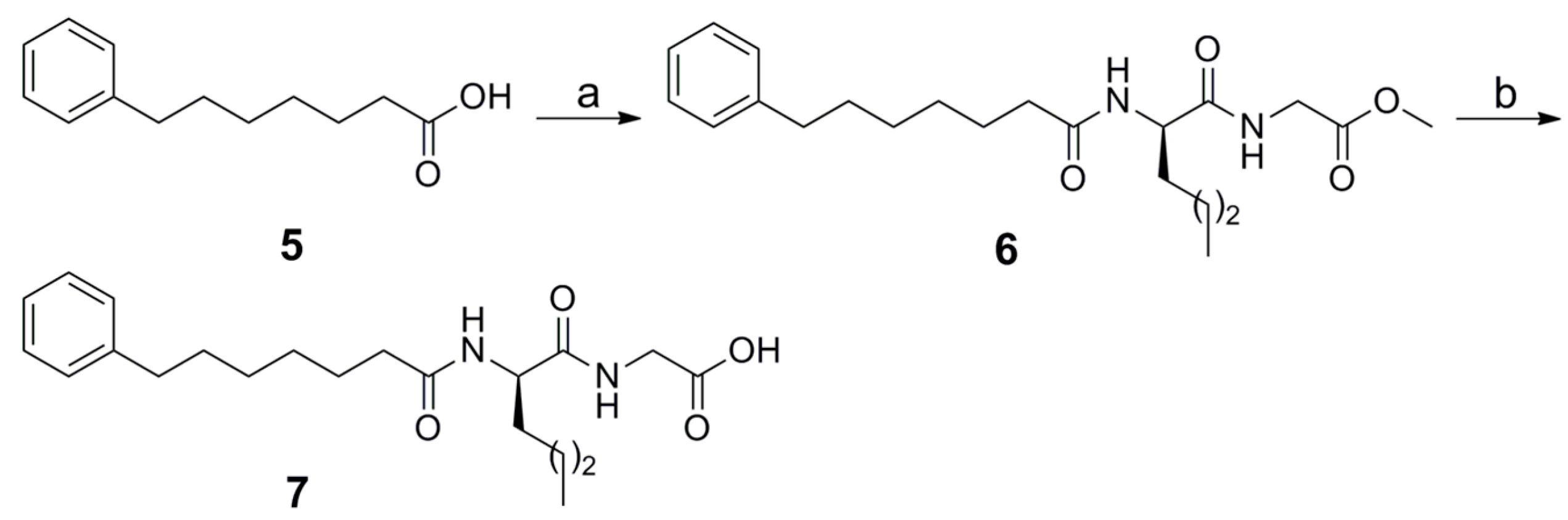
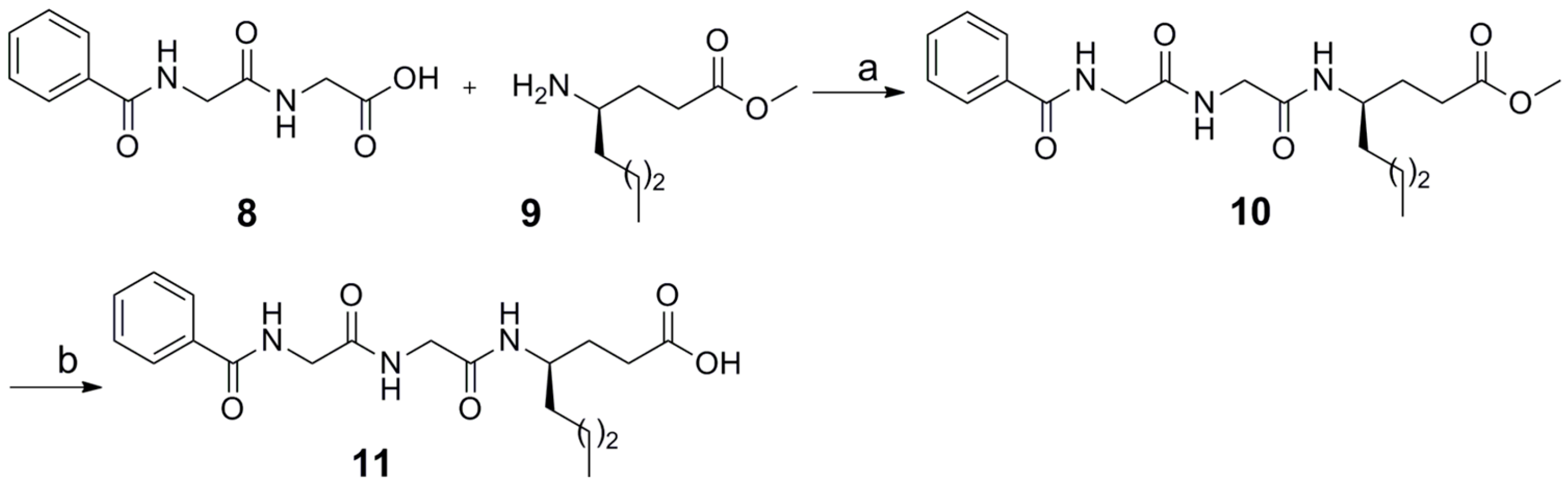
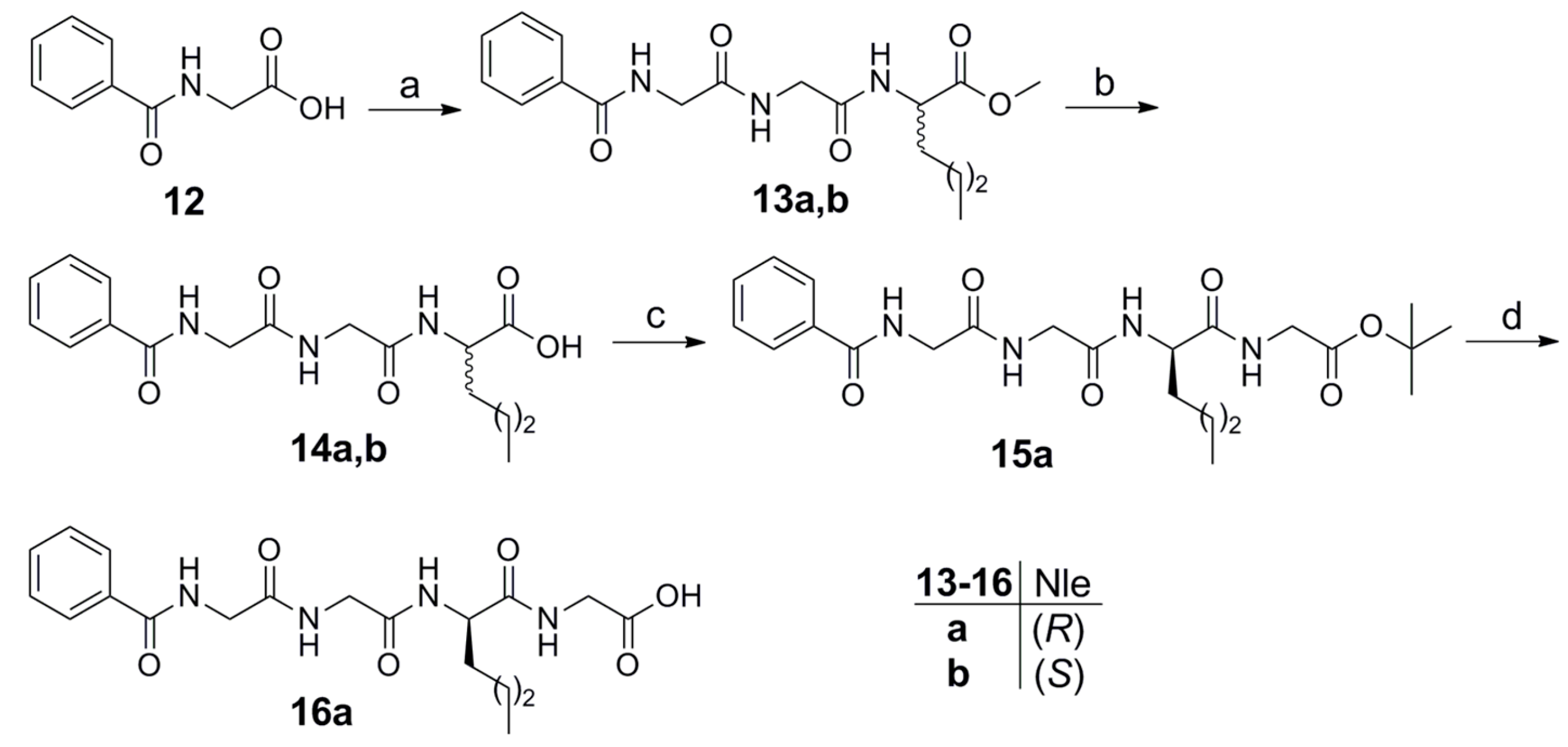
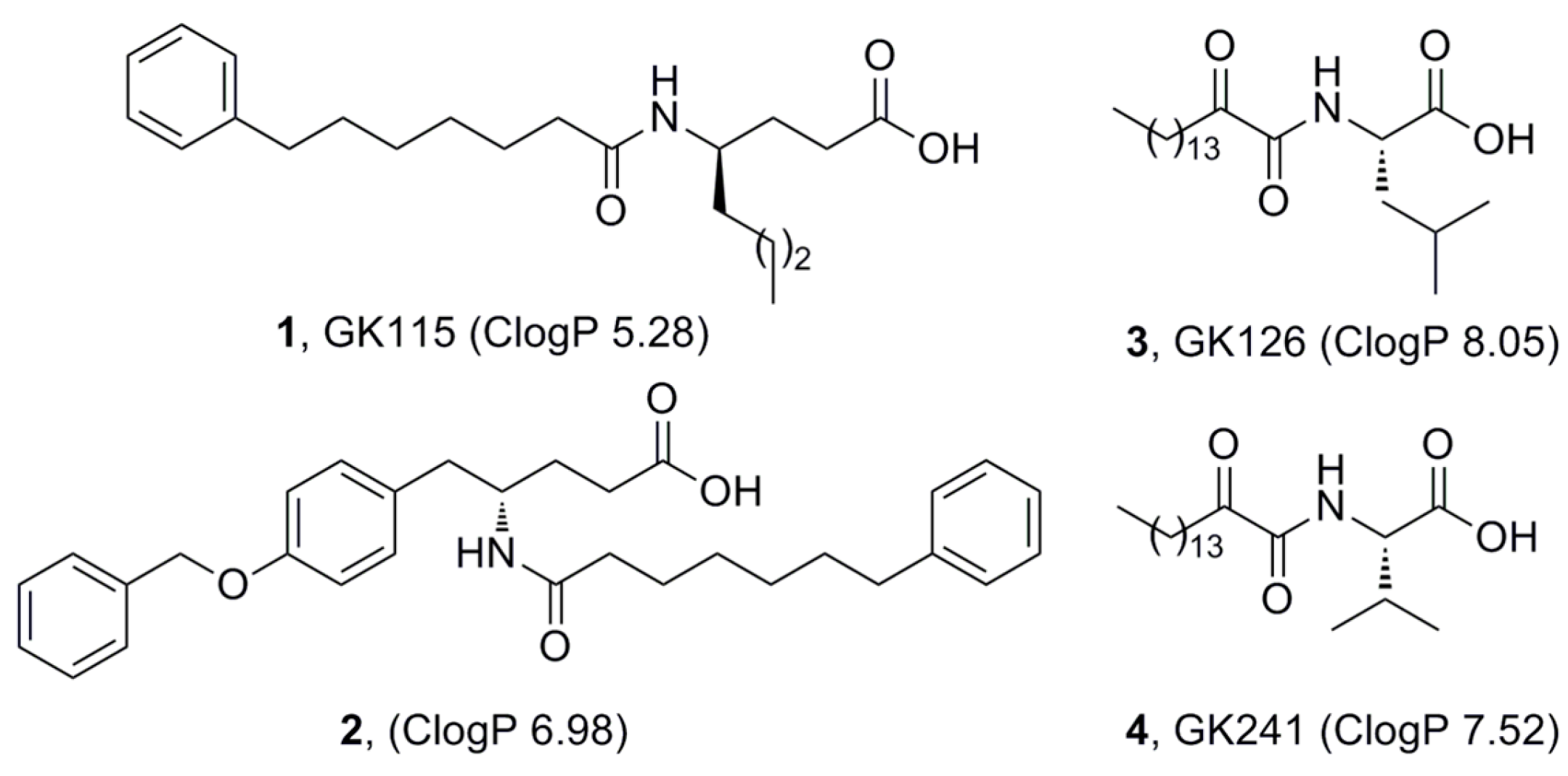
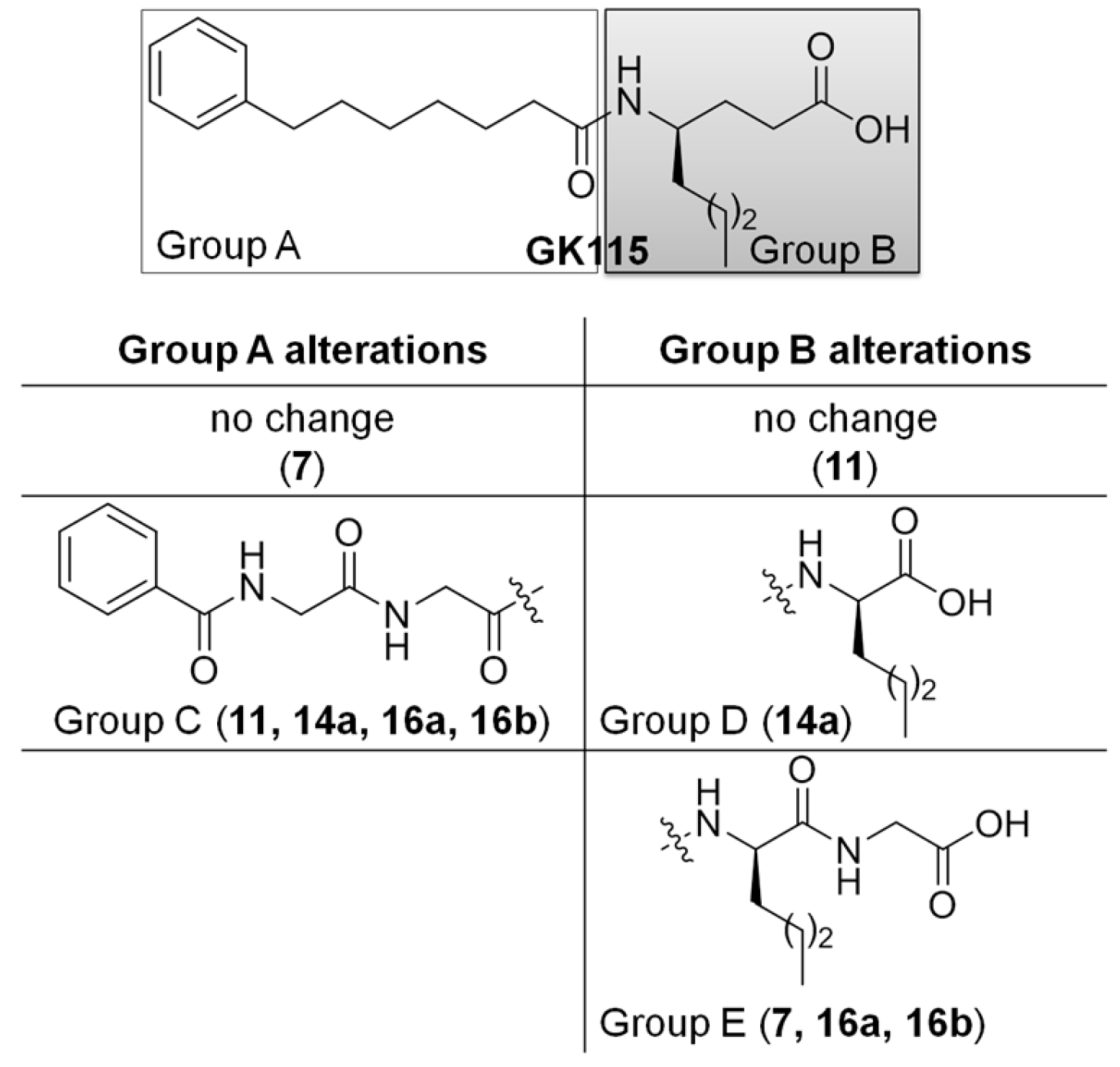
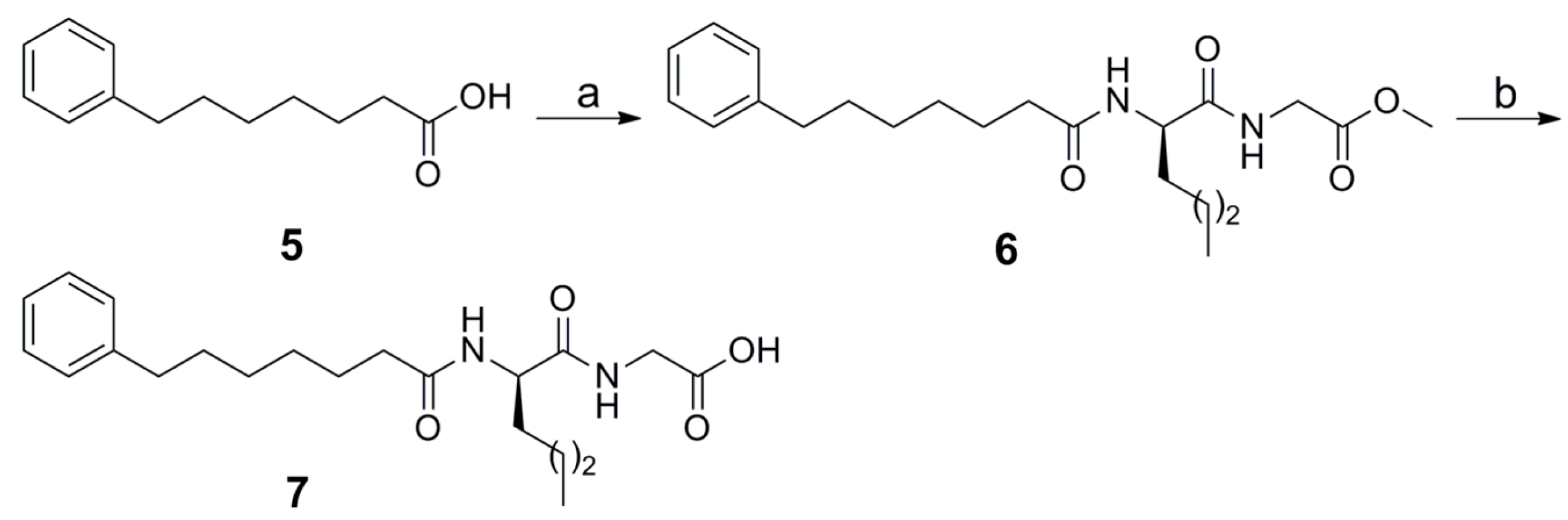
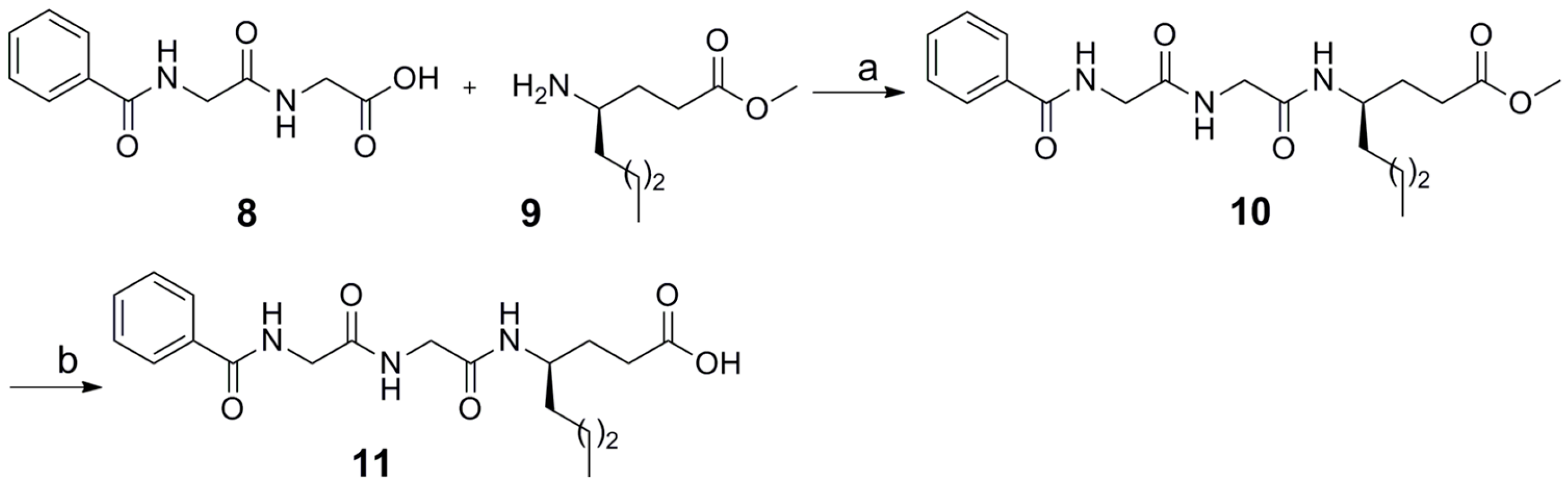
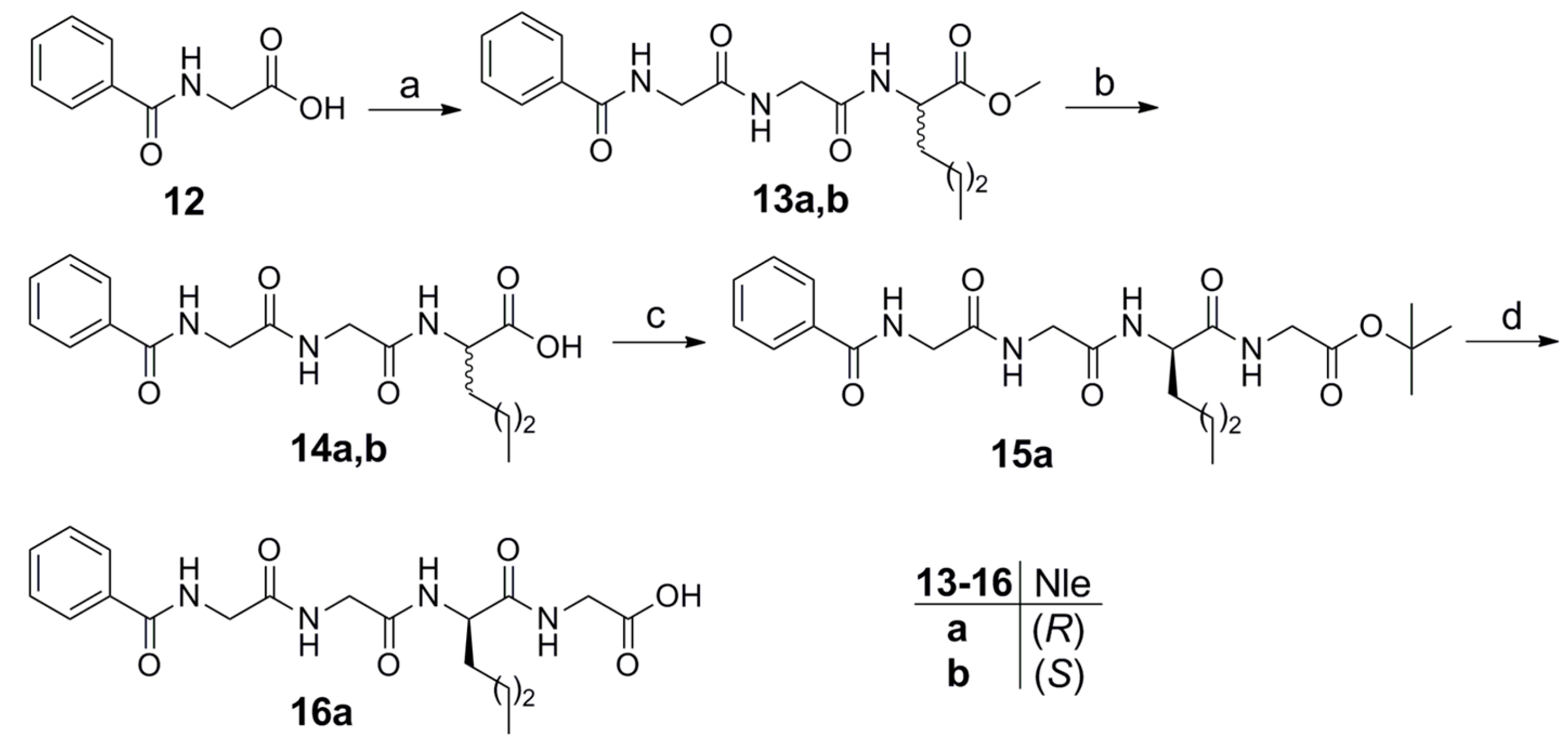
2.1. Synthesis
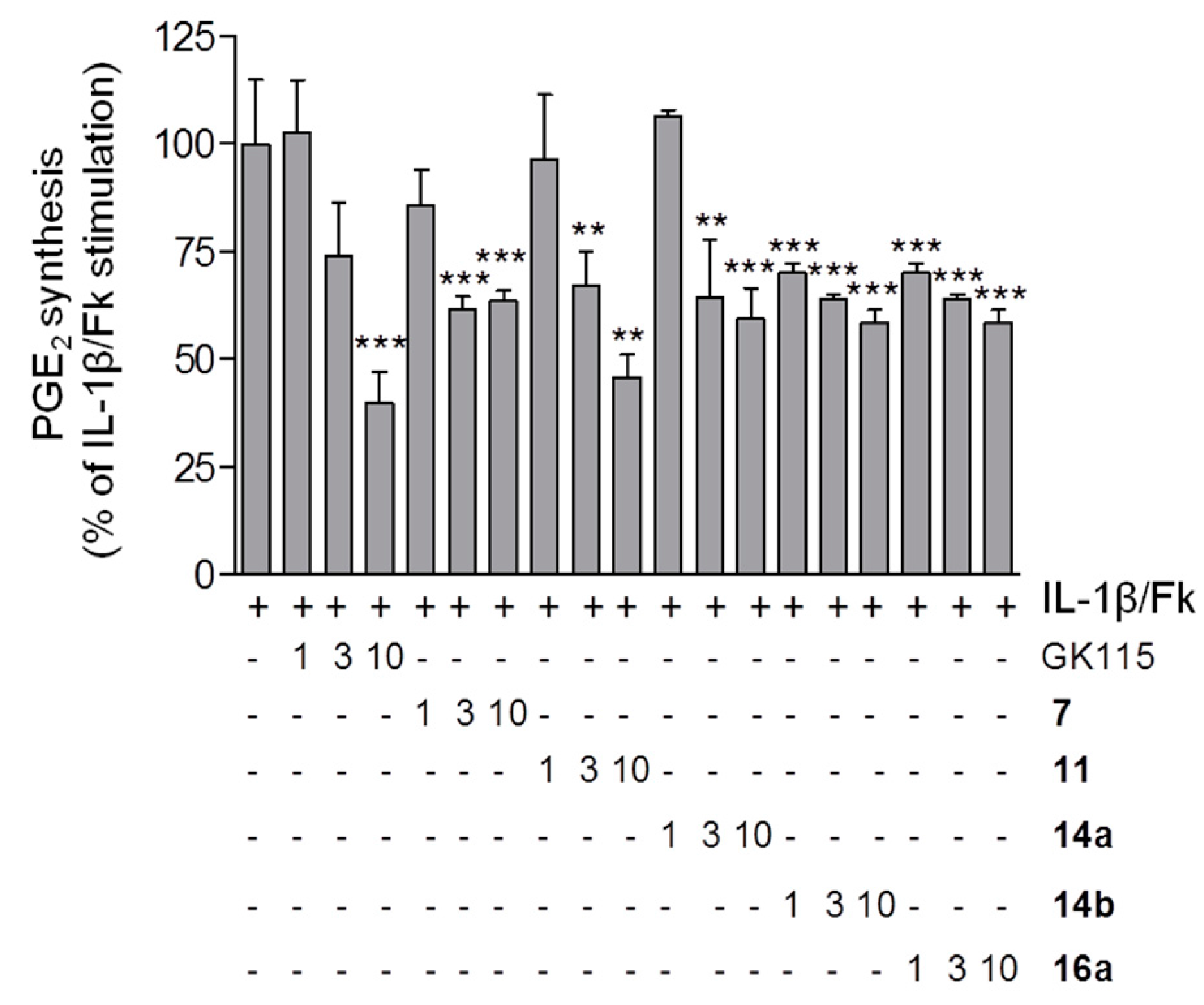
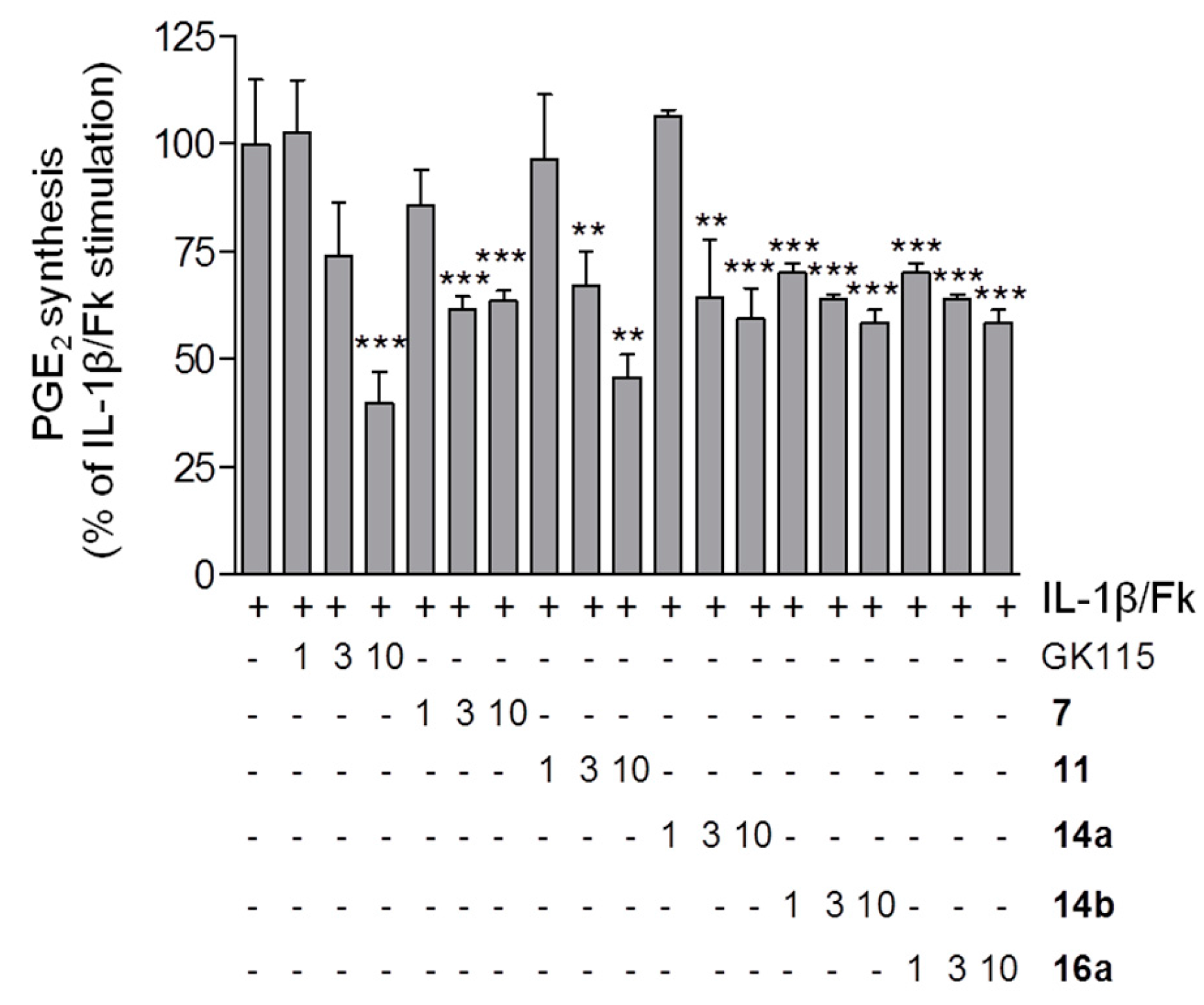
2.2. Suppression of PGE2 Release in Mesangial Cells
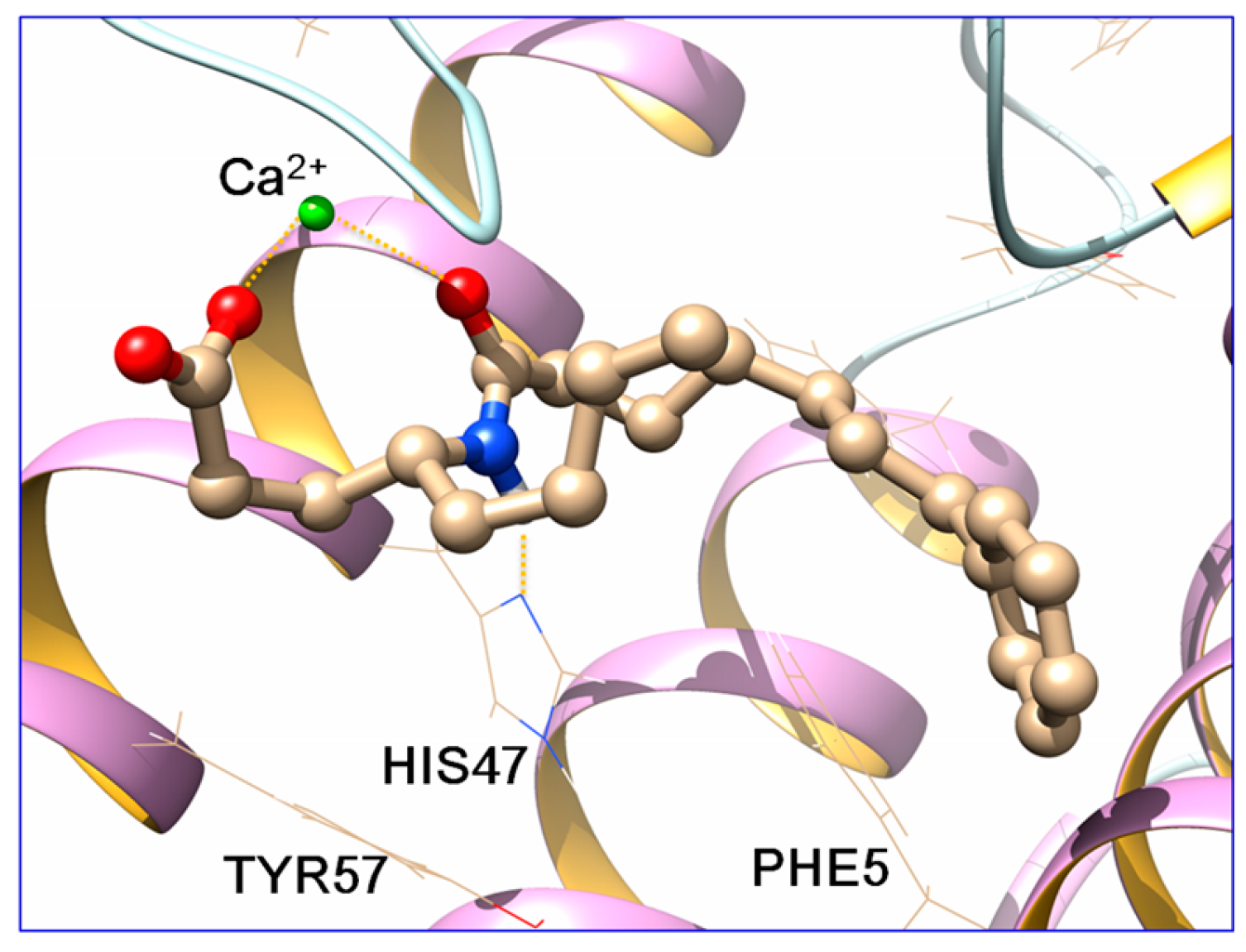
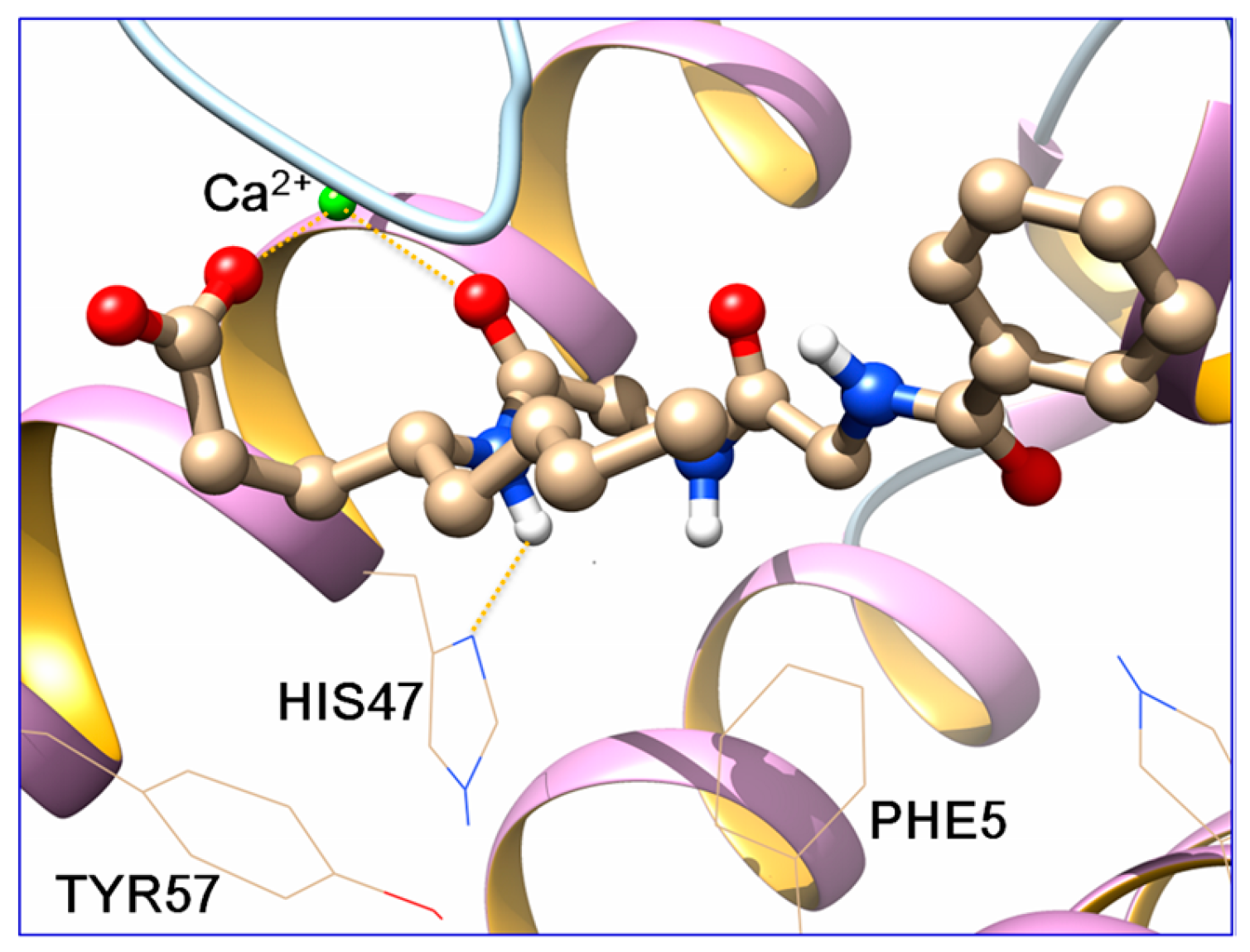
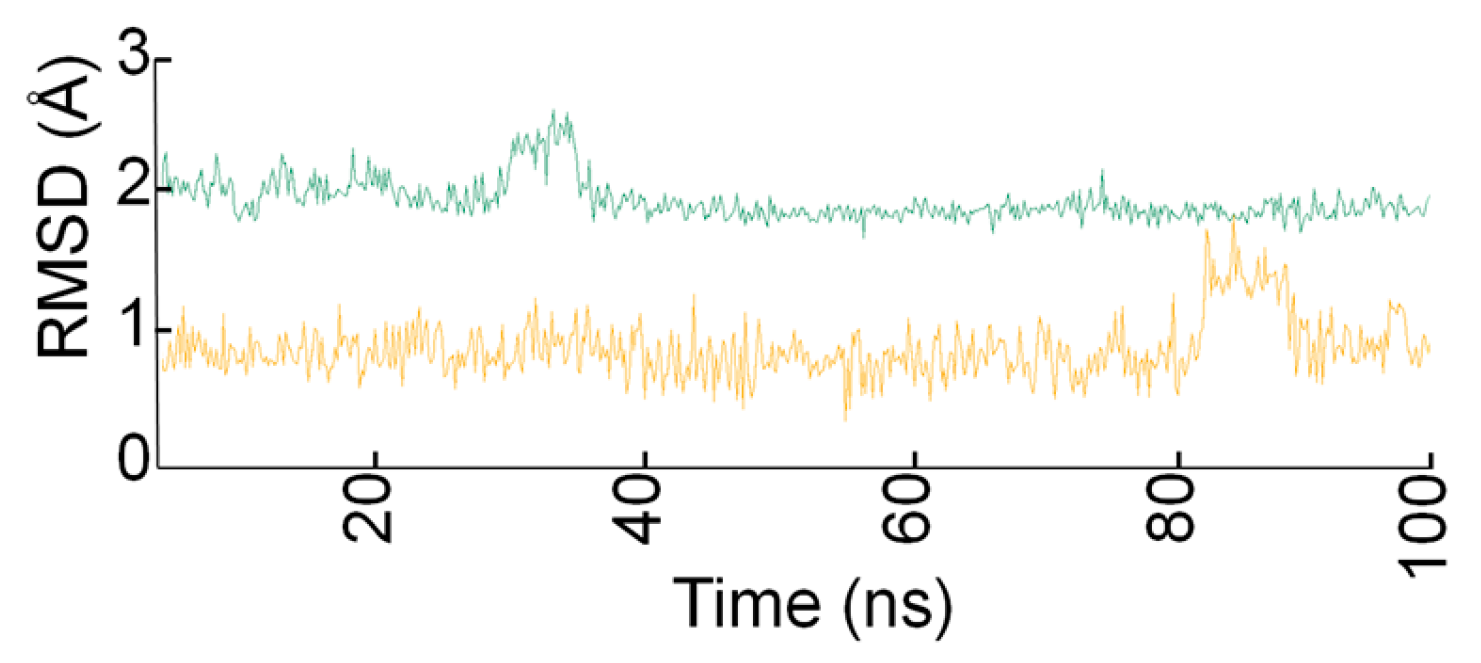
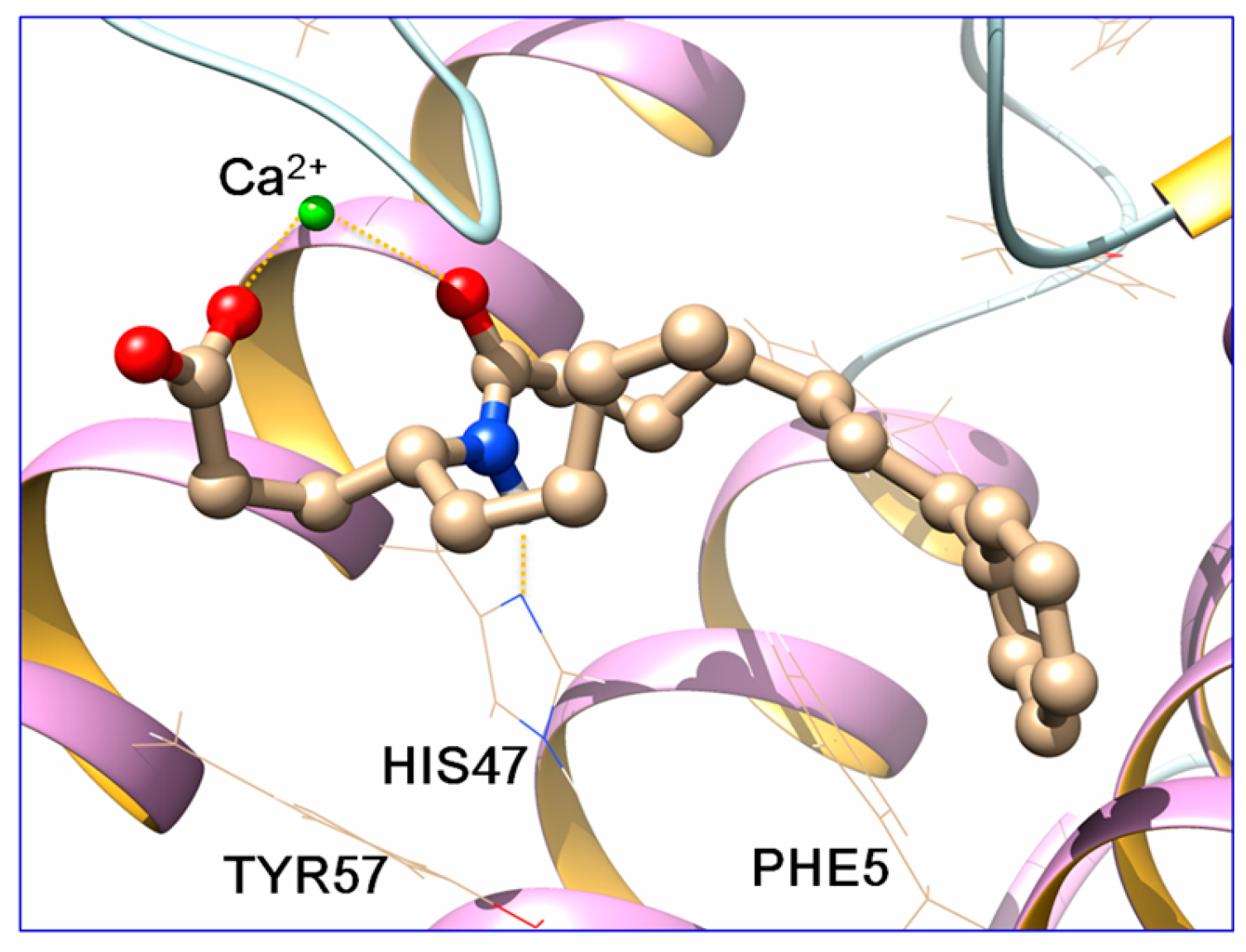
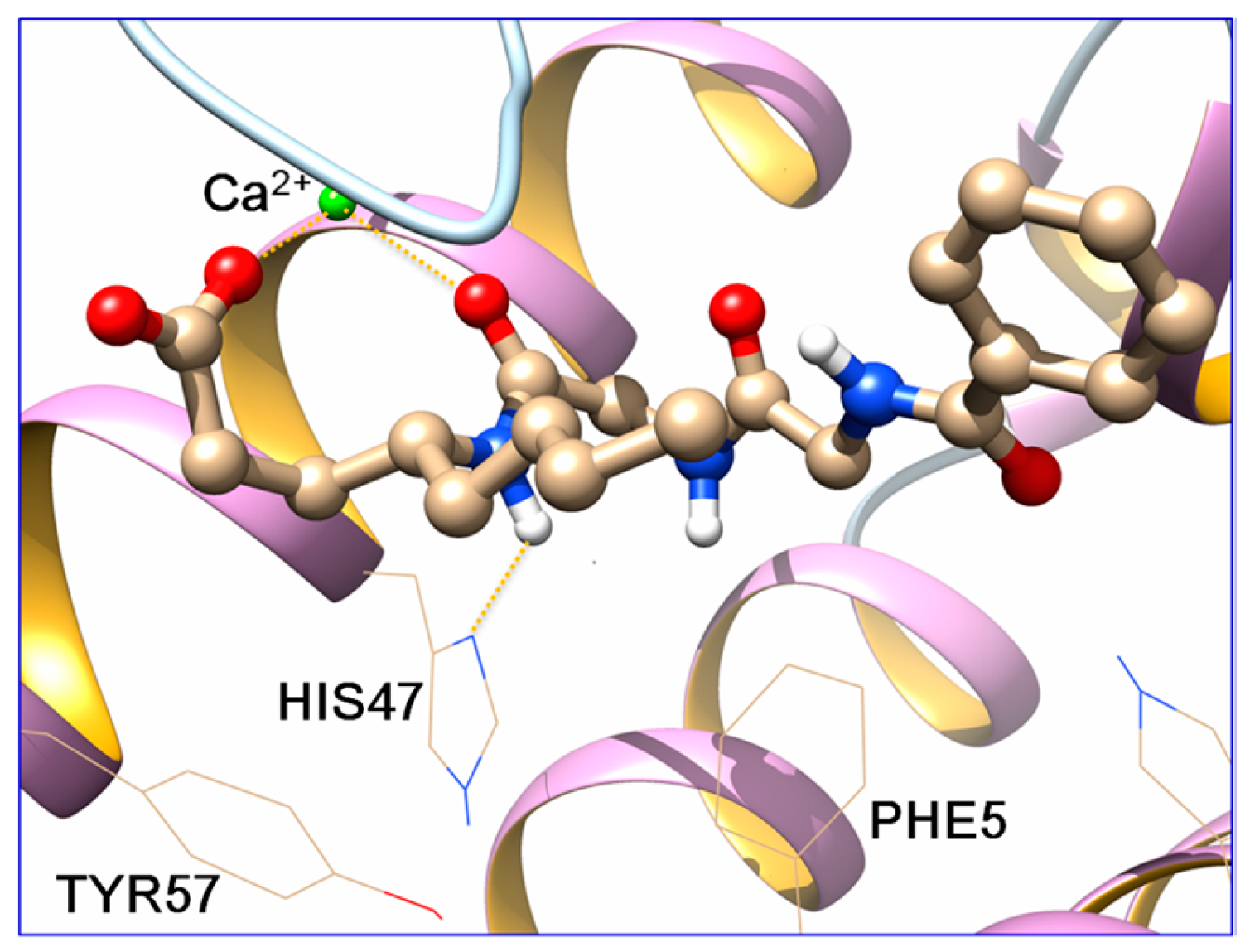
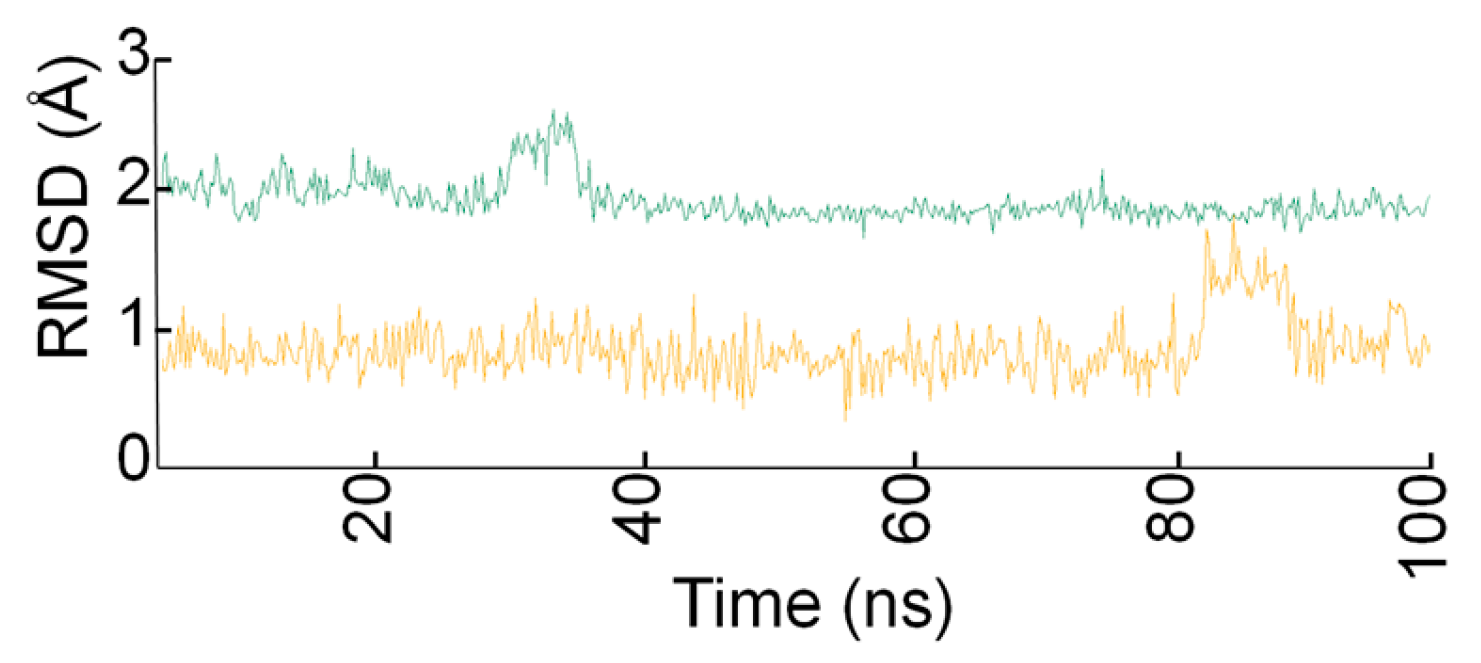
2.3. Docking Studies
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Coupling Method
4.1.2. Saponification of Methyl Esters
