Methylisoindigo and Its Bromo-Derivatives Are Selective Tyrosine Kinase Inhibitors, Repressing Cellular Stat3 Activity, and Target CD133+ Cancer Stem Cells in PDAC


Abstract
:
1. Introduction
2. Results and Discussion
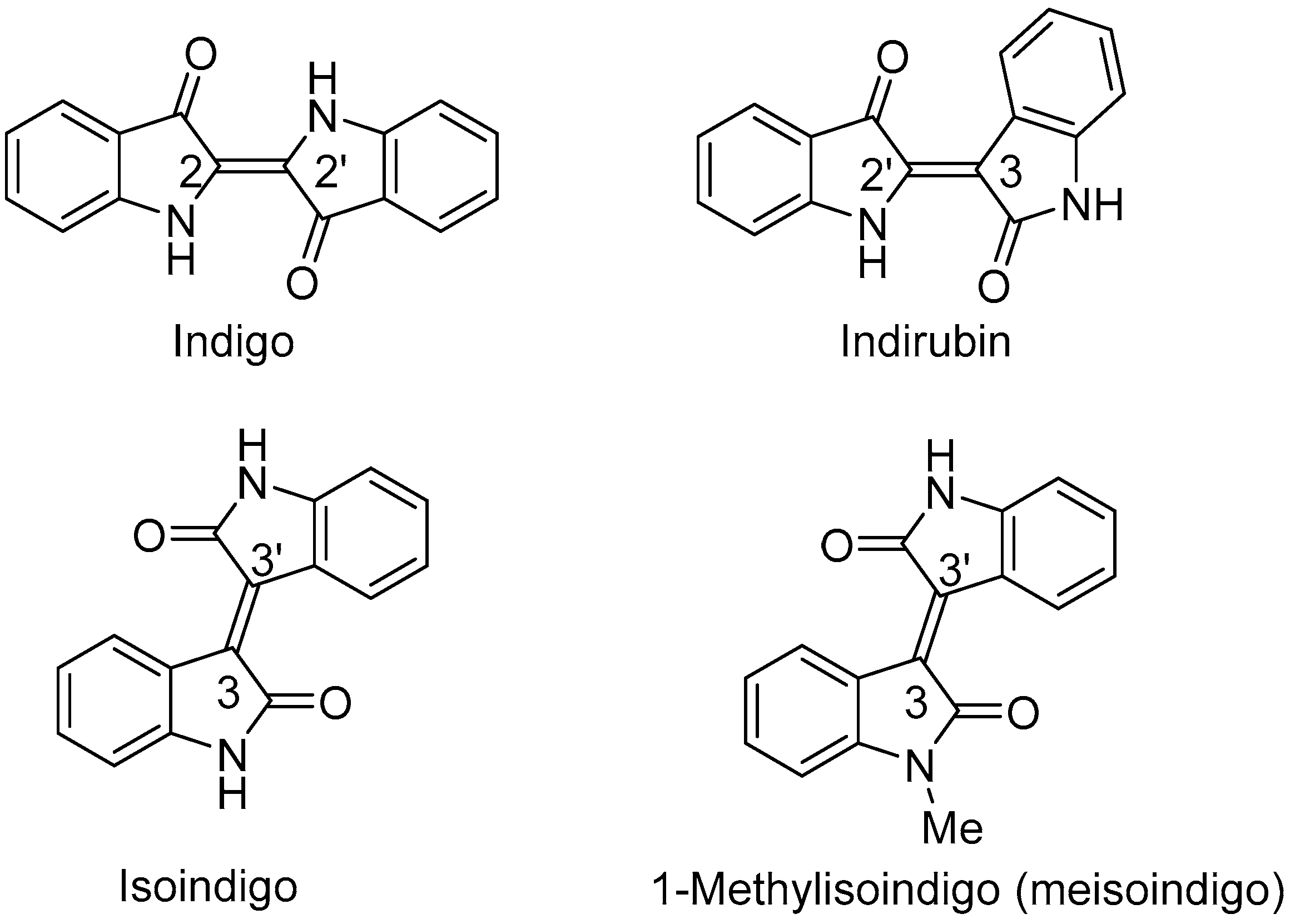
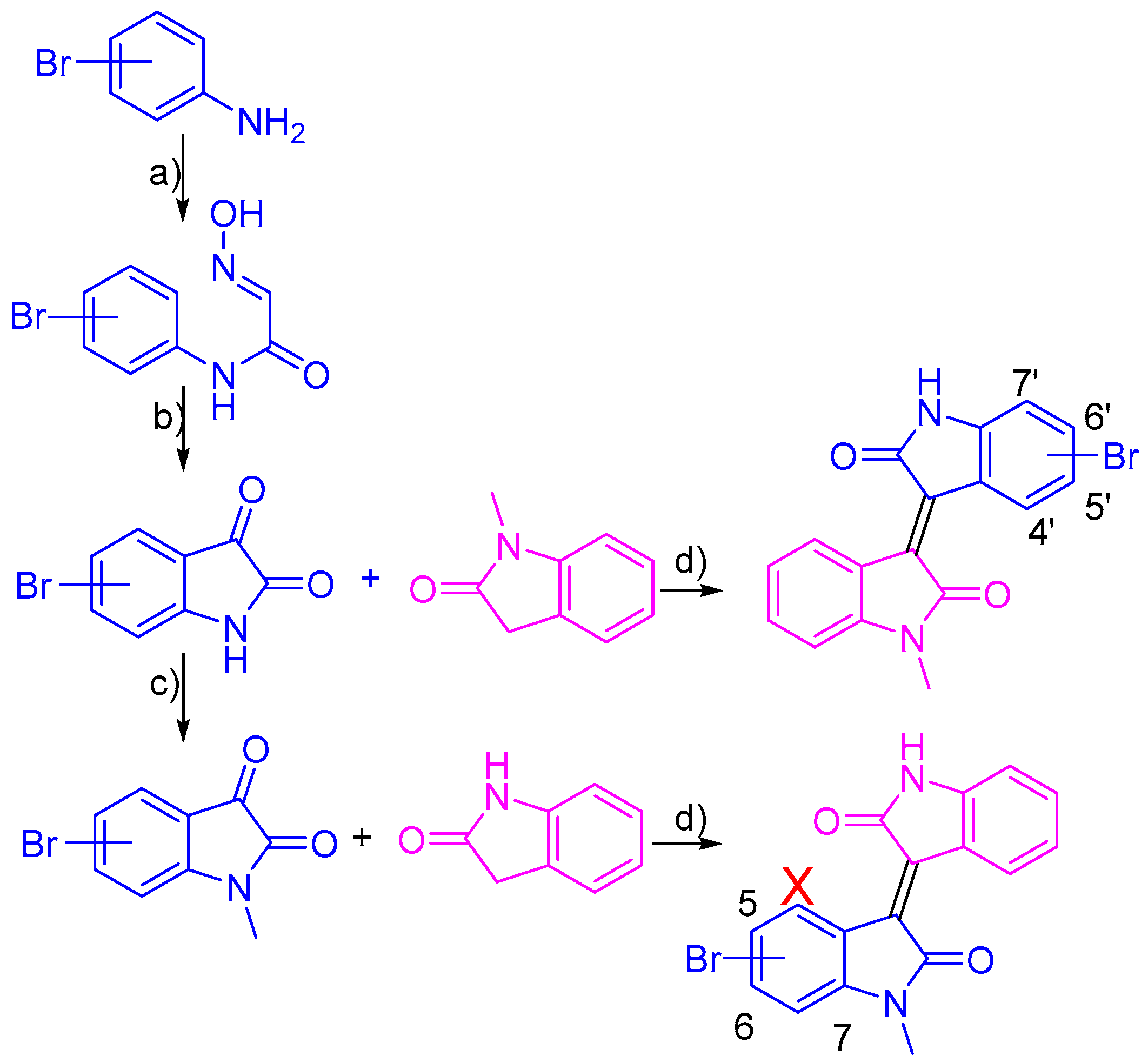
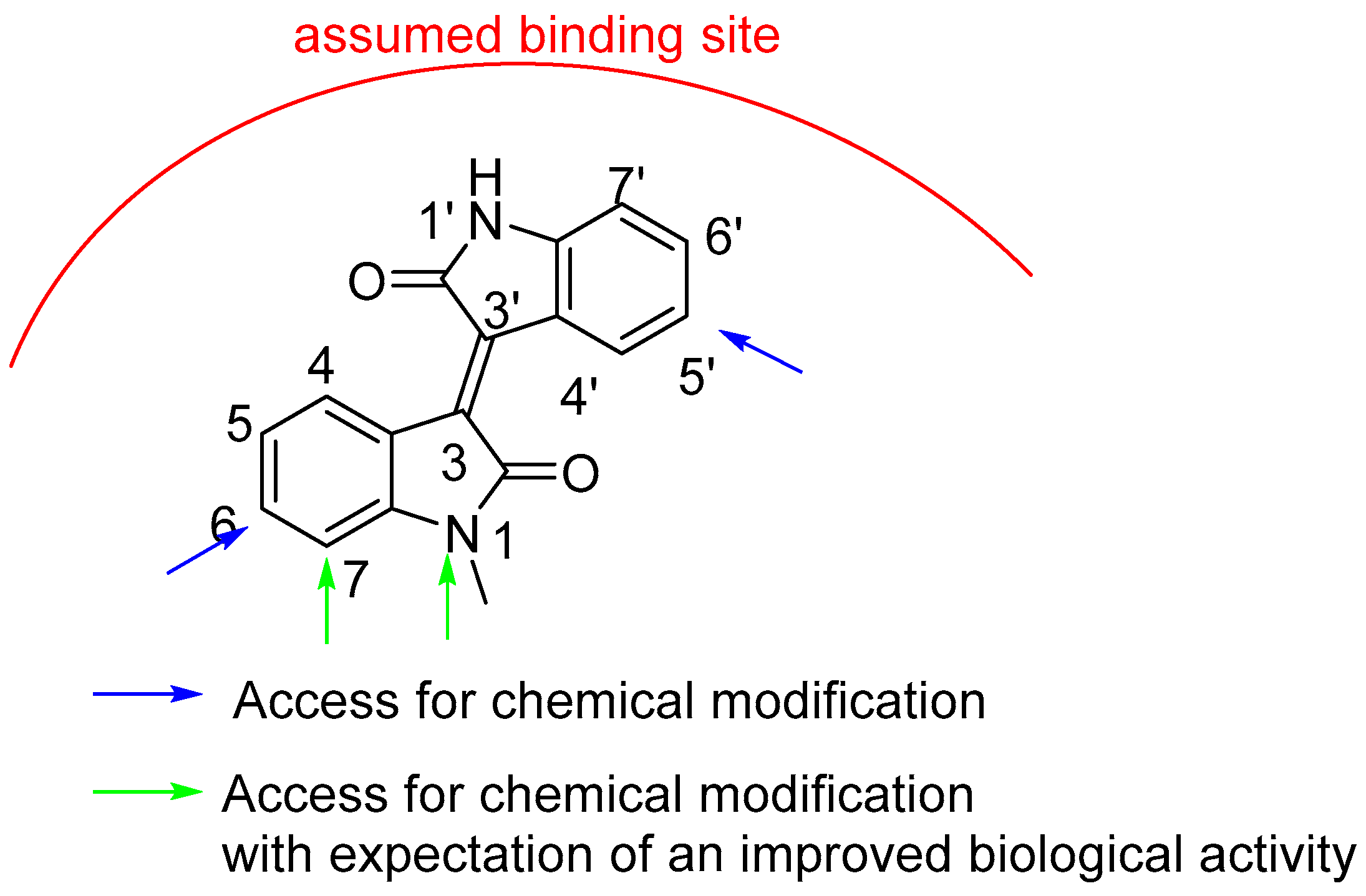
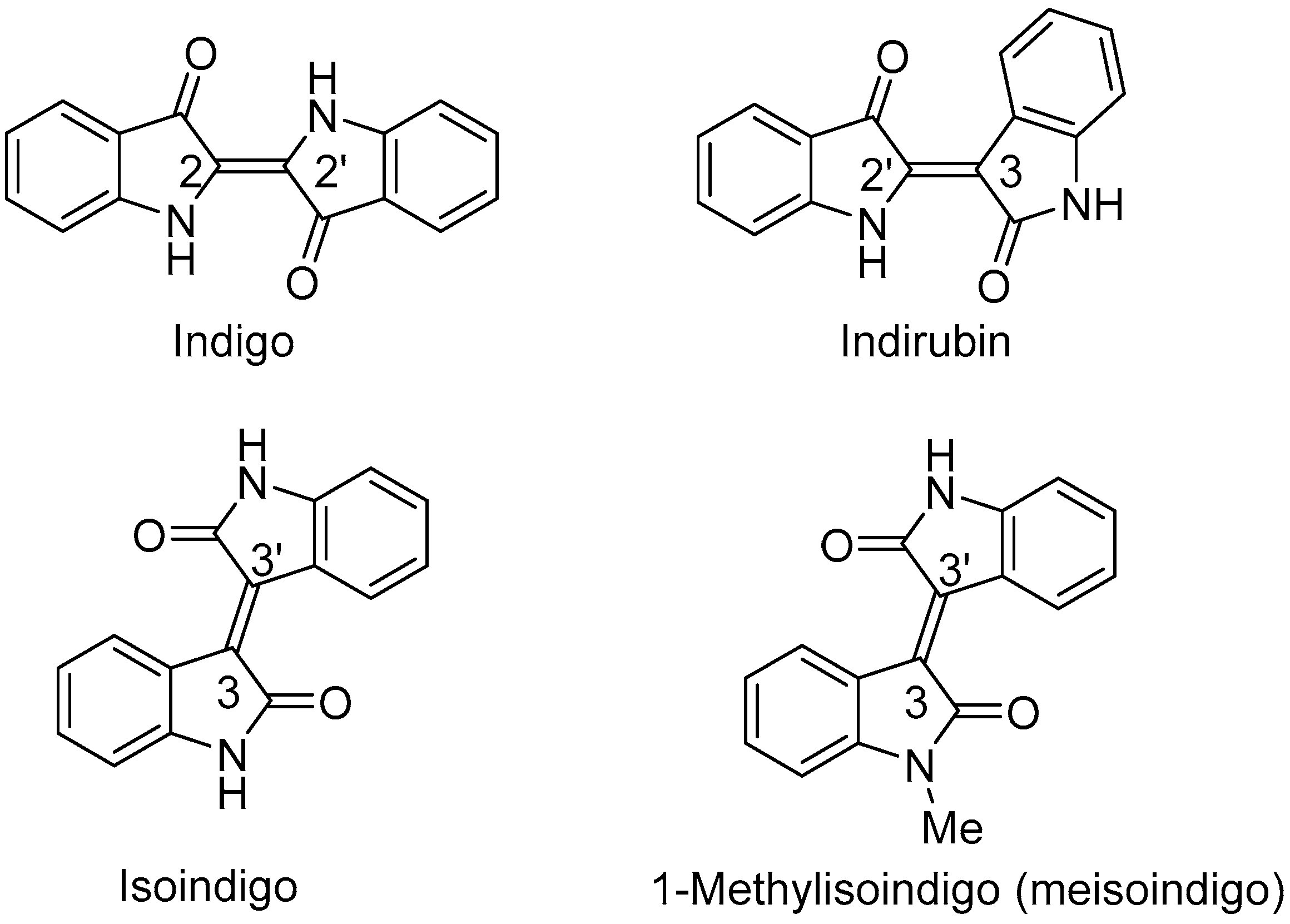
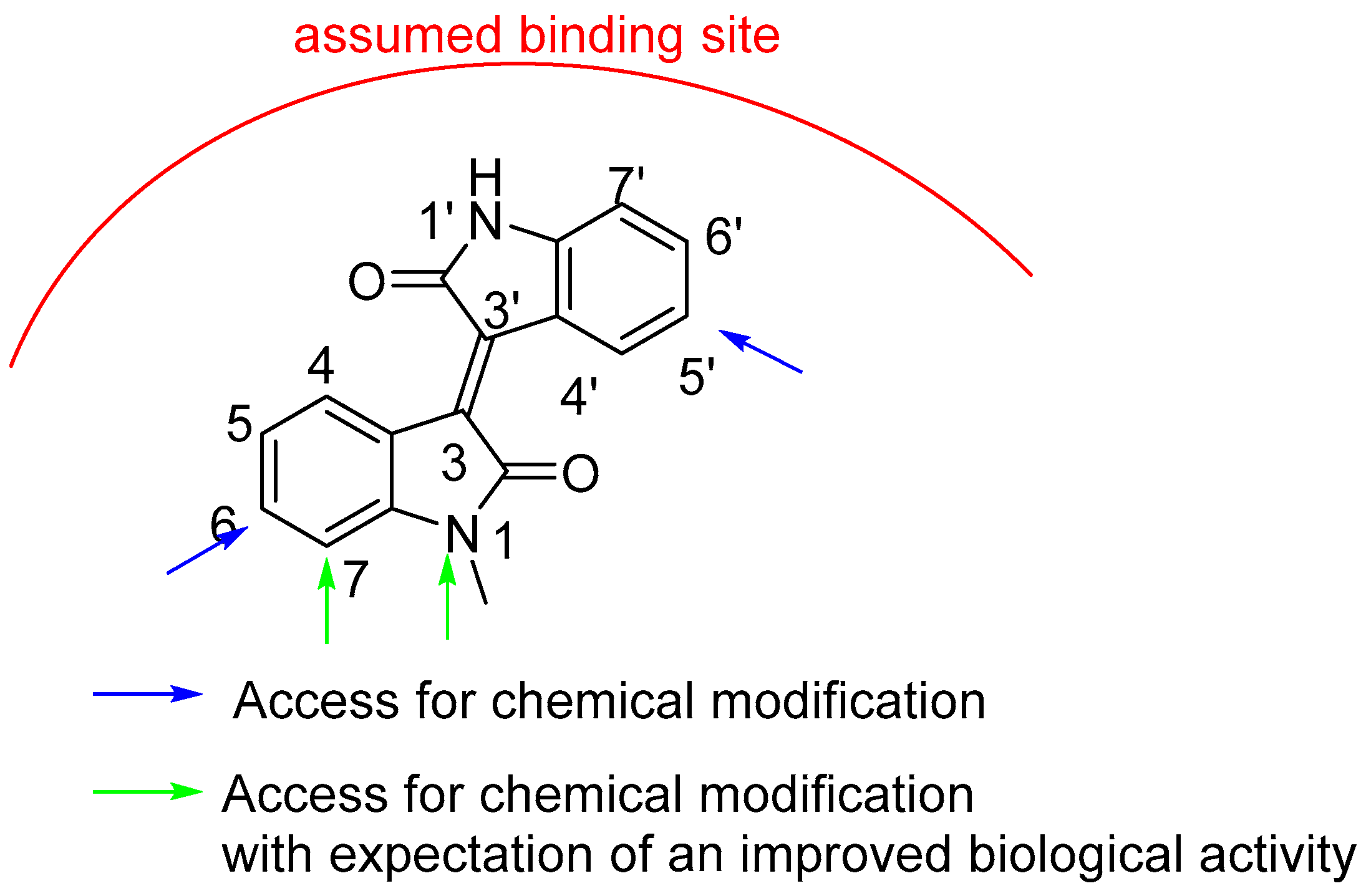
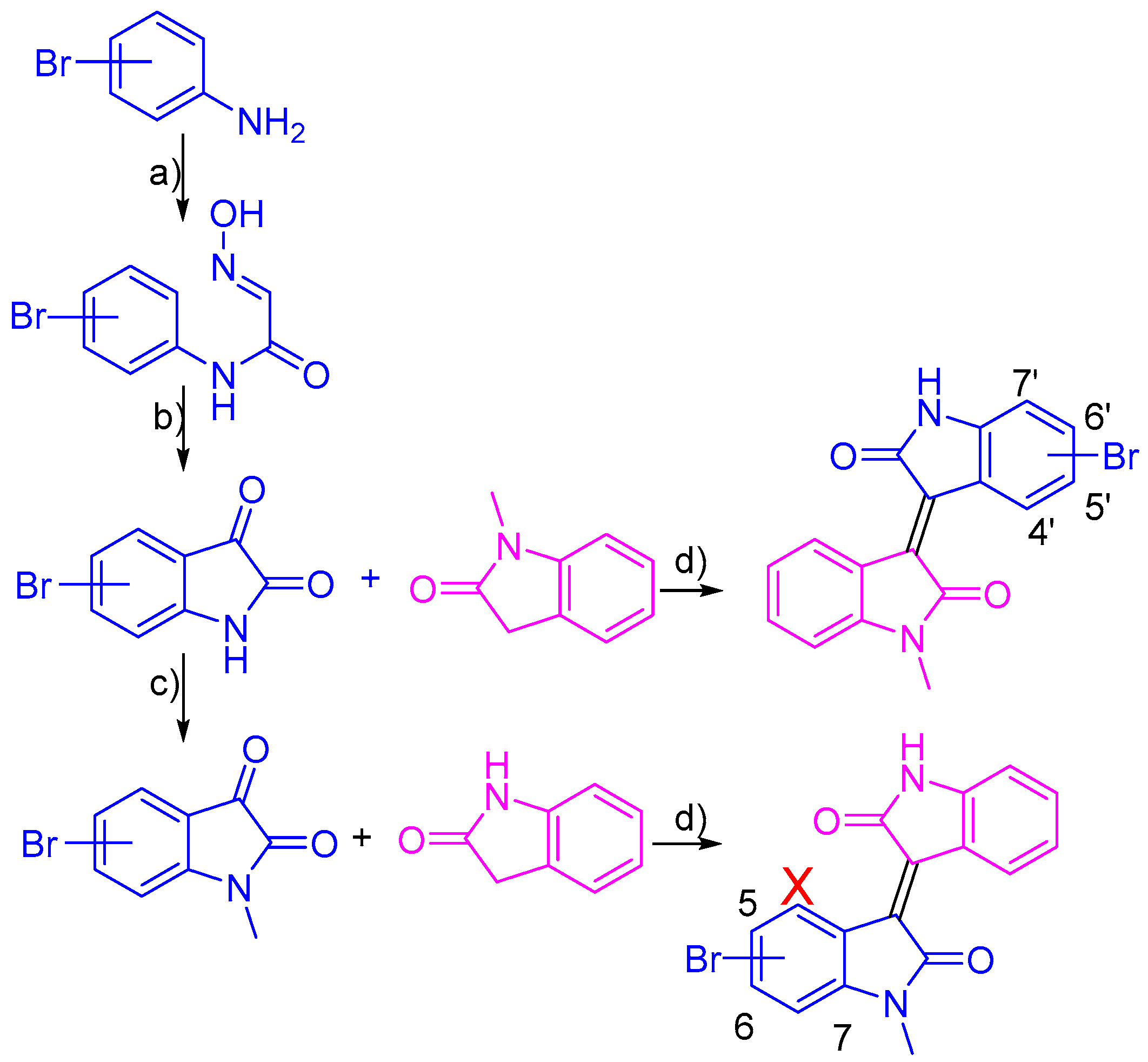
2.1. Chemistry
2.2. Biology
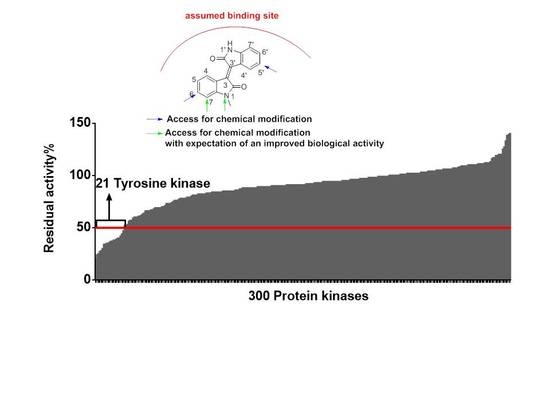
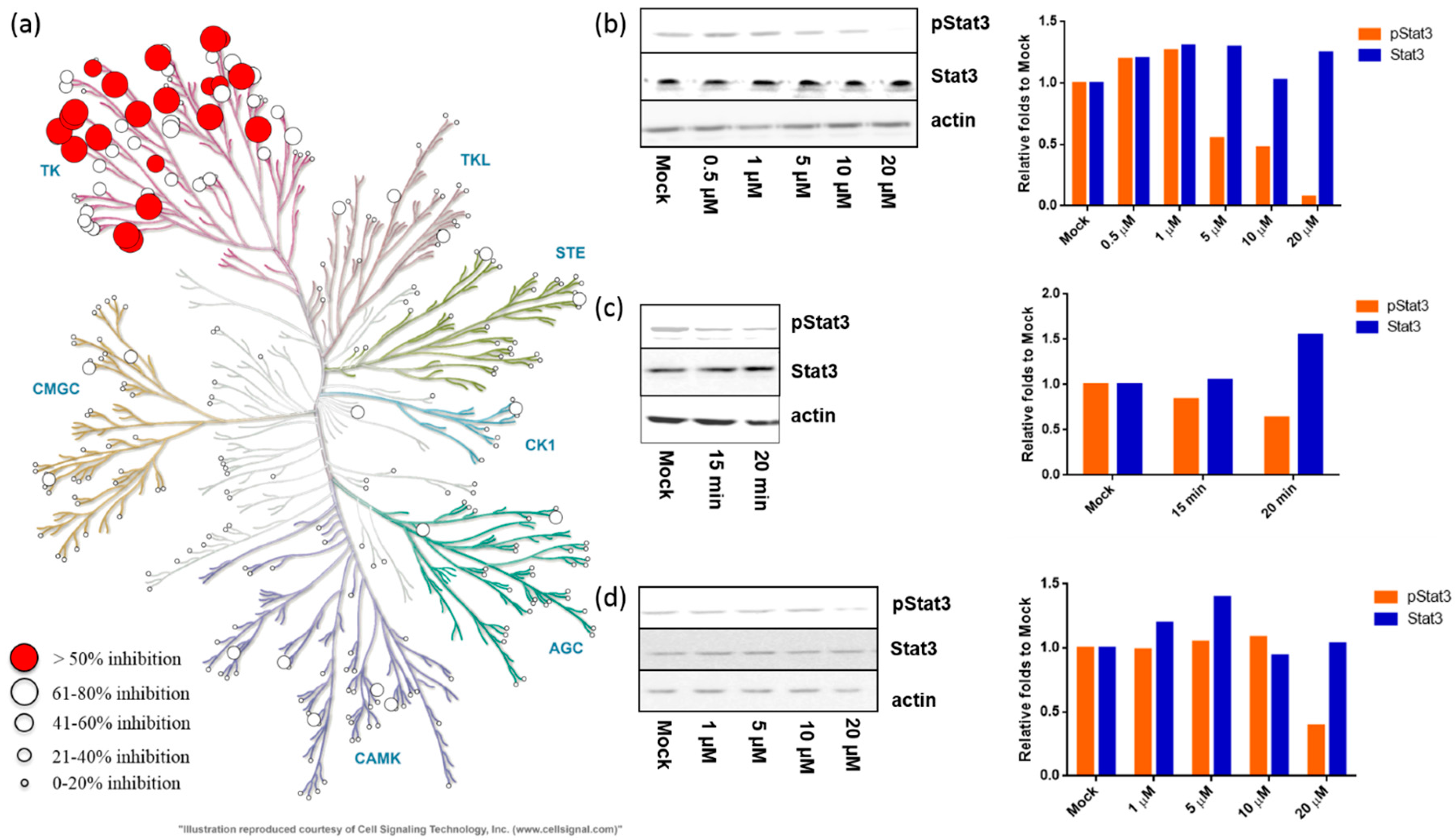
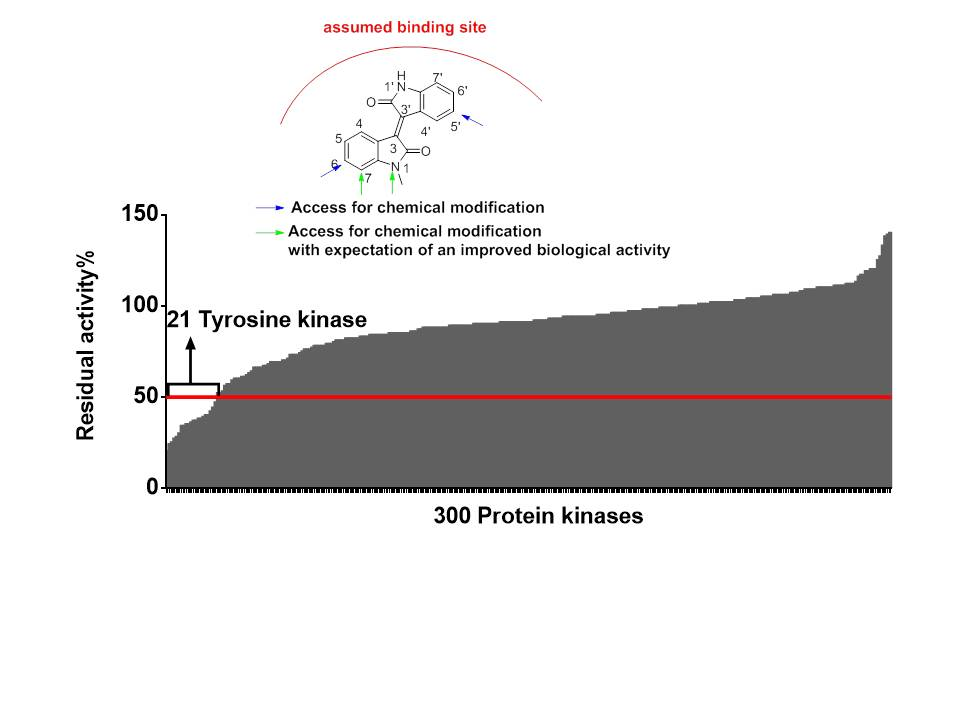
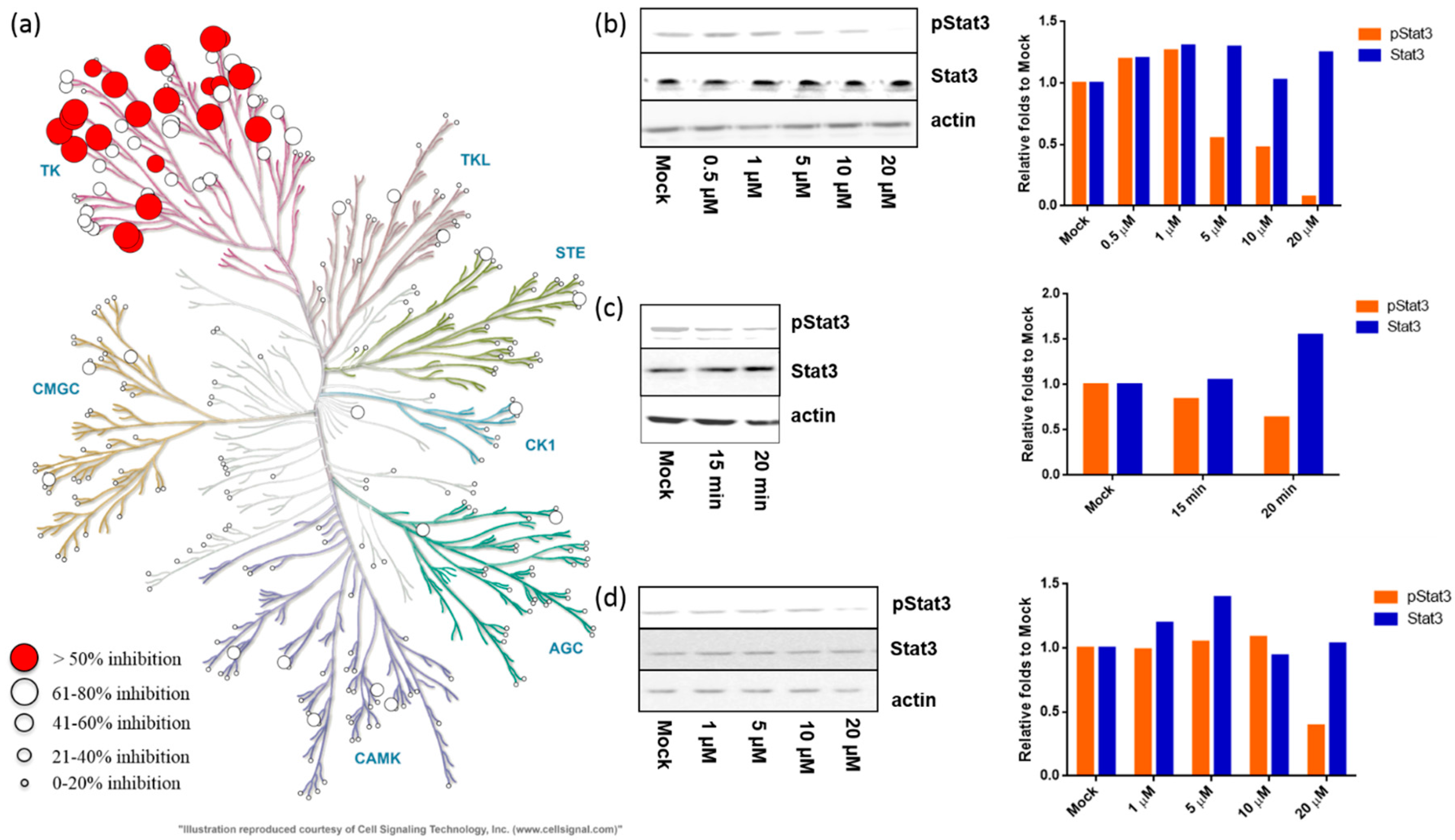
2.2.1. Meisoindigo Is a Selective Stat3-Related Tyrosine Kinase Inhibitor
2.2.2. Meisoindigo Inhibits Cellular Stat3 Activation in Time and Concentration Dependent Manner
2.2.3. Anti-Proliferative Effect of Novel Bromo-Meisoindigos
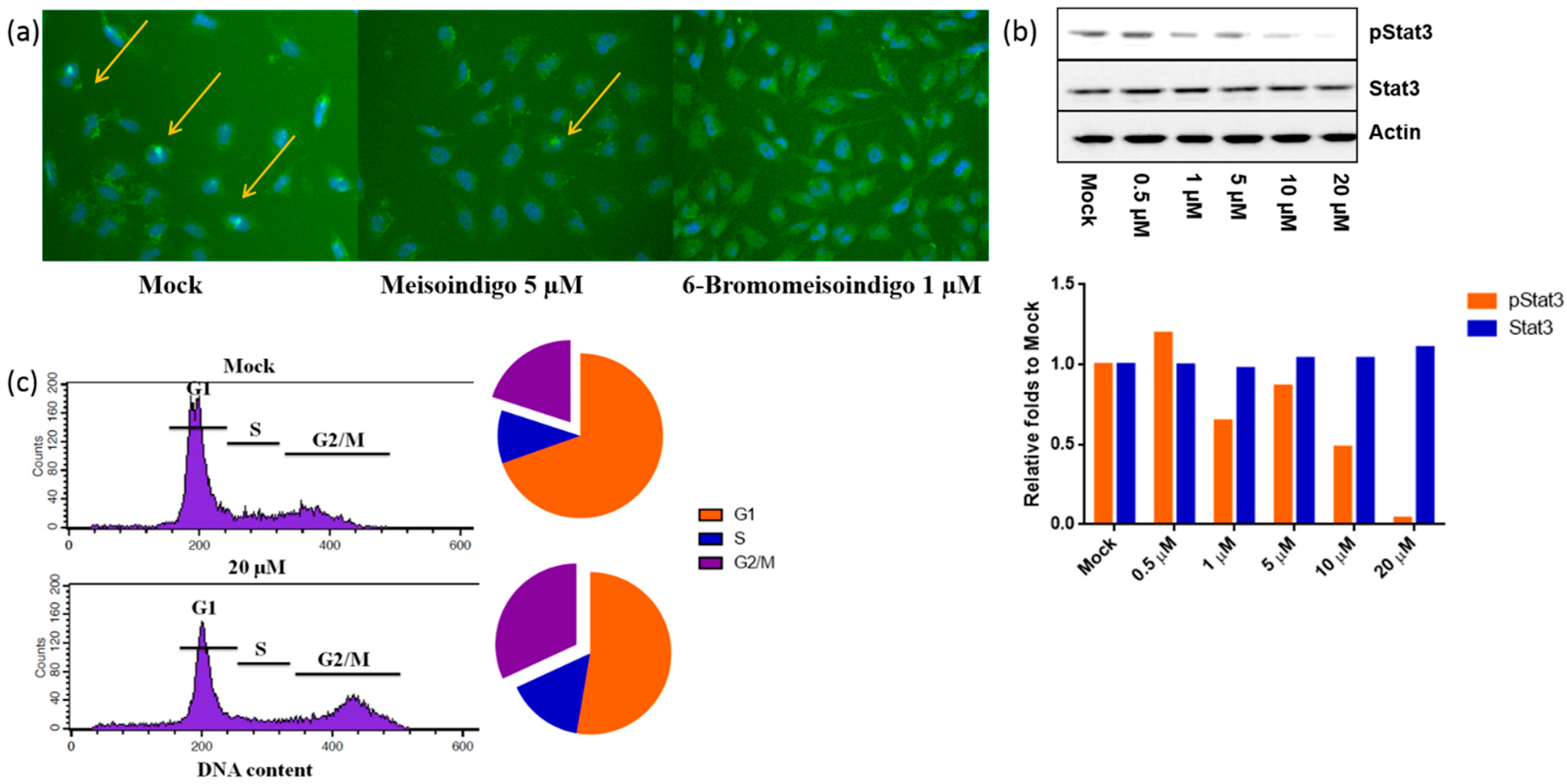
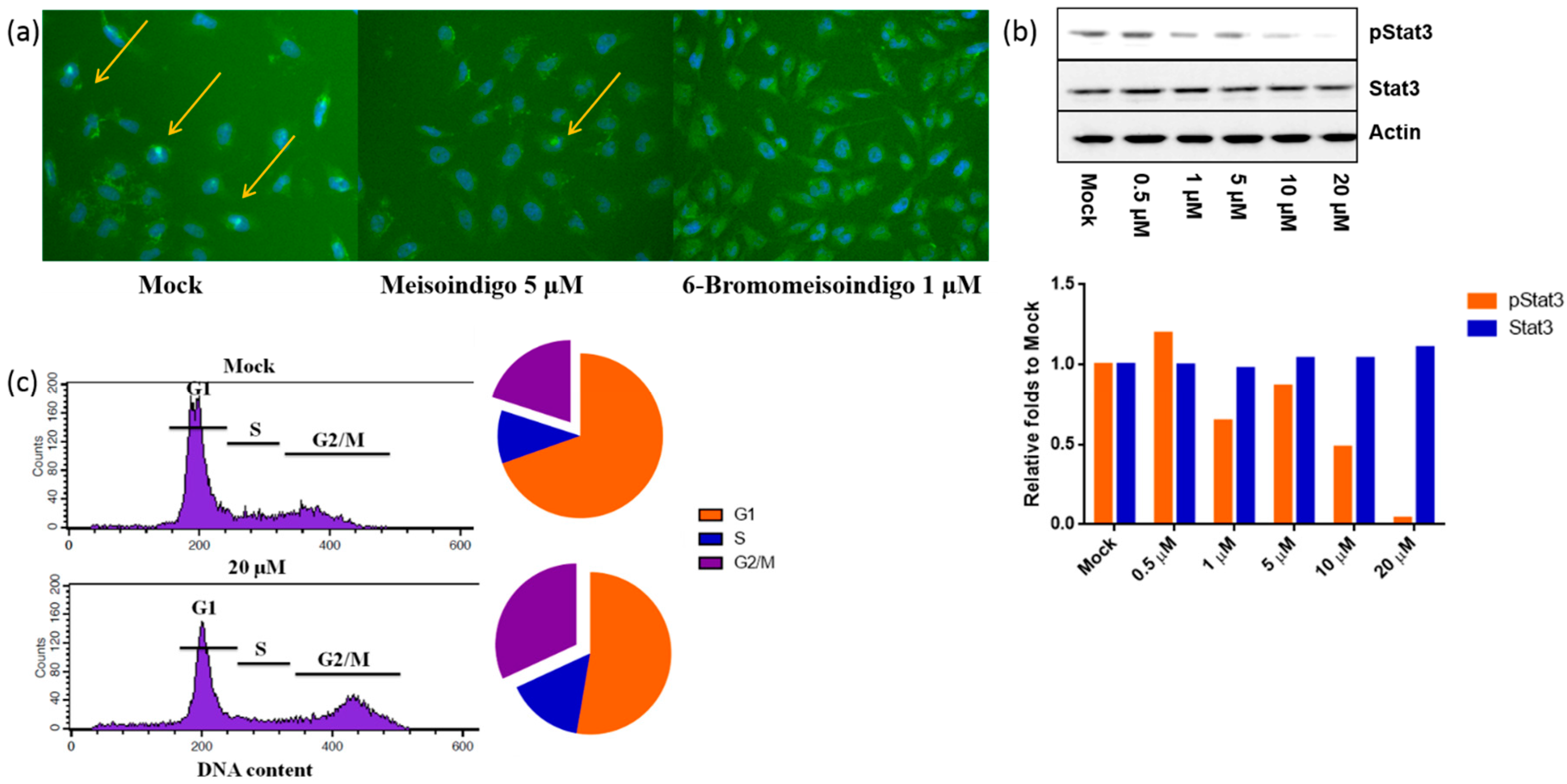
2.2.4. 6-Bromo-Meisoindigo Inhibits Stat3 Activity and Induces Cell Cycle Arrest in HeLa Cells
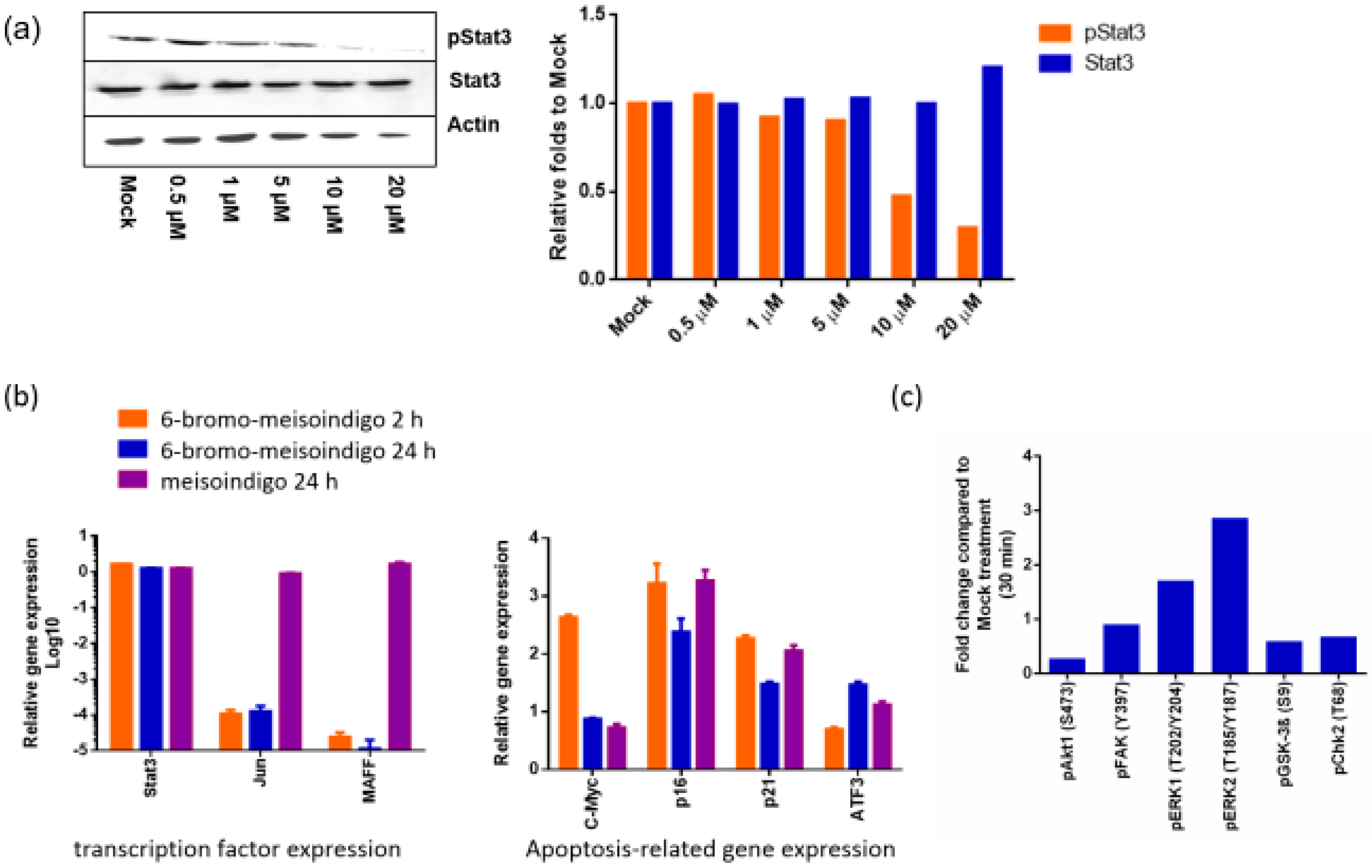
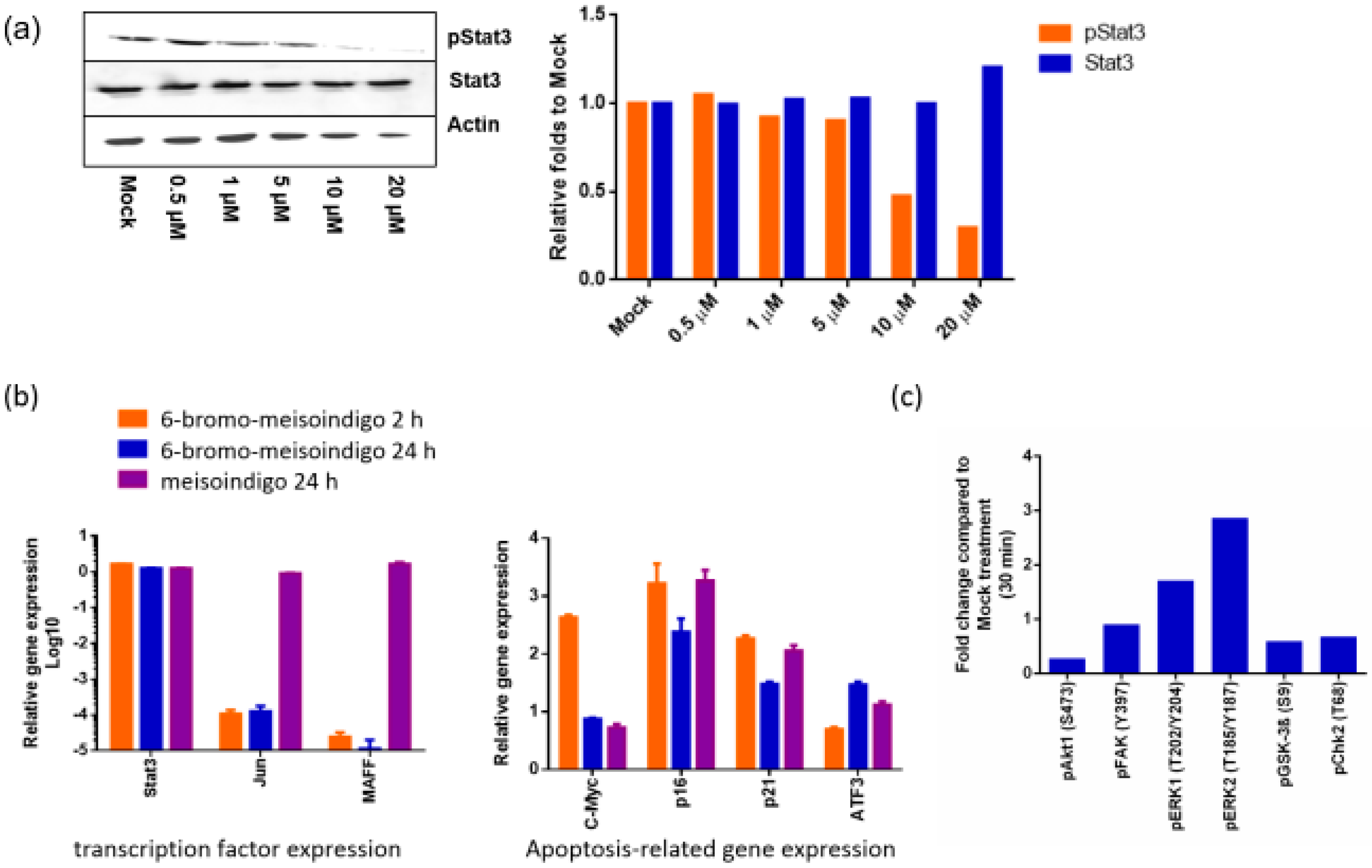
2.2.5. 6-Bromo-Meisoindigo Inhibits Stat3 Activity in Jopaca-1 Cells
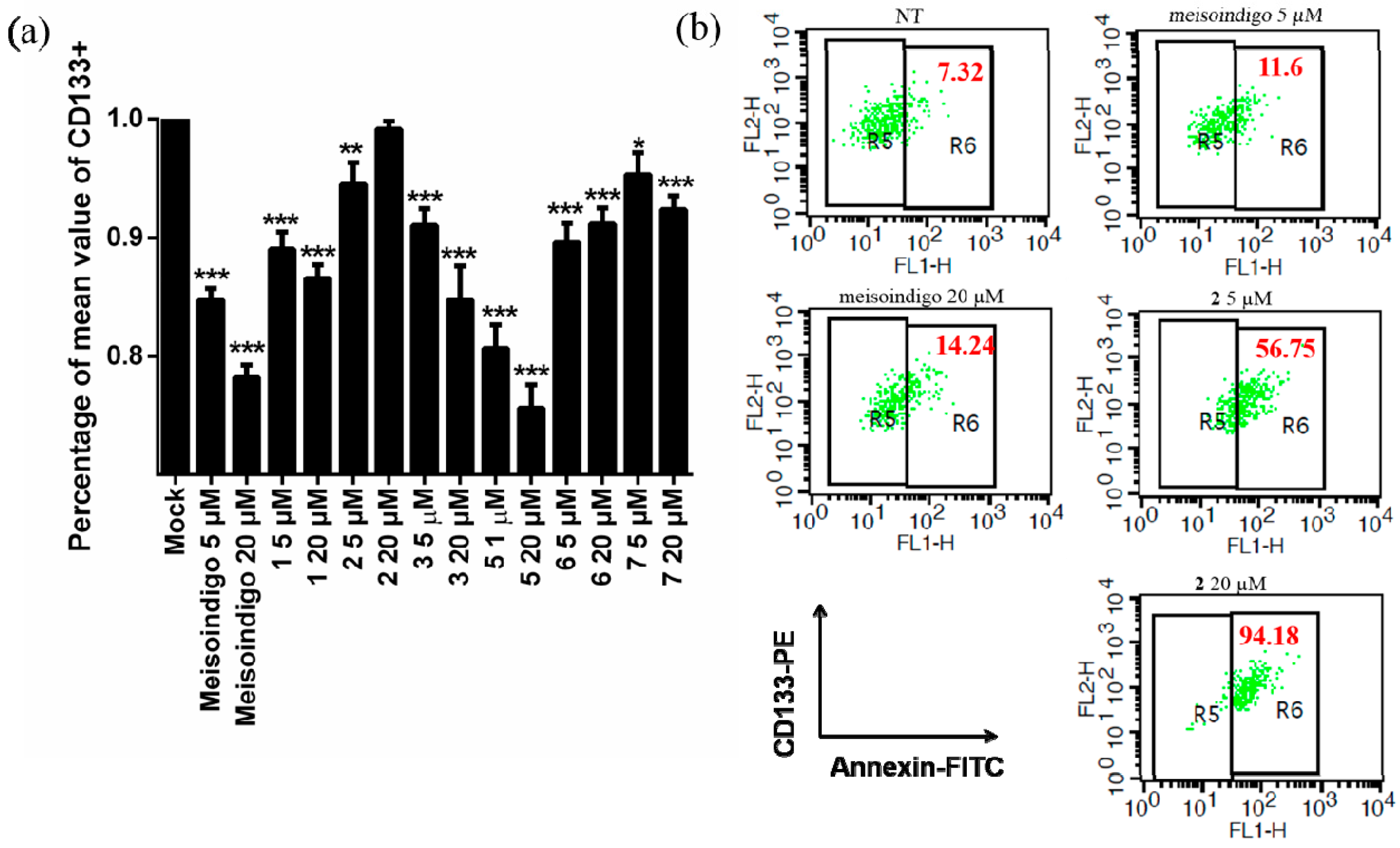
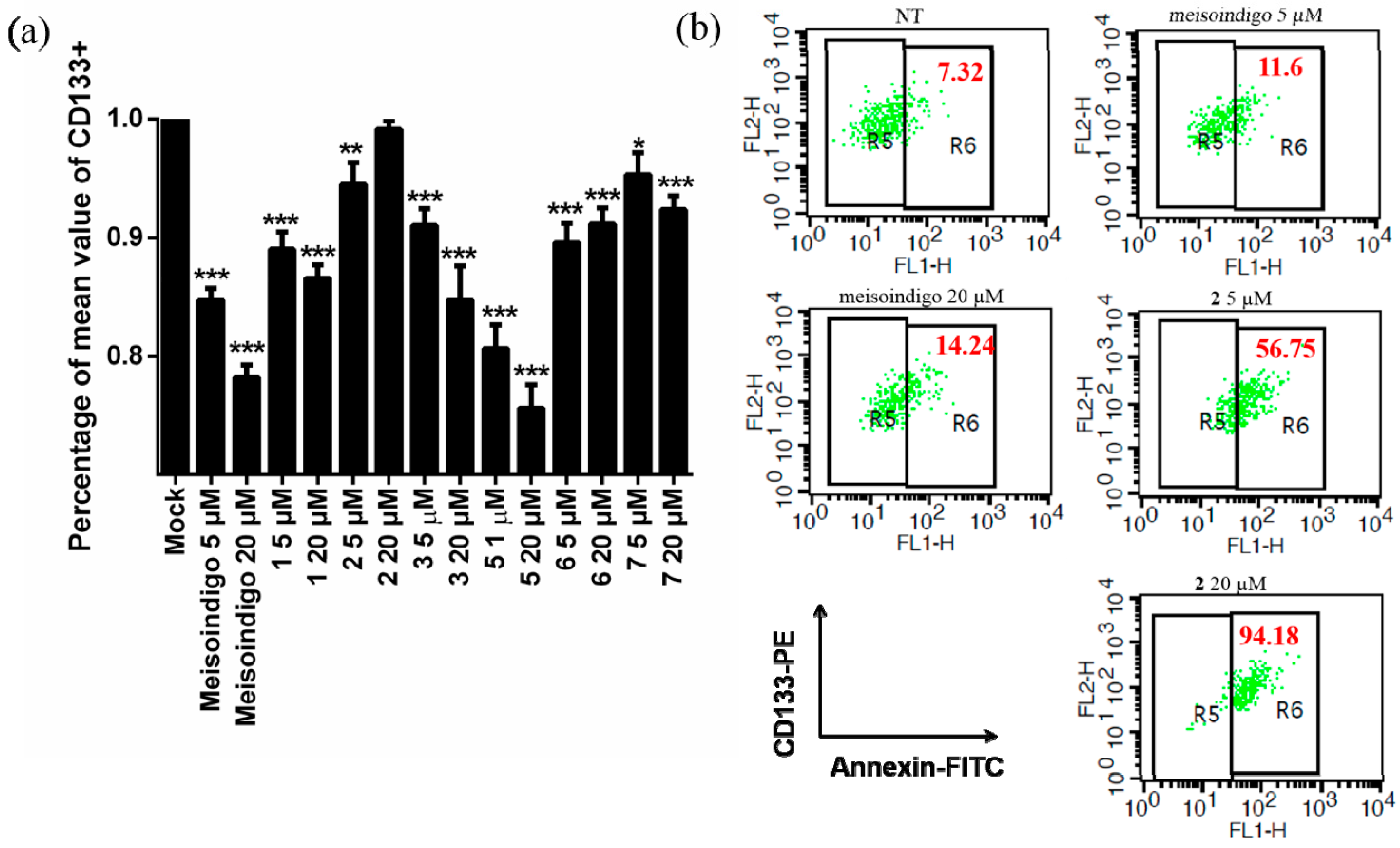
2.2.6. 6-Bromo-Meisoindigo Induces Apoptosis in CD133+ Jopaca-1 Cells
3. Experimental Section
3.1. Materials
3.2. Synthesis of Bromo-Meisoindigo Derivatives
3.2.1. 5-Bromo-1-Meisoindigo
3.2.2. 6-Bromo-1-Meisoindigo
3.2.3. 7-Bromo-1-Meisoindigo
3.2.4. 4′-Bromo-1-Meisoindigo
3.2.5. 5′-Bromo-1-Meisoindigo
3.2.6. 6′-Bromo-1-Meisoindigo
3.2.7. 7′-Bromo-1-Meisoindigo
3.3. Cell Culture
3.4. Protein Kinase Profiling
3.5. Western Blotting
3.6. Cytotoxicity Assays
3.7. CD133 and Annexin V Staining
3.8. Immunocytochemistry
3.9. qRT-PCR
3.10. Protein Microarray Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheng, X.; Merz, K.H. The role of indirubins in inflammation and associated tumorigenesis. Adv. Exp. Med. Biol. 2016, 929, 269–290. [Google Scholar] [PubMed]
