Alkaloid Constituents of the Amaryllidaceae Plant Amaryllis belladonna L

 ,
,  and
and 
Abstract
1. Introduction
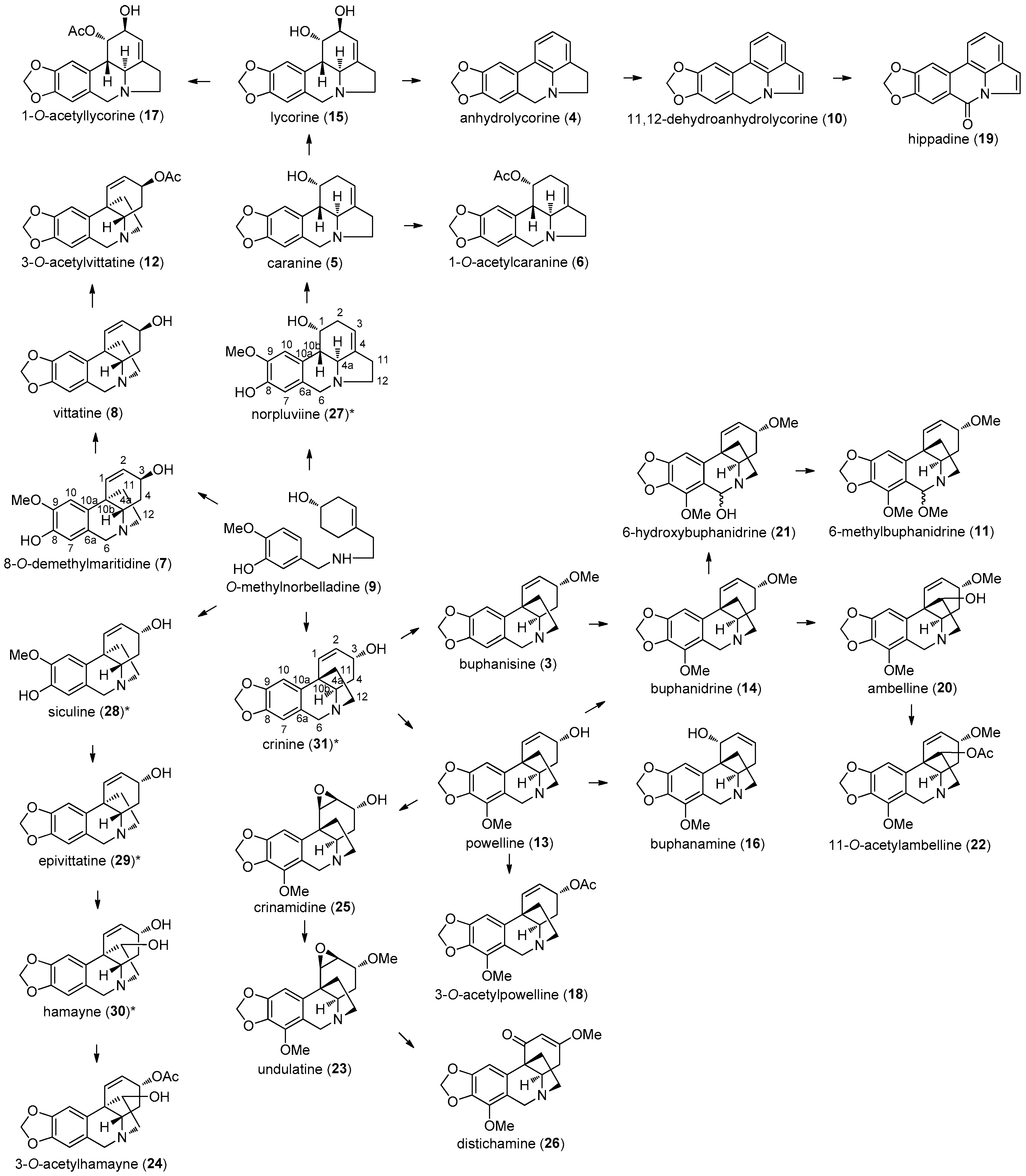
2. Results and Discussion
2.1. Alkaloids Identified in A. belladonna by GC–MS
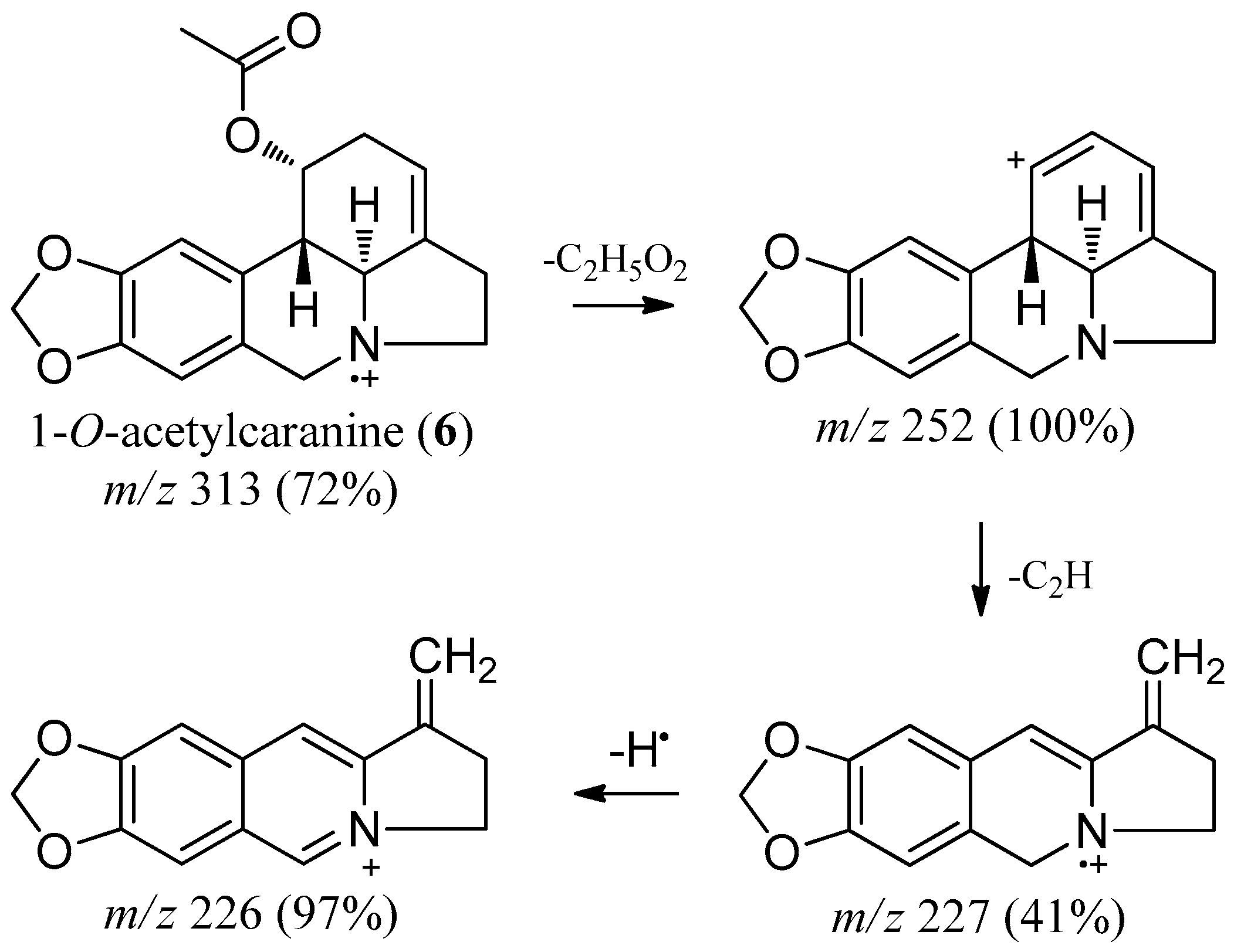
2.2. 1-O-Acetylcaranine
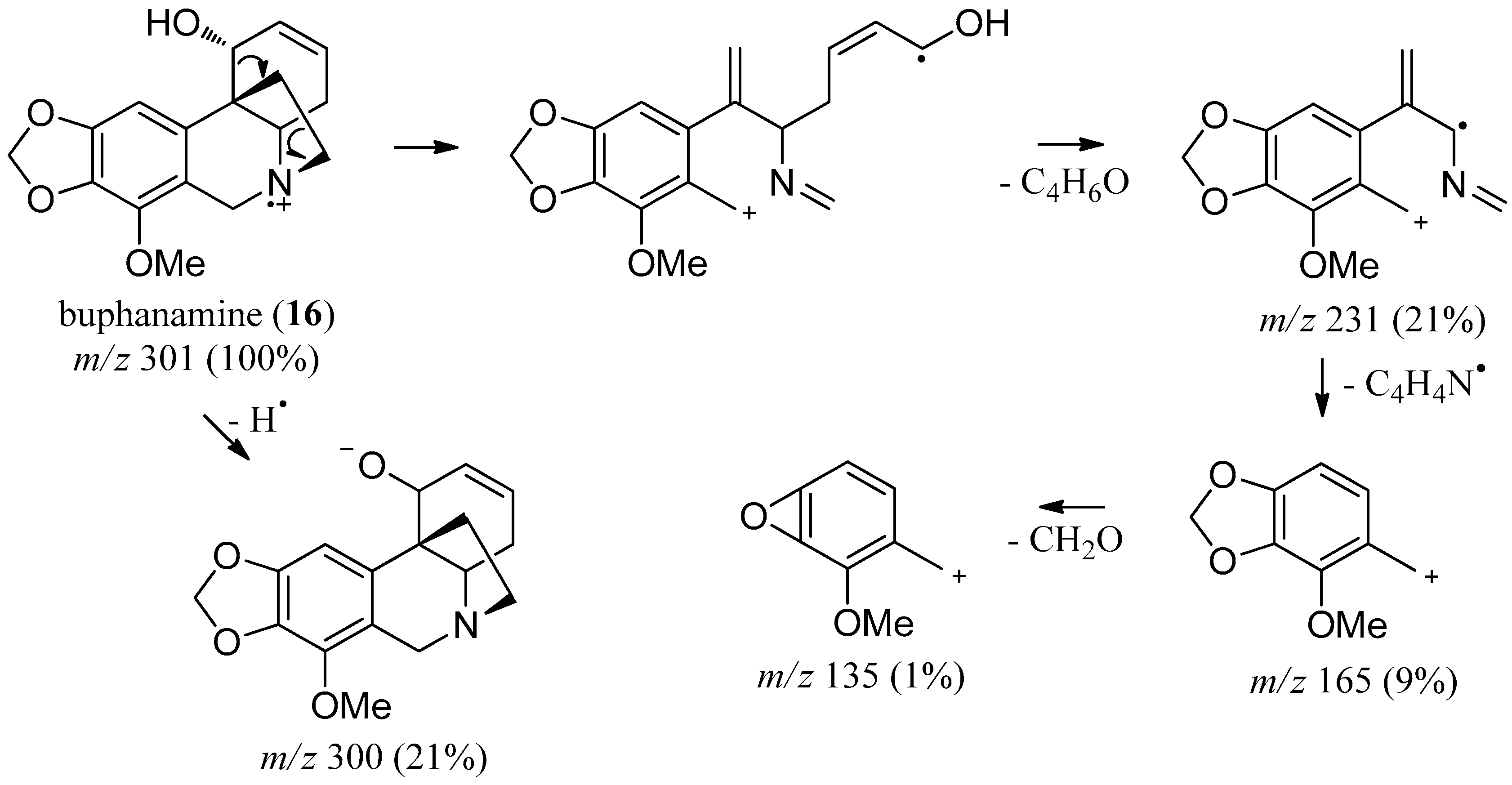
2.3. Buphanamine
2.4. Biological Activity
3. Materials and Methods
3.1. Plant Material
3.2. Equipment
3.3. Extraction
3.4. Characterization of Compounds
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bastida, J.; Lavilla, R.; Viladomat, F. Chemical and biological aspects of Narcissus alkaloids. In The Alkaloids: Chemistry and Physiology; Cordell, G.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 63, pp. 87–179. [Google Scholar]
- Pettit, G.R.; Gaddamidi, V.; Goswami, A.; Cragg, G.M. Antineoplastic Agents, 99. Amaryllis belladonna. J. Nat. Prod. 1984, 47, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.J.; Van Staden, J.; Bastida, J. Cytotoxic alkaloids constituents of the Amaryllidaceae. In Studies in Natural Products Chemistry; Rahman, A.