4.1.3. Cleavage of tert-Butyl Protecting Group
4.2. Biology
4.2.1. Cell Culture
4.2.2. Quantification of Prostaglandin E2
4.2.3. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sterzel, R.B.; Schulze-Lohoff, E.; Marx, M. Cytokines and mesangial cells. Kidney Int. Suppl. 1993, 39, S26–S31. [Google Scholar] [PubMed]
- Gomez-Guerrero, C.; Hernandez-Vargas, P.; Lopez-Franco, O.; Ortiz-Munoz, G.; Egido, J. Mesangial cells and glomerular inflammation: From the pathogenesis to novel therapeutic approaches. Curr. Drug Targets Inflamm. Allergy 2005, 4, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Limnios, D.; Nikolaou, A.; Psarra, A.; Kokotos, G. Inhibitors of phospholipase A2 and their therapeutic potential: An update on patents (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.-H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.; Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Kokotos, G. Microsomal prostaglandin E2 synthase-1 inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Gronich, J.; Konieczkowski, M.; Gelb, M.H.; Nemenoff, R.A.; Sedor, J.R. Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J. Clin. Investig. 1994, 93, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Staudt, G.; Kramer, R.M.; Pfeilschifter, J. Cross-talk between secretory phospholipase A2 and cytosolic phospholipase A2 in rat renal mesangial cells. Biochim. Biophys. Acta 1997, 1348, 257–272. [Google Scholar] [CrossRef]
- Van der Helm, H.A.; Aarsman, A.J.; Janssen, M.J.; Neys, F.W.; van den Bosch, H. Regulation of the expression of group IIA and group V secretory phospholipases A2 in rat mesangial cells. Biochim. Biophys. Acta 2000, 12, 215–224. [Google Scholar] [CrossRef]