- Xiao, Z.; Hao, Y.; Liu, B.; Qian, L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in china. Leuk. Lymphoma 2002, 43, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Merz, K.H.; Vatter, S.; Zeller, J.; Muehlbeyer, S.; Thommet, A.; Christ, J.; Wolfl, S.; Eisenbrand, G. Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. J. Med. Chem. 2017, 60, 4949–4962. [Google Scholar] [CrossRef] [PubMed]
- Vougogiannopoulou, K.; Ferandin, Y.; Bettayeb, K.; Myrianthopoulos, V.; Lozach, O.; Fan, Y.; Johnson, C.H.; Magiatis, P.; Skaltsounis, A.L.; Mikros, E.; et al. Soluble 3′,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase -3 alter circadian period. J. Med. Chem. 2008, 51, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Rasque, P.; Vatter, S.; Merz, K.H.; Eisenbrand, G. Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg. Med. Chem. 2010, 18, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Heshmati, N.; Cheng, X.; Dapat, E.; Sassene, P.; Eisenbrand, G.; Fricker, G.; Muellertz, A. In vitro and in vivo evaluations of the performance of an indirubin derivative, formulated in four different self-emulsifying drug delivery systems. J. Pharm. Pharmacol. 2014, 66, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Heshmati, N.; Cheng, X.; Eisenbrand, G.; Fricker, G. Enhancement of oral bioavailability of e804 by self-nanoemulsifying drug delivery system (snedds) in rats. J. Pharm. Sci. 2013, 102, 3792–3799. [Google Scholar] [CrossRef] [PubMed]
- Heshmati, N.; Wagner, B.; Cheng, X.; Scholz, T.; Kansy, M.; Eisenbrand, G.; Fricker, G. Physicochemical characterization and in vitro permeation of an indirubin derivative. Eur. J. Pharm. Sci. 2013, 50, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell. Biol. 1999, 1, 60–67. [Google Scholar] [PubMed]