-U., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 49, pp. 107–156. [Google Scholar]
- Battersby, A.R.; Fales, H.M.; Wildman, W.C. Biosynthesis in the Amaryllidaceae. Tyrosine and norbelladine as precursors of haemanthamine. J. Am. Chem. Soc. 1961, 83, 4098–4099. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Cohen, T. Some biogenic aspects of phenol oxidation. In Festschrift Arthur Stoll; Birkhauser, A.G., Ed.; Birkhauser: Basel, Switzerland, 1956; pp. 117–143. [Google Scholar]
- Berkov, S.; Bastida, J.; Sídjimova, B.; Viladomat, F.; Codina, C. Alkaloid diversity in Galanthus elwessi and Galanthus nivalis. Chem. Biodivers. 2011, 8, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Berkov, S.; Georgieva, L.; Kondakova, V.; Viladomat, F.; Bastida, J.; Atanassov, A.; Codina, C. The geographic isolation of Leucojum aestivum populations leads to divergation of alkaloids biosynthesis. Biochem. Syst. Ecol. 2013, 47, 1065–1073. [Google Scholar] [CrossRef]
- Takos, A.M.; Rook, F. Towards a molecular understanding of the biosynthesis of Amaryllidaceae alkaloids in support of their expanding medical use. Int. J. Mol. Sci. 2013, 14, 11713–11741. [Google Scholar] [CrossRef] [PubMed]
- Berkov, S.; Martínez-Francés, V.; Bastida, J.; Codina, C.; Ríos, S. Evolution of alkaloids biosynthesis in the genus Narcissus. Phytochemistry 2014, 99, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, J.; Takada, T.; Kita, Y.; Zenk, M.H. Biosynthesis of the Amaryllidaceae alkaloid galanthamine. Phytochemistry 1998, 49, 1037–1047. [Google Scholar] [CrossRef]
- Maelicke, A.; Samochocki, M.; Jostock, R.; Fehrenbacher, A.; Ludwig, J.; Albuquerque, E.X.; Zerlin, M. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer’s disease. Biol. Psychiatry 2001, 49, 279–288. [Google Scholar] [CrossRef]
