1. Introduction
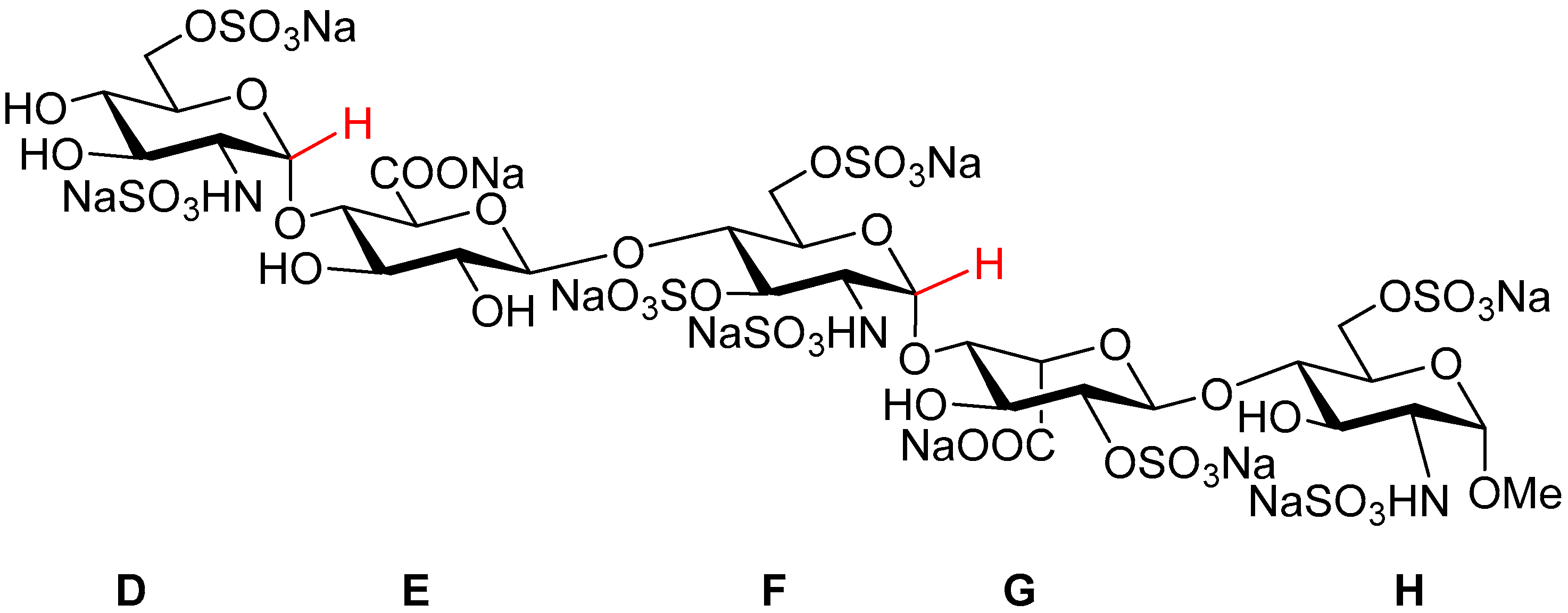
Fondaparinux sodium (referred to as ‘Fondaparinux’ in this article;
Figure 1) is a synthetic pentasaccharide [
1] derived from the high affinity antithrombin III binding site in heparin, modified by a methyl group at the reducing end. It is the active pharmaceutical ingredient of GSK’s anticoagulant drug Arixtra
® and a selective aXa inhibitor through antithrombin III [
2] and lacks aIIa activity as a result of its low molecular weight. As part of the extended physical characterization of Fondaparinux, both structure (by X-ray) and standard content (by qNMR) are described in this paper. The X-ray structures of Fondaparinux have previously been limited to co-crystals in complex with a number of proteins [
3,
4,
5,
6,
7,
8]. Also, related synthetic (penta) saccharides complexed with protein have been reported [
9,
10]. Furthermore, uncomplexed Fondaparinux (solution) structures determined by (a combination of) theoretical calculations and NMR spectroscopy have been described [
11,
12,
13,
14,
15]. In solution,
3J(
1H,
1H) coupling constants allow to determine the conformation of the pyranose residues in heparin. The glucosamine and glucuronic acids residues adopt a stable
4C
1-conformation. However, the iduronate ring adopts
1C
4 and
2S
0 conformations rapidly interconverting on the NMR time scale [
11,
12]. Complexation by proteins locks the iduronate residue in a single
2S
0 conformation both in the crystal [
5,
6,
9,
10] and in solution [
16]. Also in the crystal of uncomplexed Fondaparinux, the occurrence of a single iduronate conformation is foreseen. Here we report the X-ray single crystal structure of uncomplexed Fondaparinux and compare it to the conformation of Fondaparinux in solution, as well as complexed with proteins.