- Cheng, X.; Alborzinia, H.; Merz, K.H.; Steinbeisser, H.; Mrowka, R.; Scholl, C.; Kitanovic, I.; Eisenbrand, G.; Wolfl, S. Indirubin derivatives modulate tgfbeta/bmp signaling at different levels and trigger ubiquitin-mediated depletion of nonactivated r-smads. Chem. Biol. 2012, 19, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.X.; Li, Y.; Zhou, J.H.; Wang, H.B.; Chen, X.G. Effect of meisoindigo on wnt signal pathway in k562 and hl-60 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2010, 18, 579–582. [Google Scholar] [PubMed]
- Blazevic, T.; Heiss, E.H.; Atanasov, A.G.; Breuss, J.M.; Dirsch, V.M.; Uhrin, P. Indirubin and indirubin derivatives for counteracting proliferative diseases. Evid. Based Complement. Alternat. Med. 2015, 2015, 654098. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Merz, K.H.; Vatter, S.; Christ, J.; Wolfl, S.; Eisenbrand, G. 7,7′-diazaindirubin—A small molecule inhibitor of casein kinase 2 in vitro and in cells. Bioorg. Med. Chem. 2014, 22, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wee, X.K.; Yeo, W.K.; Zhang, B.; Tan, V.B.; Lim, K.M.; Tay, T.E.; Go, M.L. Synthesis and evaluation of functionalized isoindigos as antiproliferative agents. Bioorg. Med. Chem. 2009, 17, 7562–7571. [Google Scholar] [CrossRef] [PubMed]
- Fredebohm, J.; Boettcher, M.; Eisen, C.; Gaida, M.M.; Heller, A.; Keleg, S.; Tost, J.; Greulich-Bode, K.M.; Hotz-Wagenblatt, A.; Lathrop, M.; et al. Establishment and characterization of a highly tumourigenic and cancer stem cell enriched pancreatic cancer cell line as a well defined model system. PLoS ONE 2012, 7, e48503. [Google Scholar] [CrossRef] [PubMed]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. Ac133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kim, J.Y.; Ghafoory, S.; Duvaci, T.; Rafiee, R.; Theobald, J.; Alborzinia, H.; Holenya, P.; Fredebohm, J.; Merz, K.H.; et al. Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of lkb1 and activation of ampk in pdacs. Mol. Oncol. 2016, 10, 806–824. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kritsanida, M.; Magiatis, P.; Gaboriaud, N.; Wang, Y.; Wu, J.; Buettner, R.; Yang, F.; Nam, S.; Skaltsounis, L.; et al. A novel 7-bromoindirubin with potent anticancer activity suppresses survival of human melanoma cells associated with inhibition of stat3 and akt signaling. Cancer Biol. Ther. 2012, 13, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Nam, S.; Tian, Y.; Yang, F.; Wu, J.; Wang, Y.; Scuto, A.; Polychronopoulos, P.; Magiatis, P.; Skaltsounis, L.; et al. 6-bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011, 71, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Skaltsounis, A.L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. Gsk-3-selective inhibitors derived from tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Buettner, R.; Turkson, J.; Kim, D.; Cheng, J.Q.; Muehlbeyer, S.; Hippe, F.; Vatter, S.; Merz, K.H.; Eisenbrand, G.; et al. Indirubin derivatives inhibit stat3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5998–6003. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Scuto, A.; Yang, F.; Chen, W.; Park, S.; Yoo, H.S.; Konig, H.; Bhatia, R.; Cheng, X.; Merz, K.H.; et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of stat5 signaling. Mol. Oncol. 2012, 6, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Wen, W.; Schroeder, A.; Herrmann, A.; Yu, H.; Cheng, X.; Merz, K.H.; Eisenbrand, G.; Li, H.; Yuan, Y.C.; et al. Dual inhibition of janus and src family kinases by novel indirubin derivative blocks constitutively-activated stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013, 7, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.M. Role of stats as downstream signal transducers in src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the stat3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta. 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Hsieh, F.C.; Lieblein, J.C.; Brown, J.; Chan, C.; Wallace, J.A.; Cheng, G.; Hall, B.M.; Lin, J. