Inhibitors of the Detoxifying Enzyme of the Phytoalexin Brassinin Based on Quinoline and Isoquinoline Scaffolds



Abstract
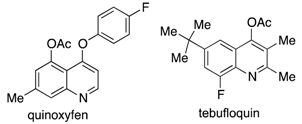
:1. Introduction
2. Results and Discussion
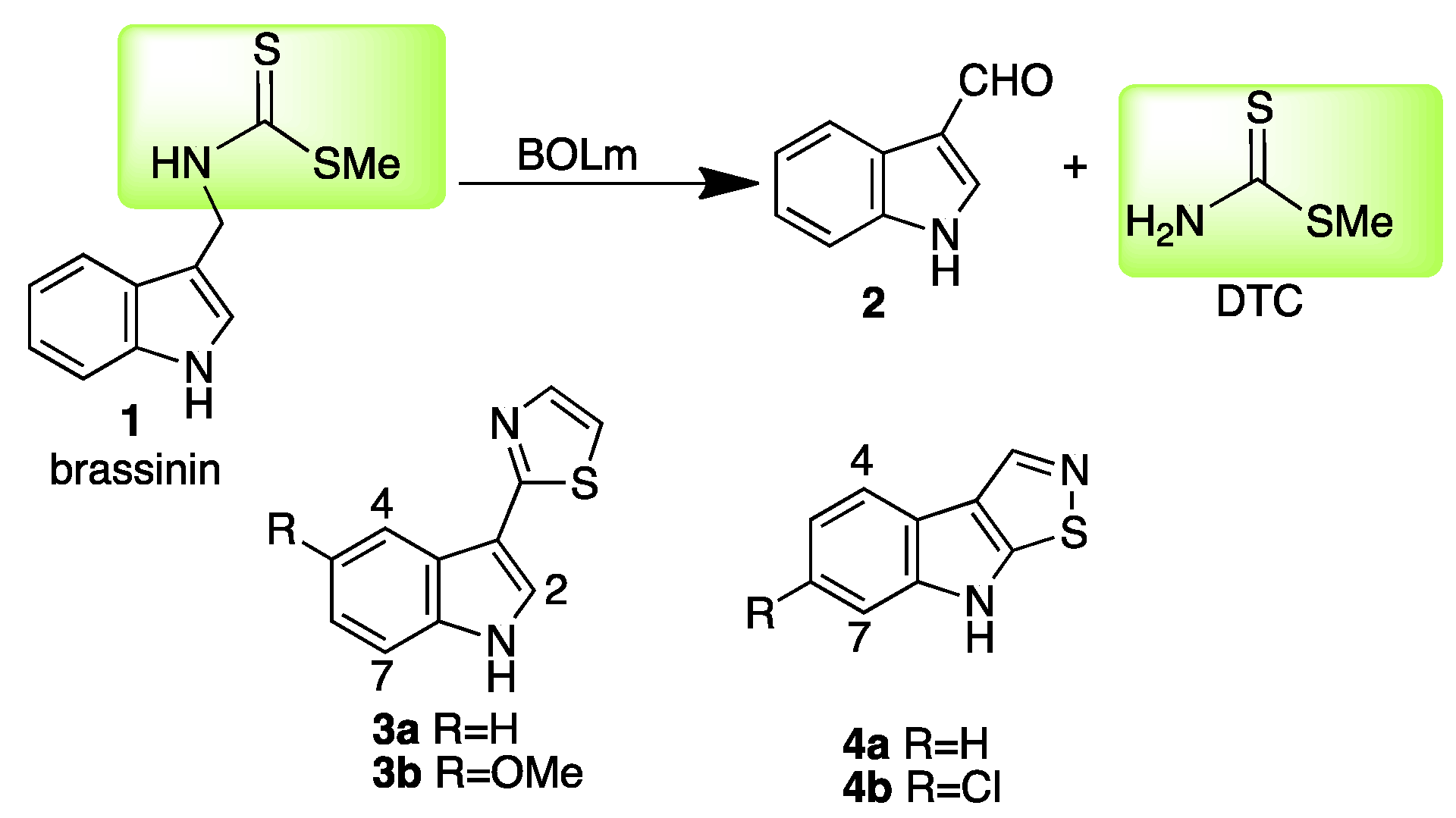