The content of ampoules of the Fondaparinux solution reference standard was determined in both 1999 and 2016 by quantitative NMR (qNMR) using an internal standard. The results of both determinations are discussed here. The ampoules of this standard are used to determine the Fondaparinux content of Fondaparinux batches. For consistency, the ability to assure the long-term stability of the standard content is crucial.
2. Results
qNMR is a well-established and accepted method for content determination [
17,
18,
19] and is described in major pharmacopoeia [
20,
21,
22]. The strength of qNMR lies in the fundamental property that, under the right conditions, the integrated response of a proton in
1H-NMR will be identical in every molecule. This implies that a content determination can be carried out relative to a completely unrelated (but well-characterized) standard. We applied qNMR for the content determination of ampoules standard solution of the relatively complicated compound Fondaparinux using a maleic acid standard.
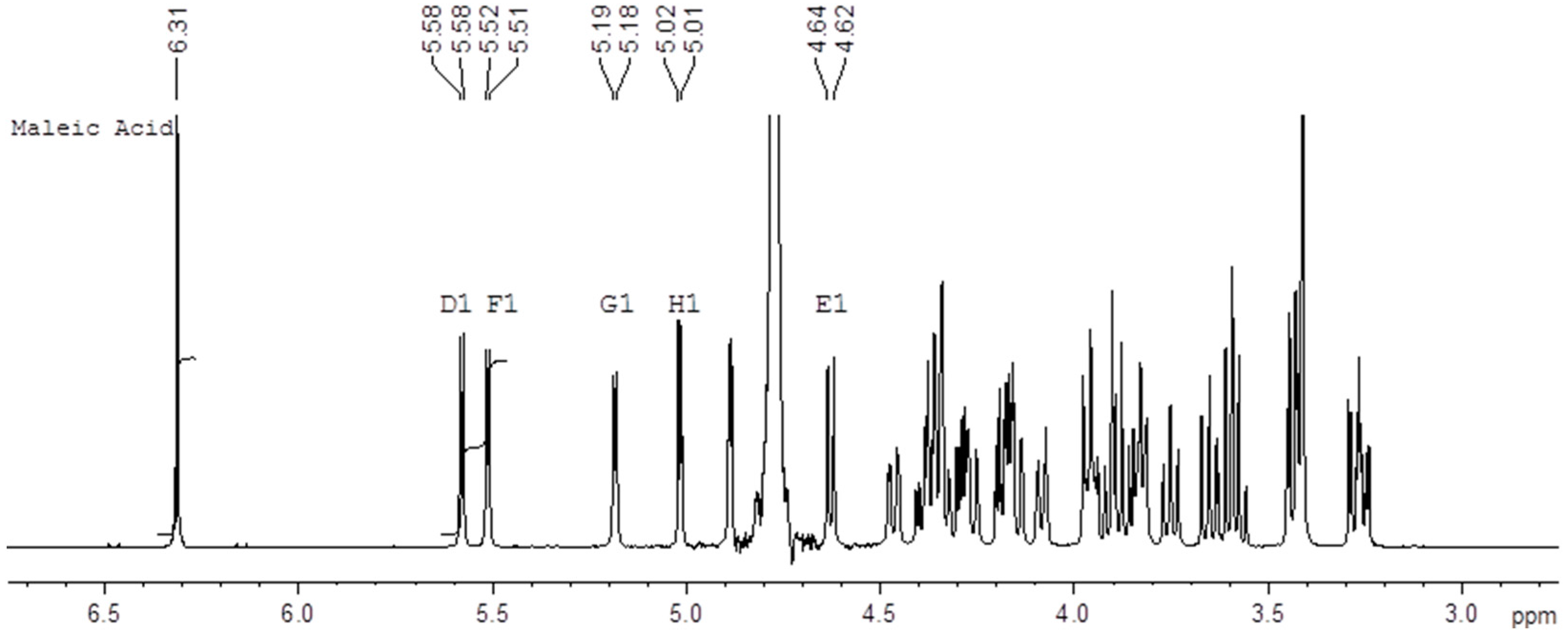
Figure 2 shows an example of the 500 MHz
1H-NMR spectrum of a mixture of the Fondaparinux standard and maleic acid in D
2O with the signals used for integration indicated. We performed these determinations on ampoules of the same standard both in 1999 and 2016 using an identical protocol which yields a unique opportunity to assess the long-term reproducibility of the qNMR method. The content determination in 1999 was performed at 400 MHz using a conventional probe whereas the content determination in 2016 was done at 500 MHz using a cryo probe. Different batches of maleic acid were used in 1996 and in 2017. For each experiment the tests were done by two different analysts and on two different days. The differences between the determinations are listed in the experimental section (Table 4).
