Targeting PDE10A GAF Domain with Small Molecules: A Way for Allosteric Modulation with Anti-Inflammatory Effects


 ,
,
Abstract
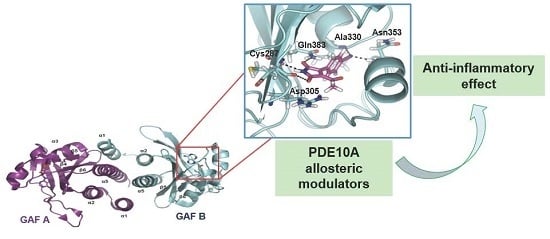
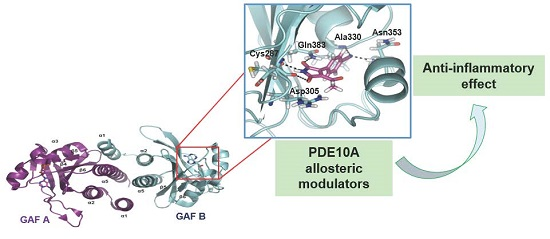
1. Introduction
2. Results and Discussion
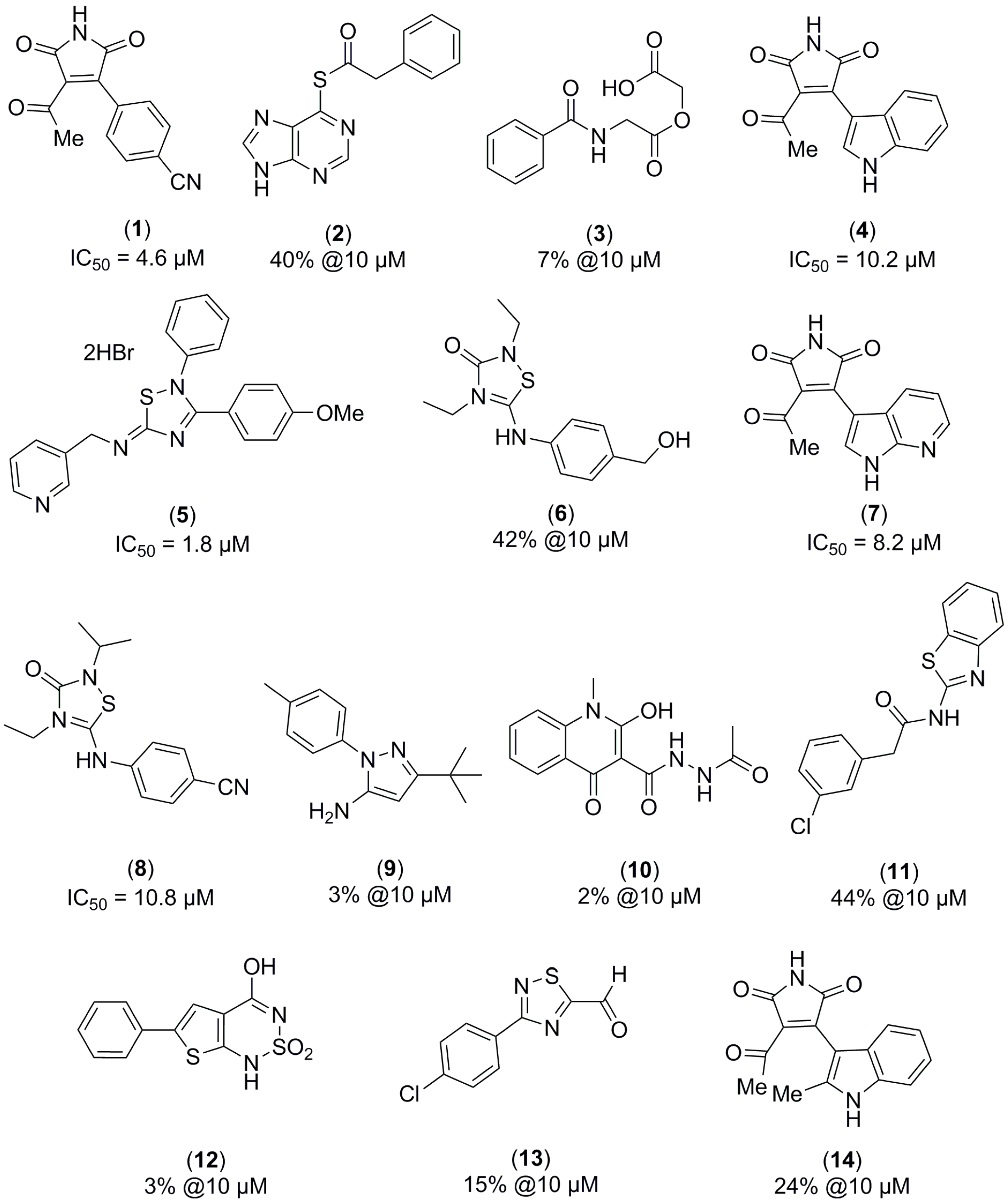
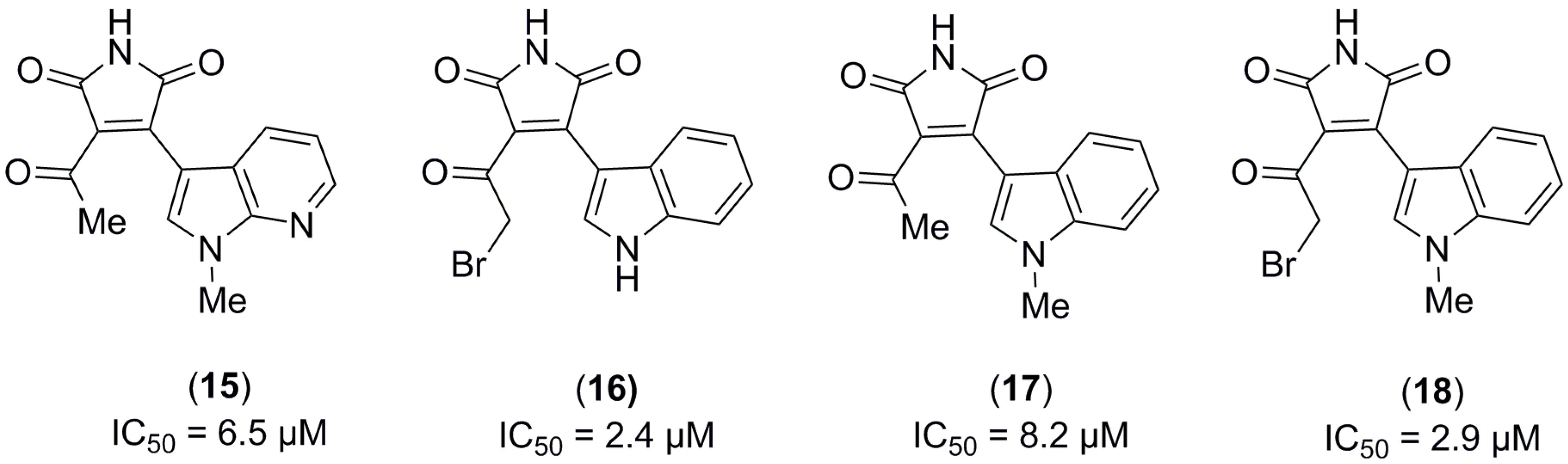
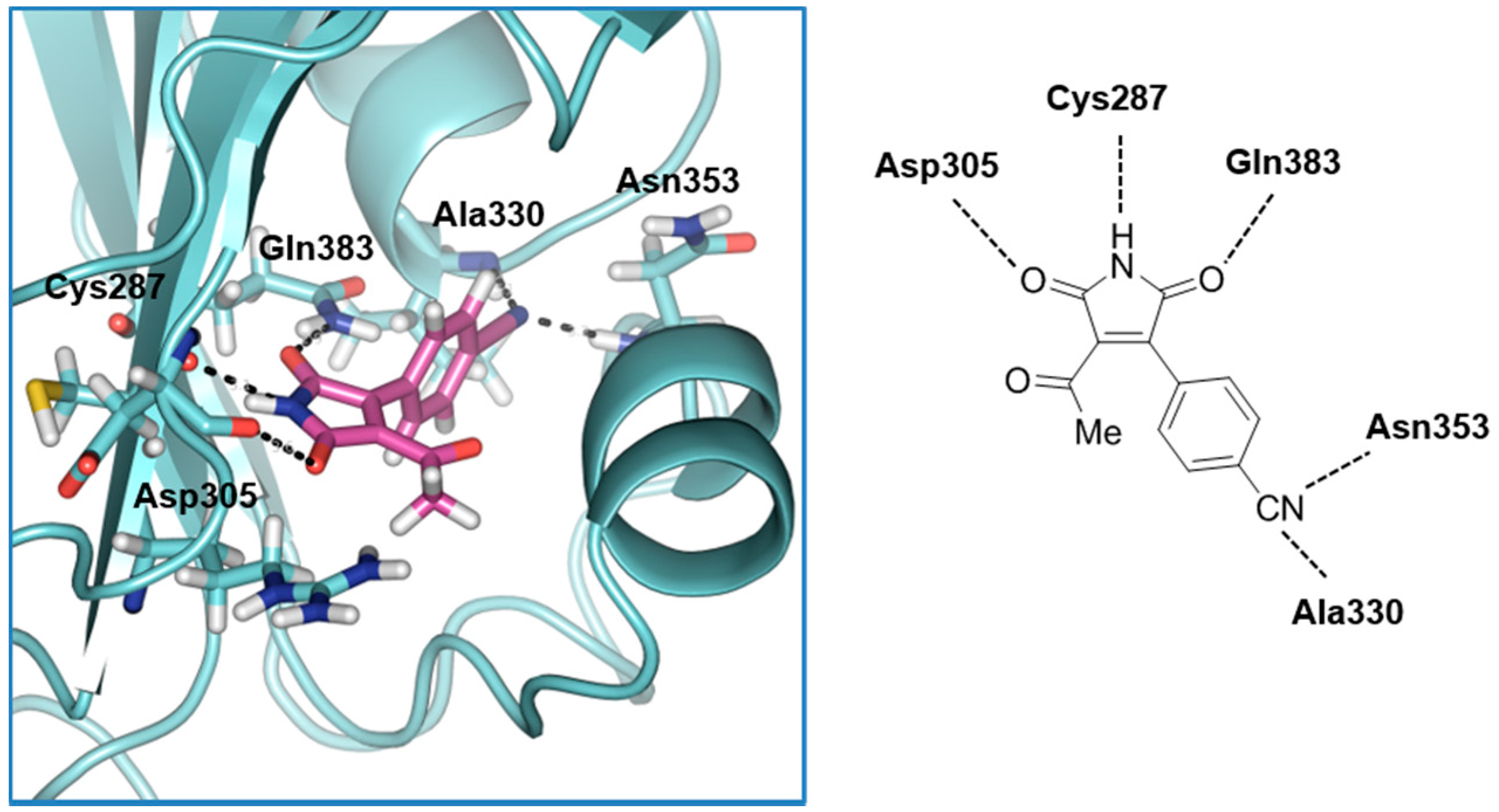
2.1. Structure-Based Virtual Screening and Ligand Identification
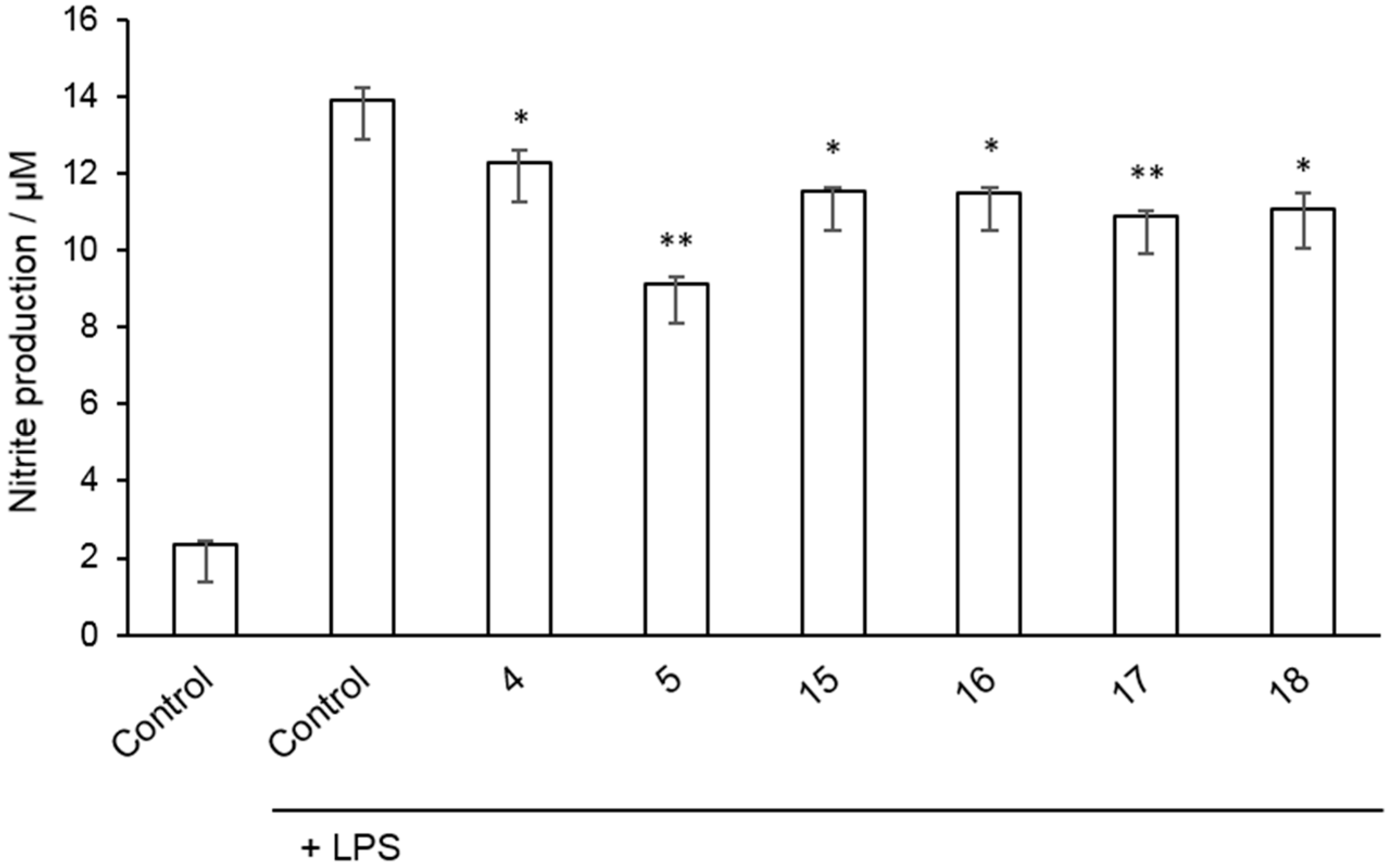
2.2. Inhibition of Nitrite Production in LPS-Stimulated Murine Macrophages
3. Materials and Methods
3.1. Computational Chemistry
3.1.1. Protein and Ligand Preparation
3.1.2. Virtual Screening Workflow
3.1.3. Comparative Analysis of Binding Modes Using Catalytic and GAF-B PDE10A Domains
3.2. Radiometric Phosphodiesterase Inhibition Assay
3.3. Cell Viability Assays
3.4. Nitrite Quantification
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- DeNinno, M.P. Future directions in phosphodiesterase drug discovery. Bioorg. Med. Chem. Lett. 2012, 22, 6794–6800. [Google Scholar] [CrossRef] [PubMed]
- Zoraghi, R.; Corbin, J.D.; Francis, S.H. Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol. Pharmacol. 2004, 65, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Gross-Langenhoff, M.; Hofbauer, K.; Weber, J.; Schultz, A.; Schultz, J.E. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 2006, 281, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Pandit, J.; Forman, M.D.; Fennell, K.F.; Dillman, K.S.; Menniti, F.S. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. USA 2009, 106, 18225–18230. [Google Scholar] [CrossRef] [PubMed]
- Handa, N.; Mizohata, E.; Kishishita, S.; Toyama, M.; Morita, S.; Uchikubo-Kamo, T.; Akasaka, R.; Omori, K.; Kotera, J.; Terada, T.; et al. Crystal structure of the GAF-B domain from human phosphodiesterase 10A complexed with its ligand, cAMP. J. Biol. Chem. 2008, 283, 19657–19664. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.E.; Dunkern, T.; Gawlitta-Gorka, E.; Sorg, G. The GAF-tandem domain of phosphodiesterase 5 as a potential drug target. Handb. Exp. Pharmacol. 2011, 151–166. [Google Scholar] [CrossRef]
- Li, N.; Lee, K.; Xi, Y.; Zhu, B.; Gary, B.D.; Ramirez-Alcantara, V.; Gurpinar, E.; Canzoneri, J.C.; Fajardo, A.; Sigler, S.; et al. Phosphodiesterase 10A: A novel target for selective inhibition of colon tumor cell growth and beta-catenin-dependent TCF transcriptional activity. Oncogene 2015, 34, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Duinen, M.V.; Reneerkens, O.A.; Lambrecht, L.; Sambeth, A.; Rutten, B.P.; Os, J.V.; Blokland, A.; Prickaerts, J. Treatment of cognitive impairment in schizophrenia: Potential value of phosphodiesterase inhibitors in prefrontal dysfunction. Curr. Pharm. Des. 2015, 21, 3813–3828. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caille, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P.; et al. Change in PDE10 across early Huntington disease assessed by [18F]MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.M.; Redondo, M.; Martinez, A.; Gil, C. Phosphodiesterase 10 inhibitors: New disease modifying drugs for Parkinson‘s disease? Curr. Med. Chem. 2014, 21, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Geerts, H.; Spiros, A.; Roberts, P. Phosphodiesterase 10 inhibitors in clinical development for CNS disorders. Expert Rev. Neurother. 2017, 17, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.M.; Martinez, A.; Gil, C. Enhancing cAMP levels as strategy for the treatment of neuropsychiatric disorders. Curr. Top. Med. Chem. 2016, 16, 3527–3535. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Perez, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE 2011, 6, e17240. [Google Scholar] [CrossRef] [PubMed]
- Sebastian-Pérez, V.; Roca, C.; Awale, M.; Reymond, J.-L.; Martínez, A.; Gil, C.; Campillo, N.E. The Medicinal and Biological Chemistry (MBC) Library: An efficient source on new hits. J. Chem. Inf. Mod. 2017. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.; Palomo, V.; Brea, J.; Perez, D.I.; Martin-Alvarez, R.; Perez, C.; Paul-Fernandez, N.; Conde, S.; Cadavid, M.I.; Loza, M.I.; et al. Identification in silico and experimental validation of novel phosphodiesterase 7 inhibitors with efficacy in experimental autoimmune encephalomyelitis mice. ACS Chem. Neurosci. 2012, 3, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, X.; Xu, J.; Rothfuss, J.; Mach, R.H.; Tu, Z. Synthesis and in vitro evaluation of new analogues as inhibitors for phosphodiesterase 10A. Eur. J. Med. Chem. 2011, 46, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.M.; Salado, I.G.; Perez, D.I.; Brea, J.; Morales-Garcia, J.A.; Gonzalez-Garcia, A.; Cadavid, M.I.; Loza, M.I.; Luque, F.J.; Perez-Castillo, A.; et al. Pharmacological tools based on imidazole scaffold proved the utility of PDE10A inhibitors for Parkinson‘s disease. Future Med. Chem. 2017, 9, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Hou, J.; Zheng, M.; Robinson, H.; Ke, H. Structural insight into substrate specificity of phosphodiesterase 10. Proc. Natl. Acad. Sci. USA 2007, 104, 5782–5787. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available upon directly request to the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scoring Function | ||||
|---|---|---|---|---|
| Protein structure | cAMP | cGMP | Compound 19 | Compound 1 |
| Catalytic domain (2OUP) | −4.133 | −2.816 | −9.386 | −4.803 |