- Kornienko, A.; Evidente, A. Chemistry, biology and medicinal potencial of narciclasine and its congeners. Chem. Rev. 2008, 108, 1982–2014. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.H.; Puschett, E.R.; Wildman, W.C. Alkaloids of the Amaryllidaceae. IV. Crystalline Alkaloids of Ammocharis coranica (Ker-Gawl.) Herb., Brunsvigia rosea (Lam.) Hannibal and Two Crinum Species. J. Am. Chem. Soc. 1955, 77, 1253–1256. [Google Scholar] [CrossRef]
- Boit, H.-G.; Ehmke, H. Alkaloide von Nerine bowdenii, Crinum powelli, Amaryllis belladonna und Pancratium maritimum (XII. Mitteil. Uber Amaryllidaceen-Alkaloids). Eur. J. Inorg. Chem. 1956, 89, 2093–2097. [Google Scholar]
- Boit, H.-G.; Döpke, W. Alkaloids from Hippeastrum brachyandrum and Hippeastrum rutilum. Chem. Ber. 1959, 92, 2582–2584. [Google Scholar] [CrossRef]
- Willaman, J.J.; Schubert, B.G. Alkaloid-Bearing Plants and Their Contained Alkaloids; Technical Bulletin 1234; Superintendent of Documents; Government Printing Office: Washington, DC, USA, 1961.