The current analysis yields a content of 9.75 mg/mL with an SD of 0.06 mg/mL for the standard. The analysis performed in 1999 yielded a content of 9.64 mg/mL with an SD of 0.08 mg/mL. The two results are therefore not significantly different. The close correspondence between the two values obtained about 17 years apart demonstrates the intrinsic robustness of the qNMR content determination. Moreover, the differences between the two values is unlikely be due to degradation since the current content is slightly higher than the one determined 17 years ago and no signals pointing at degradation are currently detected in NMR.
2.1. Fondaparinux Single Crystal X-ray Structure
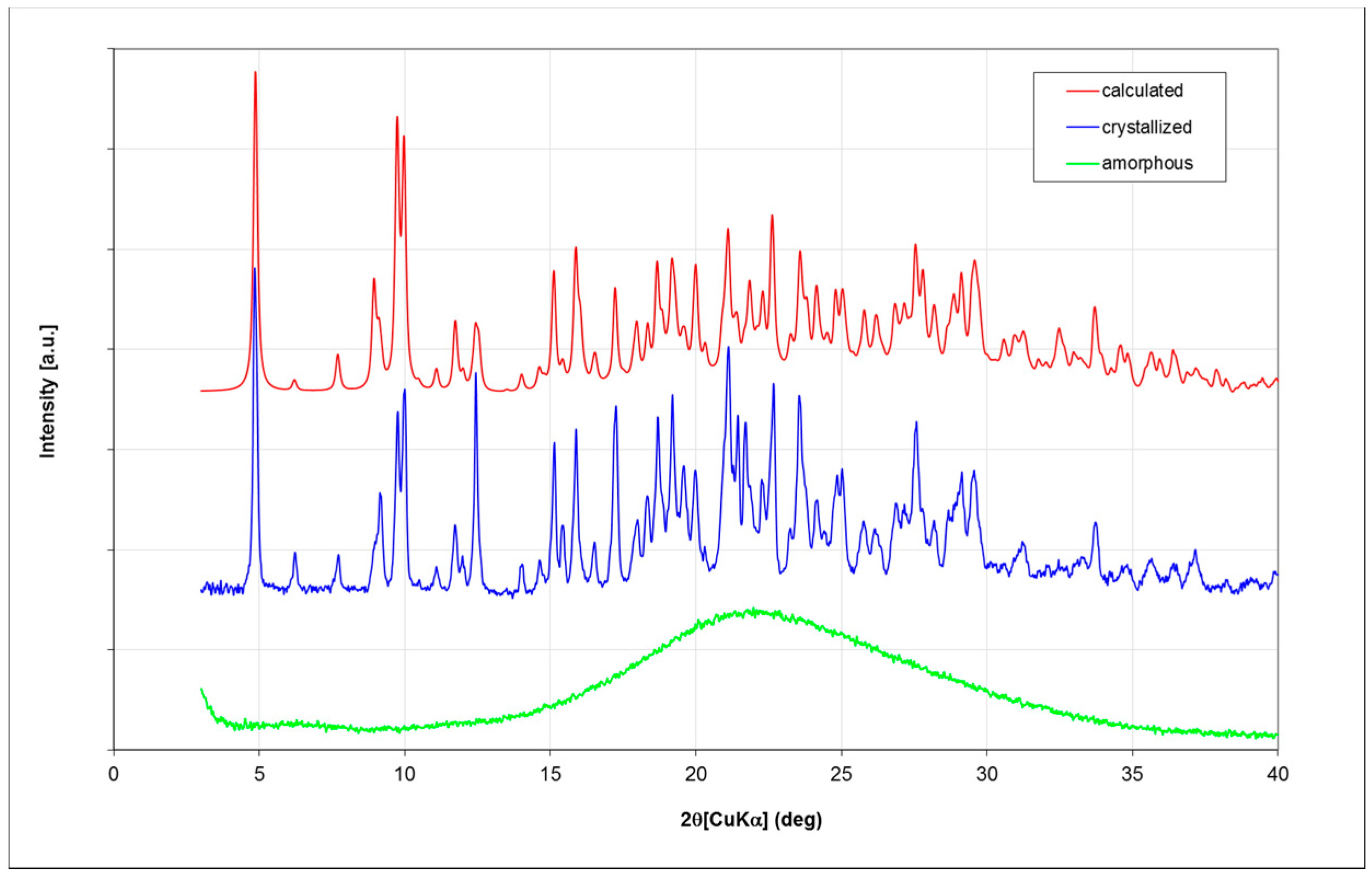