| GAF domain (2ZMF) | −8.071 | −1.040 | −1.395 | −8.898 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, A.M.; Brea, J.; González-García, A.; Pérez, C.; Cadavid, M.I.; Loza, M.I.; Martinez, A.; Gil, C. Targeting PDE10A GAF Domain with Small Molecules: A Way for Allosteric Modulation with Anti-Inflammatory Effects. Molecules 2017, 22, 1472. https://doi.org/10.3390/molecules22091472
García AM, Brea J, González-García A, Pérez C, Cadavid MI, Loza MI, Martinez A, Gil C. Targeting PDE10A GAF Domain with Small Molecules: A Way for Allosteric Modulation with Anti-Inflammatory Effects. Molecules. 2017; 22(9):1472. https://doi.org/10.3390/molecules22091472
Chicago/Turabian StyleGarcía, Ana M., José Brea, Alejandro González-García, Concepción Pérez, María Isabel Cadavid, María Isabel Loza, Ana Martinez, and Carmen Gil. 2017. "Targeting PDE10A GAF Domain with Small Molecules: A Way for Allosteric Modulation with Anti-Inflammatory Effects" Molecules 22, no. 9: 1472. https://doi.org/10.3390/molecules22091472
APA StyleGarcía, A. M., Brea, J., González-García, A., Pérez, C., Cadavid, M. I., Loza, M. I., Martinez, A., & Gil, C. (2017). Targeting PDE10A GAF Domain with Small Molecules: A Way for Allosteric Modulation with Anti-Inflammatory Effects. Molecules, 22(9), 1472. https://doi.org/10.3390/molecules22091472