- Han, W.K.; Sapirstein, A.; Hung, C.C.; Alessandrini, A.; Bonventre, J.V. Cross-talk between cytosolic phospholipase A2α (cPLA2α) and secretory phospholipase A2 (sPLA2) in hydrogen peroxide-induced arachidonic acid release in murine mesangial cells. J. Biol. Chem. 2003, 278, 24153–24163. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Lambeau, G.; Scholz-Pedretti, K.; Gelb, M.H.; Janssen, M.J.W.; Edwards, S.H.; Wilton, D.C.; Pfeilschifter, J.; Kaszkin, M. Potentiation of tumor necrosis factor α-induced secreted phospholipase A2 (sPLA2)-IIA expression in mesangial cells by an autocrine loop involving sPLA2 and peroxisome proliferator-activated receptor α activation. J. Biol. Chem. 2003, 278, 29799–29812. [Google Scholar] [CrossRef] [PubMed]
- Vasilakaki, S.; Barbayianni, E.; Magrioti, V.; Pastukhov, O.; Constantinou-Kokotou, V.; Huwiler, A.; Kokotos, G. Inhibitors of secreted phospholipase A2 suppress the release of PGE2 in renal mesangial cells. Bioorg. Med. Chem. 2016, 24, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, G.; Barbayianni, E.; Magrioti, V.; Cotton, N.; Stephens, D.; Constantinou-Kokotou, V.; Dennis, E.A.; Kokotos, G. Synthesis of 2-oxoamides based on sulfonamide analogues of γ-amino acids and their activity on phospholipase A2. Bioorg. Med. Chem. 2008, 16, 10257–10269. [Google Scholar] [CrossRef] [PubMed]
- Hansford, K.A.; Reid, R.C.; Clark, C.I.; Tyndall, J.D.A.; Whitehouse, M.W.; Guthrie, T.; McGeary, R.P.; Schafer, K.; Martin, J.L.; Fairlie, D.P. D-Tyrosine as a chiral precusor to potent inhibitors of human nonpancreatic secretory phospholipase A2 (IIa) with antiinflammatory activity. ChemBioChem 2003, 4, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Magrioti, V.; Barbayianni, E.; Cermak, N.; Oslund, R.C.; Mavromoustakos, T.M.; Gelb, M.H.; Kokotos, G. Inhibition