Fondaparinux drug substance is industrially obtained as an amorphous powder as is evident from its X-ray powder diffractogram (XRPD;
Figure 3; in green). Obtaining Fondaparinux single crystals was challenging, probably due to its inherent flexibility and linear structure. Ultimately, crystals were obtained as described in the Materials and Methods section. Several batches of crystals were produced. For a crystal from one batch, a full single-crystal structure determination was undertaken. A crystal from another batch was used for a unit-cell determination to ensure the similarity of these batches. Finally, an XRPD measurement was done for one batch of crystals see Table 7 for unit-cell comparison). In
Figure 3, the blue trace shows the XRPD patterns of crystalline Fondaparinux. The XRPD pattern calculated from the crystal structure is shown as the red trace in
Figure 3. A polarization microscopy image of representative crystals is displayed in
Figure 4.
2.2. Structural Features of the Crystal Structure
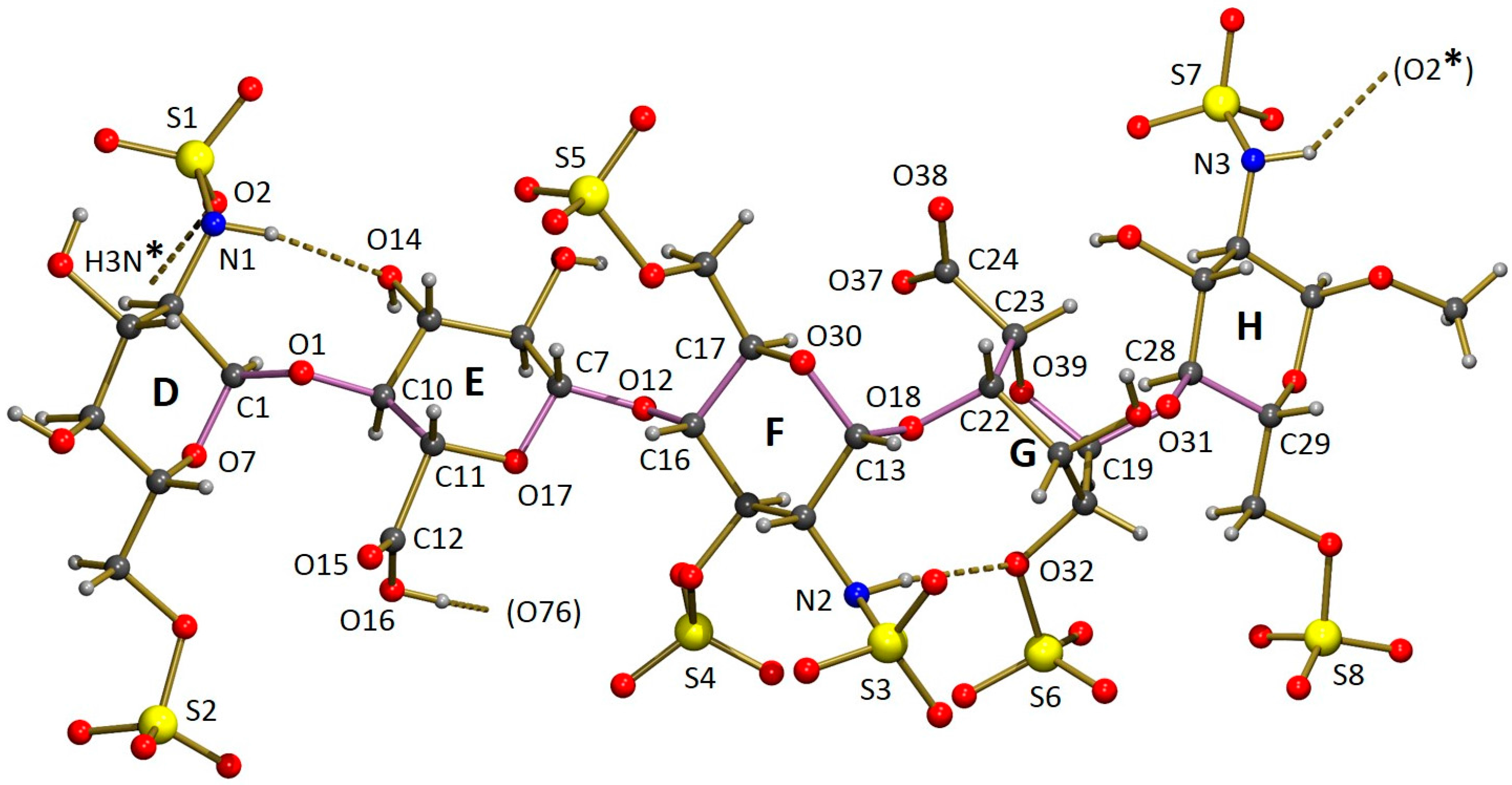
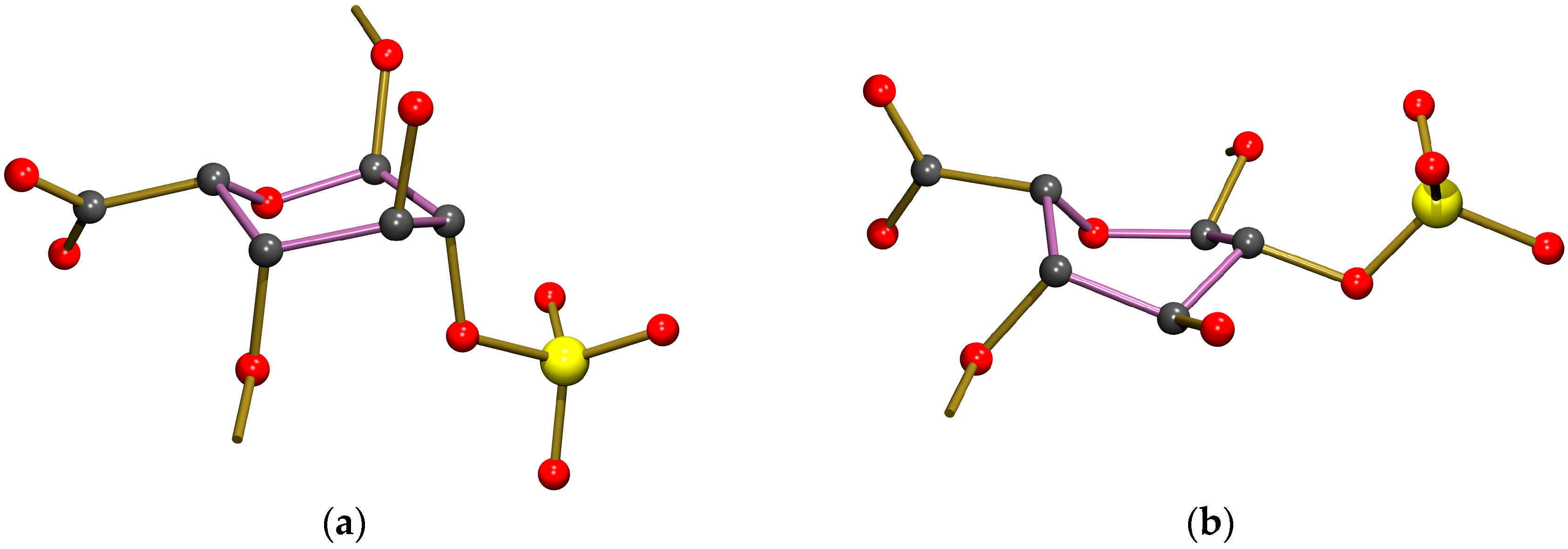
The asymmetric unit of the crystal structure of Fondaparinux contains a pentasaccharide anion, with one of the carboxylic groups protonated (vide infra), nine sodium cations, 17 water molecules coordinated to one or more sodium atoms and 15 non-coordinated water molecules. A perspective drawing of the molecular structure of the pentasaccharide Fondaparinux, along with the atomic labeling scheme of the non-hydrogen atoms and the labeling scheme of the rings is given in
Figure 5.