- Burlingame, A.L.; Fales, H.M.; Highet, R.J. The structure of amaryllisine. J. Am. Chem. Soc. 1964, 86, 4976–4979. [Google Scholar] [CrossRef]
- Hänsel, R.; Thober, H. Anhydrolycorinon, ein neus Alkaloid aus Amaryllis belladonna L. Arch. Pharm. 1982, 315, 767–768. [Google Scholar] [CrossRef]
- Queckenberg, O.R.; Frahm, A.W.; Muller-Doblies, D.; Muller-Doblies, U. Reinvestigation of Amaryllis belladonna. Phytochem. Anal. 1996, 7, 156–160. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Abou-Donia, A.H.; Touema, S.M.; Hammoda, H.M.; Shawky, E.; Motta, A. (−)-Amarbellisine, a lycorine-type alkaloid from Amaryllis belladona L. growing in Egypt. Phytochemistry 2004, 65, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Wahyuni, D.S.C.; Van Kooy, F.; Klinkhamer, P.G.L.; Verpoorte, R.; Leiss, K. The use of bio-guided fractionation to explore the use of leftover biomass in Dutch flower bulb production as allelochemicals agains weeds. Molecules 2013, 18, 4510–4525. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases; Second WHO Report on Neglected Tropical Diseases; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Osorio, E.; Arango, G.J.; Jiménez, N.; Alzate, F.; Ruiz, G.; Gutiérrez, D.; Paco, M.A.; Giménez, A.; Robledo, S. Antiprotozoal and cytotoxic activities in vitro of Colombian Annonaceae. J. Ethnopharmacol. 2007, 111, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Osorio, E.; Robledo, S.; Bastida, J. Alkaloids with antiprotozoal activity. In The Alkaloids Chemistry and Biology; Cordell, G.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 66, pp. 113–190. [Google Scholar]
- Coll, J.C.; Bowden, B.F. The application of vacuum liquid chromatography to the separation of terpene mixtures. J. Nat. Prod. 1986, 49, 934–936. [Google Scholar] [CrossRef]
- Vazquez-Flota, F.; De Carolis, E.; Alarco, A.-M.; Luca, V.D. Molecular cloning and characterization of desacetoxyvindoline-4-hydroxylase, a 2-oxoglutarate dependent-dioxygenase involved in the biosynthesis of vindoline in Catharanthus roseus (L.) G. Don. Plant Mol. Biol. 1997, 34, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Giddings, L.-A.; Liscombe, D.K.; Hamilton, J.P.; Childs, K.L.; Dellapenna, D.; Buell, C.R.; O’Connor, S.E. Stereoselective hydroxylation step of alkaloids biosynthesis by a unique cytochrome P450 in Catharanthus roseus. J. Biol. Chem. 2011, 286, 16751–16757. [Google Scholar] [CrossRef] [PubMed]
- Kirby, G.W.; Tiwari, H.P. Phenol oxidation and biosynthesis. Part IX. The biosynthesis of norpluviine and galanthamine. J. Chem. Soc. C 1966, 676–682. [Google Scholar] [CrossRef]
- Wildman, W.C.; Heimer, N.E. Alkaloid Biosynthesis and Interconversions: The conversion of caranine to lycorine. J. Am. Chem. Soc. 1967, 89, 5265–5269. [Google Scholar] [CrossRef] [PubMed]
- Fuganti, C.; Mazza, M. Stereochemistry of hydroxylation in the biosynthesis of lycorine in Clivia miniata Regel. J. Chem. Soc. 1972, 16, 936–937. [Google Scholar] [CrossRef]