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 2007, 96, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent stat3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of stat3 in cancer metastasis and translational advances. Biomed. Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Jou, D.; Wang, Y.; Ma, H.; Liu, T.; Fuchs, J.; Li, P.K.; Lu, J.; Li, C.; Lin, J. Stat3 as a potential therapeutic target in ALDH+ and CD44+/CD24+ stem cell-like pancreatic cancer cells. Int. J. Oncol. 2016, 49, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. Rnai-mediated silencing of myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013, 73, 6816–6827. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. Lif/stat3 controls es cell self-renewal and pluripotency by a myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting stat3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Rho, O.; Kim, D.J.; Kiguchi, K.; Digiovanni, J. Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol. Carcinog 2011, 50, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and Pi3k/Akt signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef] [PubMed]
- Leonis, M.A.; Thobe, M.N.; Waltz, S.E. Ron-receptor tyrosine kinase in tumorigenesis and metastasis. Future Oncol. 2007, 3, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Wojtachnio, K.; Hagens, W.; Vellenga, E.; Buys, C.H.; Hofstra, R.; Kruijer, W. Men2a-ret-induced cellular transformation by activation of stat3. Oncogene 2001, 20, 5350–5358. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Li, L.; Matsushima, G.K.; Earp, H.S.; Wang, B.; Tisch, R. A novel role for c-src and stat3 in apoptotic cell-mediated mertk-dependent immunoregulation of dendritic cells. Blood 2009, 114, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tay, A.; Guy, G.R.; Tan, Y.H. Activation and association of stat3 with src in v-src-transformed cell lines. Mol. Cell. Biol. 1996, 16, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Werdich, X.Q.; Penn, J.S. Src, fyn and yes play differential roles in vegf-mediated endothelial cell events. Angiogenesis 2005, 8, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zoubeidi, A.; Rocha, J.; Zouanat, F.Z.; Hamel, L.; Scarlata, E.; Aprikian, A.G.; Chevalier, S. The fer tyrosine kinase cooperates with interleukin-6 to activate signal transducer and activator of transcription 3 and promote human prostate cancer cell growth. Mol. Cancer Res. 2009, 7, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.L.; Rogers, J.A.; Bowman, T.L.; Jove, R.; Smithgall, T.E. Activation of stat3 by the c-fes protein-tyrosine kinase. J. Biol. Chem. 1998, 273, 7072–7077. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gao, Y.; Qiu, H.; Miller, W.T.; Poli, V.; Reich, N.C. Identification of stat3 as a specific substrate of breast tumor kinase. Oncogene 2006, 25, 4904–4912. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.P.; Cao, X. Structure, function, and regulation of stat proteins. Mol. Biosyst 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. Stat3 target genes relevant to human cancers. Cancers (Basel) 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.-S.; Lee, Y.-S.; Kim, S.-H.; Lu, J. Involvement of c-jun n-terminal kinase in g2/m arrest and caspase-mediated apoptosis induced by sulforaphane in du145 prostate cancer cells. Nutr. Cancer 2005, 52, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, R.; Johnson, R.S.; Moore, C. C-jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.B.; Solovieva, V.; Blank, V. The small maf transcription factors maff, mafg and mafk: Current knowledge and perspectives. Biochim. Biophys. Acta. 2012, 1823, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Williams, J.; Goga, A. In vivo reprogramming of cancer metabolism by myc. Front. Cell Dev. Biol. 