The sodium ions are coordinated by numerous oxygen atoms from the pentasaccharide and extensively co-crystallized water molecules. The seven ordered sodium ions are coordinated by six oxygen atoms each. For the ordered ions, most Na...O distances fall in the range 2.3–2.7 Å. However, a small number of Na...O distances up to 2.9 Å are observed. The two disordered sodium ions are also surrounded by a total of six oxygen atoms that can be assigned to the coordination shell, but this shell is somewhat larger than that observed for the ordered sodium atoms. As a result of this, the sodium can take up several positions and some of the individual disorder components are only directly coordinated by five oxygen atoms. Full details of the observed coordination distances are given in the deposited Crystallographic Information File. Through these coordinative bonds an infinite three-dimensional structure is formed. A perspective drawing of the infinite substructure formed by sodium counter-ions, sulfate groups and coordinated water molecules is included in the
Supporting Information. The final model contained no solvent accessible void. DFT calculations also indicated extensive coordination to both sodium ions and water (e.g., [
13,
15]).
3. Discussion
The individual glucosamine (D, F and H) rings have adopted the conventional stable
4C
1 chair conformation. The glucuronic acid ring (E) is in a slightly distorted
4C
1 chair-conformation. The iduronic acid G ring conformation has been the subject of intense debate [
11,
12,
13,
14,
15,
23,
24,
25,
26]. Uncomplexed in solution, its structure cannot be described by a single conformer but is an equilibrium of
1C
4, chair and
2S
0. Skew-boat conformations with the equilibrium shifted to the
2S
0 conformation [
11,
12,
13,
14,
15,
23,
24,
25,
26]. The conformation is
2S
0 in complex with antithrombin III [
5,
6,
16].
In our crystal structure, the iduronic acid ring (G) is in a chair conformation, heavily distorted towards a half-chair conformation with the best-fitting local twofold rotation axis running through the midpoint of the bond ring O–C5 (crystal structure numbering O39–C23; see
Table 1 for quantification using Cremer & Pople puckering parameters [
27]). The iduronic ring conformation is therefore clearly different from the conformation reported by Johnson et al. [
5], who found a skew-boat conformation (related Cremer & Pople puckering parameters are included in
Table 1). A comparison of the conformations of the iduronic acid G ring in both structures is given in
Figure 6. Interestingly, in the co-crystal of heparin and teichoic acid α-glycosyl transferase the iduronic acid adopts a chair conformation according to the coordinates reported by Sobhanifar et al. ([
8], PDB entry 4X7R), related Cremer & Pople puckering parameters are also included in
Table 1.
The overall conformation of the pentasaccharide can be described in terms of the torsion angles of the glycosidic links between the carbohydrate rings. An alternative description uses the dihedral angles between least-squares planes fitted through the ring atoms. Numerical data for both descriptions are given in
Table 2.
Table 2 contains the same set of descriptors for the pentasaccharide molecules co-crystallized with antithrombin S195A, platelet factor 4 and a glycosyl transerase as well as for the structure in solution determined by NMR. A comparison of the free and co-crystallized pentasaccharide shows only relatively small differences when the glycosidic links between adjacent sugar rings are considered. In most cases the range of torsion angles observed is approximately 30 °C. However, in combination with the conformational change of the iduronic acid, these differences add up to a substantial difference for the overall conformation of the pentasaccharide. This can be convincingly illustrated by the angle between the least-squares planes fitted through rings D and H, which shows a difference of almost 60 °C. This difference in dihedral angles (which have a defined range of 0–90 °C) corresponds to the mean planes of rings D and H being almost parallel in the crystal structure of Fondaparinux while they are almost perpendicular in complex 2GD4.
The intramolecular hydrogen bond between rings D and E, found in the Fondaparinux crystal structure, is also present in the co-crystal with antithrombin. In the latter, an even shorter donor-acceptor distance of 2.98 Å is found, suggesting a reasonably strong hydrogen bond. The intramolecular hydrogen bond between residues F and G (iduronic acid), found in the single-crystal structure of Fondaparinux, is not present in the pentasaccharide unit in the co-crystal. The conformational change of the iduronic acid to a twist-boat has moved the accepting sulfate out of reach of the sulfamido group. In the co-crystal, this sulfamido group rotated around the bond to which it is attached to the sugar ring so that an intramolecular hydrogen bond to the neighbouring sulfate group attached to the same sugar ring can be formed (donor-acceptor is 2.98 Å, also suggesting the presence of a reasonably strong intramolecular hydrogen bond).