- Miyakado, M.; Kato, T.; Ohno, N.; Koshimizu, K. Alkaloids of Urginea altissima and their antimicrobial activity against Phytophthora capsici. Phytochemistry 1975, 14, 2717. [Google Scholar] [CrossRef]
- Lamoral-Theys, D.; Adolfi, A.; Goietsenoven, G.V.; Cimmino, A.; Calvé, B.L.; Wauthoz, N.; Mégalizzi, V.; Gras, T.; Bruyère, C.; Dubois, J.; et al. Lycorine, the phananthridine Amaryllidaceae alkaloid, exhibits significant antitumor activity in cancer cell that display resistence to proapoptotic stimuli: An investigation of structure—Activity relationship and mechanistic insight. J. Med. Chem. 2009, 52, 6244–6256. [Google Scholar] [CrossRef] [PubMed]
- Longevialle, P.; Smith, D.H.; Burlingame, A.L.; Fales, M.H.; Highet, R.J. High resolution mass spectrometry in molecular structure studies V: The fragmentation of Amaryllis alkaloids in the crinine series. Org. Mass Spectrom. 1973, 7, 401–415. [Google Scholar] [CrossRef]
- Sandager, M.; Nielsen, N.D.; Stafford, G.I.; Van Staden, J.; Jager, A.K. Alkaloids from Boophane disticha with affinity to the serotonin transporter in rat brain. J. Ethnopharmacol. 2005, 98, 367–370. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, G.G.; Wildman, W.C. Circular dichroism studies—I: A Quadrant rule for the optically active aromatic chromophore in rigid polycyclic systems. Tetrahedron 1969, 25, 5099–5112. [Google Scholar] [CrossRef]
- Orhan, I.; Şener, B.; Kaiser, M.; Brun, R.; Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 2010, 8, 47–58. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkaloid | RI | M+ | MS |
|---|---|---|---|
| ismine (1) | 2288.1 | 257 (28) | 239 (20), 238 (100), 225 (7), 211 (7), 196 (9), 180 (8), 139 (8) |
| trisphaeridine (2) | 2305.0 | 223 (100) | 224 (15), 222 (38), 193 (2), 164 (15), 138 (26), 111 (14) |
| buphanisine (3) | 2447.5 | 285 (100) | 270 (34), 254 (34), 242 (12), 227 (24), 215 (85), 201 (23), 185 (22), 172 (22), 157 (33), 128 (33) |
| anhydrolycorine (4) | 2470.2 | 251 (42) | 250 (100), 220 (3), 192 (18), 163 (3), 125 (2) |
| caranine (5) | 2560.9 | 271 (59) | 270 (37), 252 (53), 240 (10), 227 (44), 226 (100), 212 (6), 181 (1) |
| 1-O-acetylcaranine (6) | 2569.6 | 313 (72) | 270 (3), 252 (100), 240 (9), 227 (41), 226 (97)181 (2) |
| 8-O-demethylmaritidine (7) | 2575.5 | 273 (100) | 256 (13), 244 (12), 230 (26), 216 (14), 201 (90), 189 (61), 175 (28), , 157 (19), 141 (11), 128 (24) |
| vittatine (8) | 2578.0 | 271 (100) | 254 (11), 242 (11), 228 (22), 214 (13), 199 (58), 187 (63), 173 (24), 157 (18), 141(13), 128 (29) |
| O-methylnorbelladine (9) | 2578.5 | 273 (-) | 166 (36), 137 (100), 122 (8), 94 (6) |
| 11,12-dehydroanhydrolycorine (10) | 2638.1 | 249 (59) | 248 (100), 218 (1),190 (26), 163 (8), 137 (1), 123 (5), 95 (13) |
| 6-methoxylbuphanidrine (11) | 2656.8 | 345 (58) | 330 (33), 298 (14), 287 (32), 259 (68), 227 (30), 181 (10), 145 (13) |