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.F.; Ao, J.H.; Zhang, J.; Su, Y.M.; Yang, R.Y. Atf3 activates stat3 phosphorylation through inhibition of p53 expression in skin cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 7439–7444. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dimou, E.; Alborzinia, H.; Wenke, F.; Gohring, A.; Reuter, S.; Mah, N.; Fuchs, H.; Andrade-Navarro, M.A.; Adjaye, J.; et al. Identification of 2-[4-[(4-methoxyphenyl)methoxy]-phenyl]acetonitrile and derivatives as potent Oct3/4 inducers. J. Med. Chem. 2015, 58, 4976–4983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Yoshida, H.; Raoofi, D.; Saleh, S.; Alborzinia, H.; Wenke, F.; Gohring, A.; Reuter, S.; Mah, N.; Fuchs, H.; et al. Ethyl 2-((4-chlorophenyl)amino)thiazole-4-carboxylate and derivatives are potent inducers of oct3/4. J. Med. Chem. 2015, 58, 5742–5750. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(t) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Holenya, P.; Can, S.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wolfl, S. A TrxR inhibiting gold(i) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Molecular Cancer 2014, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Holenya, P.; Can, S.; Rubbiani, R.; Alborzinia, H.; Junger, A.; Cheng, X.; Ott, I.; Wolfl, S. Detailed analysis of pro-apoptotic signaling and metabolic adaptation triggered by a n-heterocyclic carbene-gold(i) complex. Metallomics 2014, 6, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through stat3 expression of immunosuppressive protein CD274 (PD-l1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of meisoindigo derivatives are available are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
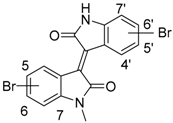
| Nr | Compounds | Nr. | Compounds |
|---|---|---|---|
| 4 | 4′-bromo-meisoindigo | ||
| 1 | 5-bromo-meisoindigo | 5 | 5′-bromo-meisoindigo |
| 2 | 6-bromo-meisoindigo | 6 | 6′-bromo-meisoindigo |
| 3 | 7-bromo-meisoindigo | 7 | 7′-bromo-meisoindigo |