The sulfamido groups play an important role in stabilizing the pentasaccharide molecule in a relatively linear conformation by forming two N-H...O intramolecular hydrogen bonds. The NH hydrogen of the sulfamido group in carbohydrate residue F is involved in an interresidue hydrogen bond to an oxygen of the 2-sulfate group residue G, as shown in
Figure 5. This hydrogen bond is also found in solution [
15]. Previously, in solution, an intraresidue hydrogen bond between the NH hydrogen of the sulfamido group in carbohydrate residue F and the adjacent 3-sulfo group [
15,
28] was found. This hydrogen bond is not observed in the crystal, possibly as a consequence of the different conformation of the iduronic acid G discussed above. Geometric details are given in
Table 3.
Besides the items listed in
Table 3, the crystal structure displays numerous hydrogen bonds, which further strengthen the infinite three-dimensional network formed by the pentasaccharide ions, sodium ions and water molecules. As can be expected in view of their acidic nature, all SO
3H groups are deprotonated, which is supported by the observed equality of the S–O bond lengths.
Interestingly, the geometry of the carboxylic acid group bonded to ring E strongly suggests a protonated, neutral acid group (
d(C12–O15) = 1.09(2) Å,
d(C12–O16) = 1.40(3) Å), in spite of the fact that the crystallization was carried out at neutral pH. Due to high anisotropic displacement parameters, the C–O distances appear shortened. The anisotropicity is also reflected by the accuracy of these bond lengths, which is significantly lower than that of similar bond lengths in other sections of the pentasaccharide anion. The presence of a hydrogen bond acceptor at hydrogen bonding distance from O16 (see
Table 3) supports the model of a protonated acid group. However, the unfavorable value of the
D–H
…A angle (131 deg) does not suggest this is a particularly strong hydrogen bond.
The acid group bonded to ring G (iduronic acid) is deprotonated as expected, as is clearly shown by the similar lengths of the C–O bonds: d(C24–O37) = 1.242(11) Å and d(C24–O38) = 1.267(10) Å.
Based on the covalent angles between their substituents, the nitrogen atoms N1, N2 and N3 can be considered sp3 hybridized, which is common for this type of group. The coordinates of the hydrogen atoms bonded to the nitrogen atoms were derived from the availability of hydrogen bond acceptors at reasonable distances from the nitrogen donor atoms (see “Crystal Structure Determination and Refinement”).
Absolute Configuration
The Flack
x-parameter and its standard uncertainty
u(
x) can be used to assess absolute structure and absolute configuration [
29,
30]. A structure determination is considered to have strong inversion-distinguishing power if
u(
x) < 0.04. If the enantiopurity of a sample is certain, a less strict criterion is applied. When
u(
x) < 0.08, the experiment is considered to have enantiopure sufficient inversion-distinguishing power. If one of these criteria is met, a numerical value of
x that lies within statistical fluctuation of zero, i.e., |
x| < 2
u(
x), assures a valid absolute structure (and absolute configuration) determination from a single crystal which is not twinned by inversion. The cited criteria are taken from Flack and Bernardinelli [
31].
In this study, a value of −0.02(11) was found by classical fit to all intensities, which does not satisfy the criterion for enantiopure sufficient inversion-distinguishing power. However, as argued by Parsons et al. [
32] anomalous dispersion differences can be obscured by other effects, such as absorption and extinction. If these effects are filtered out as proposed by these authors, and
x-value of 0.017(14) is obtained, we can classify the current structure determination as having strong inversion-distinguishing power.
As a final alternative to assess the absolute configuration, the method of Hooft et al. [
33] was considered. Using Bayesian statistics, these authors offer a method that gives reliable absolute structure determinations even in cases where only weak anomalous scatterers are present. According to this method, the probability that the absolute configuration was determined correctly is calculated to be 1.00, based on the assumption of an enantiopure sample. In case the measured crystal is a racemic twin, the twin ratio, expressed as the
y-parameter, comparable to Flack’s
x-parameter, amounts to −0.002(14), further confirming the correct assignment of the absolute configuration of the enantiopure sample.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}