| 3-O-acetylvittatine (12) | 2690.6 | 313 (100) | 298 (1), 284 (6), 270 (27), 254 (75), 252 (61), 224 (38), 216 (60), 198 (36), 187 (36), 139 (15), 128 (33) |
| powelline (13) | 2702.7 | 301 (100) | 284 (10), 272 (14), 258 (39), 244 (24), 229 (8), 217 (7), 202 (22), 187 (23), 159 (13), 143 (11), 127 (19) |
| buphanidrine (14) | 2705.2 | 315 (100) | 300 (31), 284 (35), 272 (9), 260 (39), 245 (69), 228 (24), 215 (21), 202 (20), 187 (18), 130 (18) |
| lycorine (15) | 2722.0 | 287 (18) | 286 (10), 268 (19), 250 (16), 238 (7), 227 (78), 226 (100), 211 (6), 147 (5), 119 (3) |
| buphanamine (16) | 2726.5 | 301 (100) | 282 (21), 272 (13), 256 (22), 244 (11), 190 (7), 165 (9) |
| 1-O-acetyllycorine (17) | 2754.9 | 329 (31) | 268 (31), 250 (20), 227 (64), 226 (100), 211 (6), 192 (3), 167 (3), 147 (6) |
| 3-O-acetylpowelline (18) | 2768.7 | 343 (100) | 314 (6), 300 (25), 284 (78), 283 (24), 268 (9), 254 (21), 246 (81), 228 (39), 217 (18), 202 (6), 183 (9) |
| hippadine (19) | 2775.0 | 263 (100) | 233 (2), 205 (7), 177 (24), 150 (13), 131 (9), 111 (2), 75 (7) |
| ambelline (20) | 2814.6 | 331 (86) | 316 (8), 299 (44), 287 (100), 270 (33), 260 (76), 257 (58), 239 (52), 228 (20), 211 (62), 190 (39) |
| 6-hydroxybuphanidrine (21) | 2828.4 | 331 (44) | 314 (5), 300 (10), 282 (7), 276 (100), 261 (33), 258 (13), 243 (12), 228 (8), 217 (21), 216 (22) |
| 11-O-acetylambelline (22) | 2850.2 | 373 (100) | 314 (58), 313 (50), 282 (46), 258 (48), 255 (36), 254 (63), 241 (38), 240 (35), 218 (44) |
| undulatine (23) | 2855.2 | 331 (100) | 316 (9), 300 (9), 286 (15), 258 (33), 244 (14), 232 (17), 217 (28), 205 (60), 189 (32), 173 (30) |
| 3-O-acetylhamayne (24) | 2907.7 | 329 (3) | 300 (100), 269 (6), 240 (16), 224 (8), 212 (34), 211 (14), 199 (6), 181 (54), 167 (3), 153 (18), 128 (10) |
| crinamidine (25) | 2954.3 | 317 (59) | 300 (2), 288 (100), 258 (21), 244 (27), 230 (16), 217 (35), 203 (34), 189 (20), 173 (40), 145 (17) |
| distichamine (26) | 2984.3 | 329 (100) | 328 (25), 314 (15), 300 (9), 286 (9), 269 (5), 256 (3), 231 (20), 204 (13), 190 (5), 130 (2) |
| Position | δC, Type | δH (J in Hz) | HMBC |
|---|---|---|---|
| 1 | 66.60 d | 5.84 br dd (4.5, 2.0) | C-3, C-4a, C-10b, OCOCH3 |
| 2α | 33.55 t | 2.39 m | |
| 2β | 33.55 t | 2.64 m | |
| 3 | 114.43 d | 5.39 dd (4.9, 2.4) | C-4a, C-1 |
| 4 | 139.56 s | ||
| 4a | 61.51 d | 2.82 d (10.8) | |
| 6α | 57.09 t | 3.54 d (14.3) | C-10a |
| 6β | 57.09 t | 4.13 d (14.1) | C-4a, C-7, C-10a |
| 6a | 129.54 s | ||
| 7 | 107.34 d | 6.57 br s | C-6, C-9, C10a |
| 8 | 146.24 s | ||
| 9 | 146.49 s | ||
| 10 | 105.25 d | 6.72 d (1.0) | C-6a, C-8, C-10b |
| 10a | 127.79 s | ||
| 10b | 43.56 d | 2.67 m | |
| 11α | 28.70 t | 2.61 m | |
| 11β | 28.70 t | 2.61 m | |
| 12α | 53.83 t | 2.39 m | C-6 |
| 12β | 53.83 t | 3.33 ddd (9.4, 4.9, 4.4) | C-4, C-4a, C-6 |
| OCH2O | 101.16 t | 5.91 d (1.5)–5.92 d (1.5) | |