of secreted phospholipases A2 by 2-oxoamides based on α-amino acids: Synthesis, in vitro evaluation and molecular docking calculations. Bioorg. Med. Chem. 2011, 19, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Vasilakaki, S.; Barbayianni, E.; Leonis, G.; Papadopoulos, M.G.; Mavromoustakos, T.; Gelb, M.H.; Kokotos, G. Development of a potent 2-oxoamide inhibitor of secreted phospholipase A2 guided by molecular docking calculations and molecular dynamics simulations. Bioorg. Med. Chem. 2016, 24, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Feuerherm, A.J.; Sakem, B.; Pastukhov, O.; Filipenko, I.; Nguyen, T.; Johansen, B. The ω3-polyunsaturated fatty acid derivatives AVX001 and AVX002 directly inhibit cytosolic phospholipase A2 and suppress PGE2 formation in mesangial cells. Br. J. Pharmacol. 2012, 167, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Frackenpohl, J.; Arvidsson, P.I.; Schreiber, J.V.; Seebach, D. The outstanding biological stability of β- and γ-peptides toward proteolytic enzymes: An in vitro investigation with fifteen peptidases. ChemBioChem 2001, 2, 445–455. [Google Scholar] [CrossRef]
- SYBYL Molecular Modeling Software Packages, version 8.0; Tripos: St. Louis, MO, USA, 2007.
- Schrödinger Suite 2009 Prime, version 2.1; Schrödinger, LLC: New York, NY, USA, 2009.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; van Rossum, G.; Wartmann, M.; Pfeilschifter, J. Stimulation by extracellular ATP and UTP of the stress-activated protein kinase cascade in rat renal mesangial cells. Br. J. Pharmacol. 1997, 120, 807–812. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilakaki, S.; Pastukhov, O.; Mavromoustakos, T.; Huwiler, A.; Kokotos, G. Small Peptides Able to Suppress Prostaglandin E2 Generation in Renal Mesangial Cells. Molecules 2018, 23, 158. https://doi.org/10.3390/molecules23010158
Vasilakaki S, Pastukhov O, Mavromoustakos T, Huwiler A, Kokotos G. Small Peptides Able to Suppress Prostaglandin E2 Generation in Renal Mesangial Cells. Molecules. 2018; 23(1):158. https://doi.org/10.3390/molecules23010158
Chicago/Turabian StyleVasilakaki, Sofia, Oleksandr Pastukhov, Thomas Mavromoustakos, Andrea Huwiler, and George Kokotos. 2018. "Small Peptides Able to Suppress Prostaglandin E2 Generation in Renal Mesangial Cells" Molecules 23, no. 1: 158. https://doi.org/10.3390/molecules23010158
APA StyleVasilakaki, S., Pastukhov, O., Mavromoustakos, T., Huwiler, A., & Kokotos, G. (2018). Small Peptides Able to Suppress Prostaglandin E2 Generation in Renal Mesangial Cells. Molecules, 23(1), 158. https://doi.org/10.3390/molecules23010158