| IC50/Cell Line | Meisoindigo | 5-Bromo-Meisoindigo | 6-Bromo-Meisoindigo | 7-Bromo-Meisoindigo | 4′-Bromo-Meisoindigo | 5′-Bromo-Meisoindigo | 6′-Bromo-Meisoindigo | 7′-Bromo-Meisoindigo | |
|---|---|---|---|---|---|---|---|---|---|
| HeLa | 72 h | 9.53 ± 1.09 | >100 | 5.84 ± 0.40 | 24.69 ± 6.64 | >50 | 18.32 ± 3.22 | 17.71 ± 1.98 | >100 |
| HCT116 | 72 h | 9.09 ± 1.13 | >100 | 7.55 ± 1.05 | 26.34 ± 9.63 | n.t. | 21.96 ± 3.22 | 16.17 ± 1.73 | >100 |
| JoPaca-1 | 72 h | 13.92 ± 1.39 | >100 | 10.52 ± 1.67 | 13.15 ± 0.99 | >50 | 13.04 ± 1.49 | >100 | >100 |
| HF | 72 h | >50 | n.t. | >50 | n.t. | n.t. | n.t. | n.t. | n.t. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tegethoff, J.; Bischoff, R.; Saleh, S.; Blagojevic, B.; Merz, K.-H.; Cheng, X. Methylisoindigo and Its Bromo-Derivatives Are Selective Tyrosine Kinase Inhibitors, Repressing Cellular Stat3 Activity, and Target CD133+ Cancer Stem Cells in PDAC. Molecules 2017, 22, 1546. https://doi.org/10.3390/molecules22091546
Tegethoff J, Bischoff R, Saleh S, Blagojevic B, Merz K-H, Cheng X. Methylisoindigo and Its Bromo-Derivatives Are Selective Tyrosine Kinase Inhibitors, Repressing Cellular Stat3 Activity, and Target CD133+ Cancer Stem Cells in PDAC. Molecules. 2017; 22(9):1546. https://doi.org/10.3390/molecules22091546
Chicago/Turabian StyleTegethoff, Jana, Roland Bischoff, Sawsan Saleh, Biljana Blagojevic, Karl-Heinz Merz, and Xinlai Cheng. 2017. "Methylisoindigo and Its Bromo-Derivatives Are Selective Tyrosine Kinase Inhibitors, Repressing Cellular Stat3 Activity, and Target CD133+ Cancer Stem Cells in PDAC" Molecules 22, no. 9: 1546. https://doi.org/10.3390/molecules22091546
APA StyleTegethoff, J., Bischoff, R., Saleh, S., Blagojevic, B., Merz, K.-H., & Cheng, X. (2017). Methylisoindigo and Its Bromo-Derivatives Are Selective Tyrosine Kinase Inhibitors, Repressing Cellular Stat3 Activity, and Target CD133+ Cancer Stem Cells in PDAC. Molecules, 22(9), 1546. https://doi.org/10.3390/molecules22091546