| OCOCH3 | 171.03 q | 1.93 s | |
| OCOCH3 | 21.53 q | 1.93 s |
| Position | δC, Type | δH (J in Hz) | HMBC |
|---|---|---|---|
| 1 | 64.5 d | 4.74 d (5.5) | C-3, C-4a, C-10a, C-11 |
| 2 | 125.5 d | 6.00 dddd (10.0, 5.5, 2.8, 1.8) | C-10b |
| 3 | 128.9 d | 5.86 ddd (10.0, 4.5, 2.8) | C-4a |
| 4α | 28.2 t | 2.57 dddd (19.7, 8.1, 4.5, 1.9) | C-2, C-10b |
| 4β | 28.2 t | 1.99 dddd (19.8, 8.6, 1.1, 0.3) | C-2, C-10b |
| 4a | 59.2 d | 3.43 t (8.3) | C-10a |
| 6a | 117.9 s | ||
| 6α | 56.9 t | 4.17 d (17.2) | C-4a, C-7, C-10a |
| 6β | 56.9 t | 3.81 d (17.2) | C-4a, C-7, C-10a |
| 7 | 140.8 s | ||
| 8 | 133.6 s | ||
| 9 | 148.6 s | ||
| 10 | 98.0 d | 6.59 s | C-6a, C-8, C-10b |
| 10a | 137.0 s | ||
| 10b | 48.3 s | ||
| 11endo | 38.7 t | 1.85 dddd (11.9, 8.8, 3.1, 1.3) | C-4a, C-10a |
| 11exo | 38.7 t | 1.92 ddd (12.1, 10.0, 7.3) | C-4a, C-10a |
| 12endo | 51.4 t | 2.75 ddd (13.0, 8.8, 7.2) | C-4a, C-6 |
| 12exo | 51.4 t | 3.40 ddd (13.0, 10.0, 3.0) | C-6, C-10b |
| OCH2O | 100.7 t | 5.88 d (1.5)–5.89 d (1.5) | C-8, C-9 |
| OCH3 | 59.1 q | 3.98 s | C-7 |
| Parasite | T. b. rhodesiense | T. cruzi | L. donovani | P. falciparum | Cytotoxicity |
|---|---|---|---|---|---|
| Stage | Trypamastigotes | Amastigotes | Amastigotes | IEF (Intraerythrocytic) | |
| Strain | STIB 900 (IC50 a) | Tulahuen C4 LacZ (IC50 a) | MHOM-ET-67/L82 (IC50 a) | NF54 (IC50 a) | L6 (IC50 a) |
| melarsoprol | 0.0035 b and 0.0010 c | ||||
| benznidazole | 0.660 b and 1.080 c | ||||
| miltefosine | 0.085 b and 0.091 c | ||||
| chloroquine | 0.002 b,c | ||||
| podophyllotoxin | 0.005 b and 0.007 c | ||||
| 1-O-acetylcaranine (6) | 1.97 | 35.9 | >100 | 3.21 | 14.2 |
| 3-O-acetylhamayne (24) | 1.51 | 8.3 | 17.9 | 1.14 | 1.72 |
| buphanamine (16) | 28.2 | 62.9 | >100 | 25.9 | >100 |
| crude extract | 4.67 | 34.9 | >100 | 1.17 | 34.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallini, L.R.; Andrade, J.P.d.; Kaiser, M.; Viladomat, F.; Nair, J.J.; Zuanazzi, J.A.S.; Bastida, J. Alkaloid Constituents of the Amaryllidaceae Plant Amaryllis belladonna L. Molecules 2017, 22, 1437. https://doi.org/10.3390/molecules22091437
Tallini LR, Andrade JPd, Kaiser M, Viladomat F, Nair JJ, Zuanazzi JAS, Bastida J. Alkaloid Constituents of the Amaryllidaceae Plant Amaryllis belladonna L. Molecules. 2017; 22(9):1437. https://doi.org/10.3390/molecules22091437
Chicago/Turabian StyleTallini, Luciana R., Jean Paulo de Andrade, Marcel Kaiser, Francesc Viladomat, Jerald J. Nair, José Angelo S. Zuanazzi, and Jaume Bastida. 2017. "Alkaloid Constituents of the Amaryllidaceae Plant Amaryllis belladonna L" Molecules 22, no. 9: 1437. https://doi.org/10.3390/molecules22091437
APA StyleTallini, L. R., Andrade, J. P. d., Kaiser, M., Viladomat, F., Nair, J. J., Zuanazzi, J. A. S., & Bastida, J. (2017). Alkaloid Constituents of the Amaryllidaceae Plant Amaryllis belladonna L. Molecules, 22(9), 1437. https://doi.org/10.3390/molecules22091437