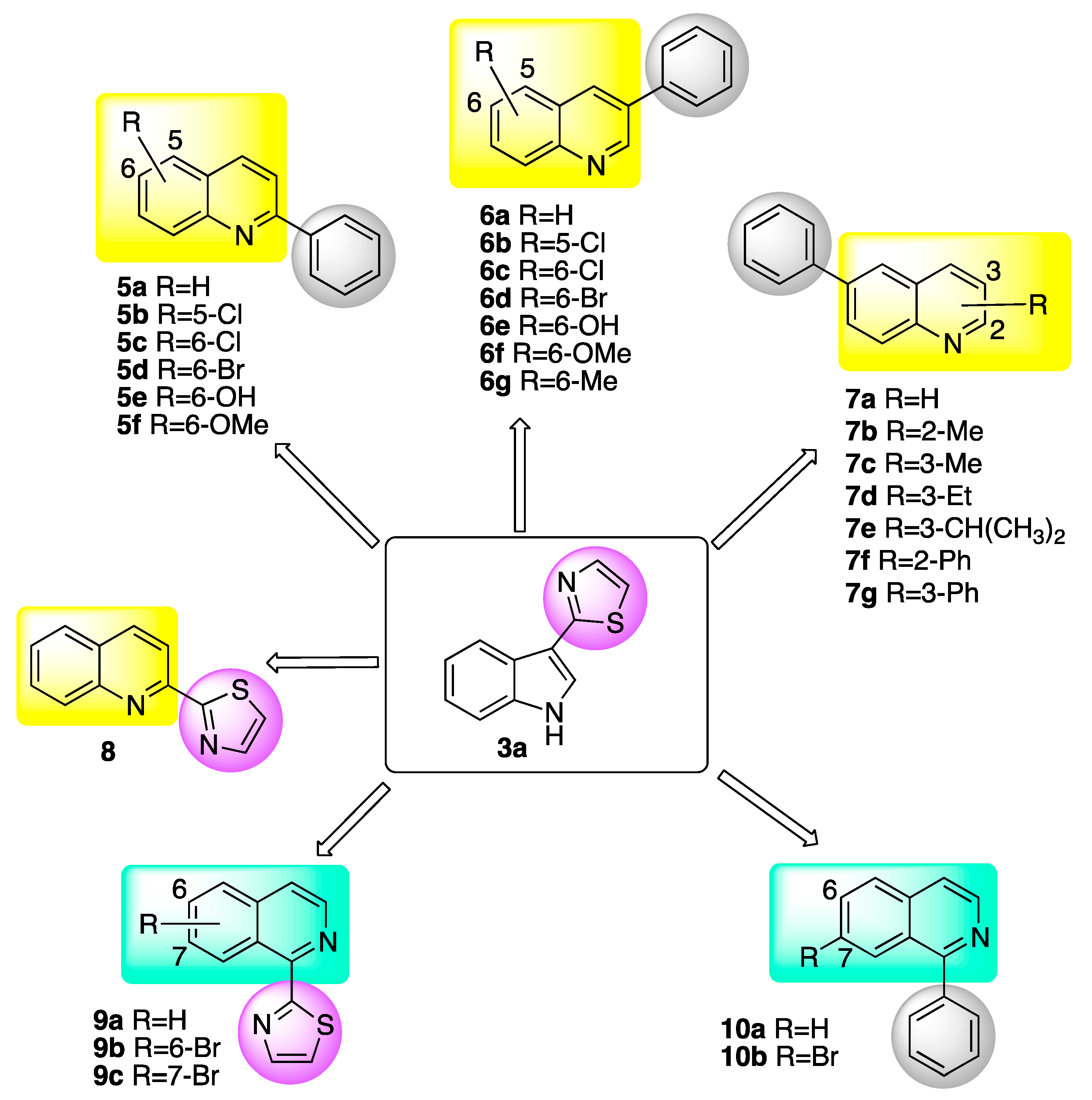
2.1. Design and Synthesis of Potential Inhibitors of BOLm
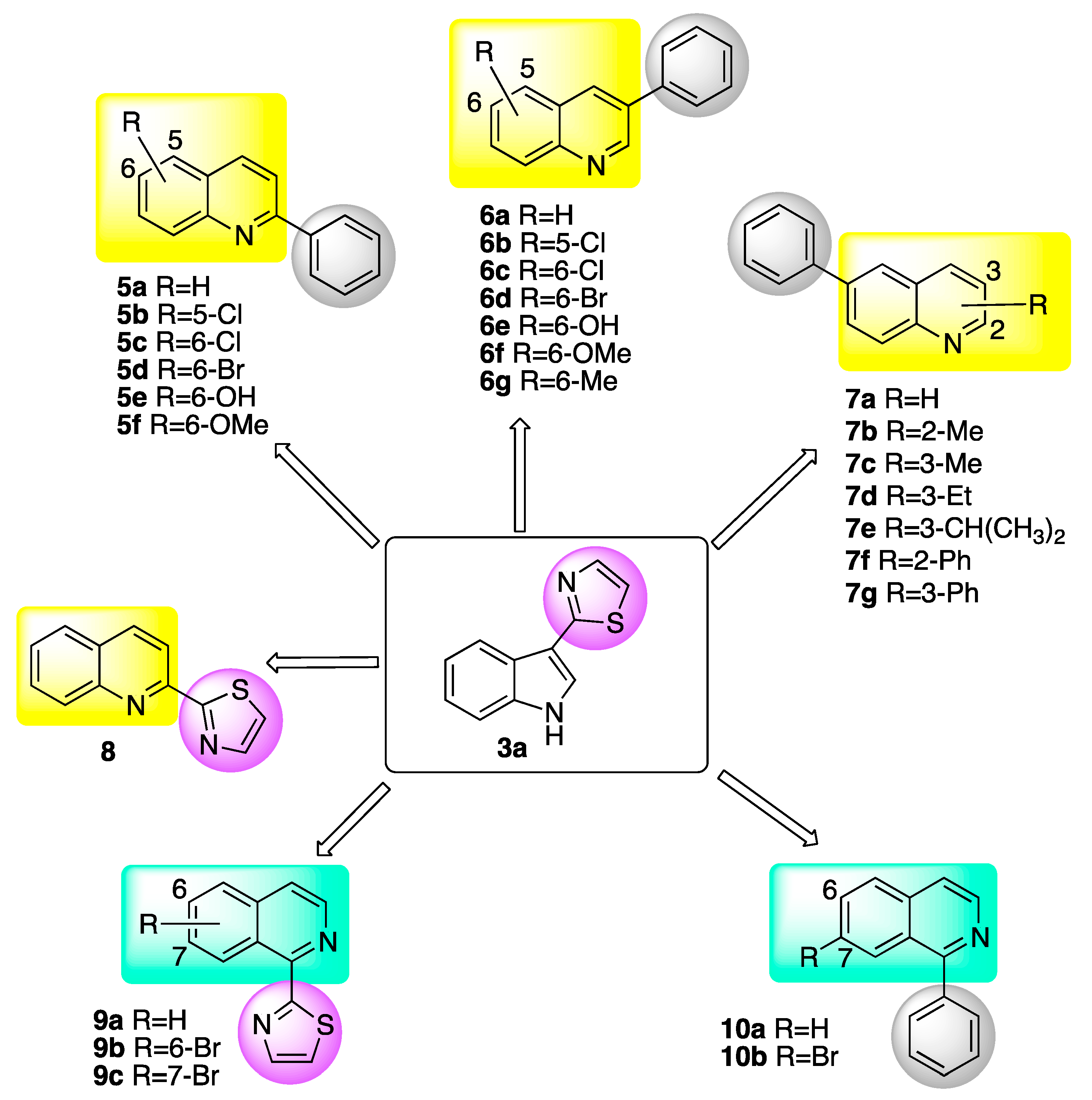
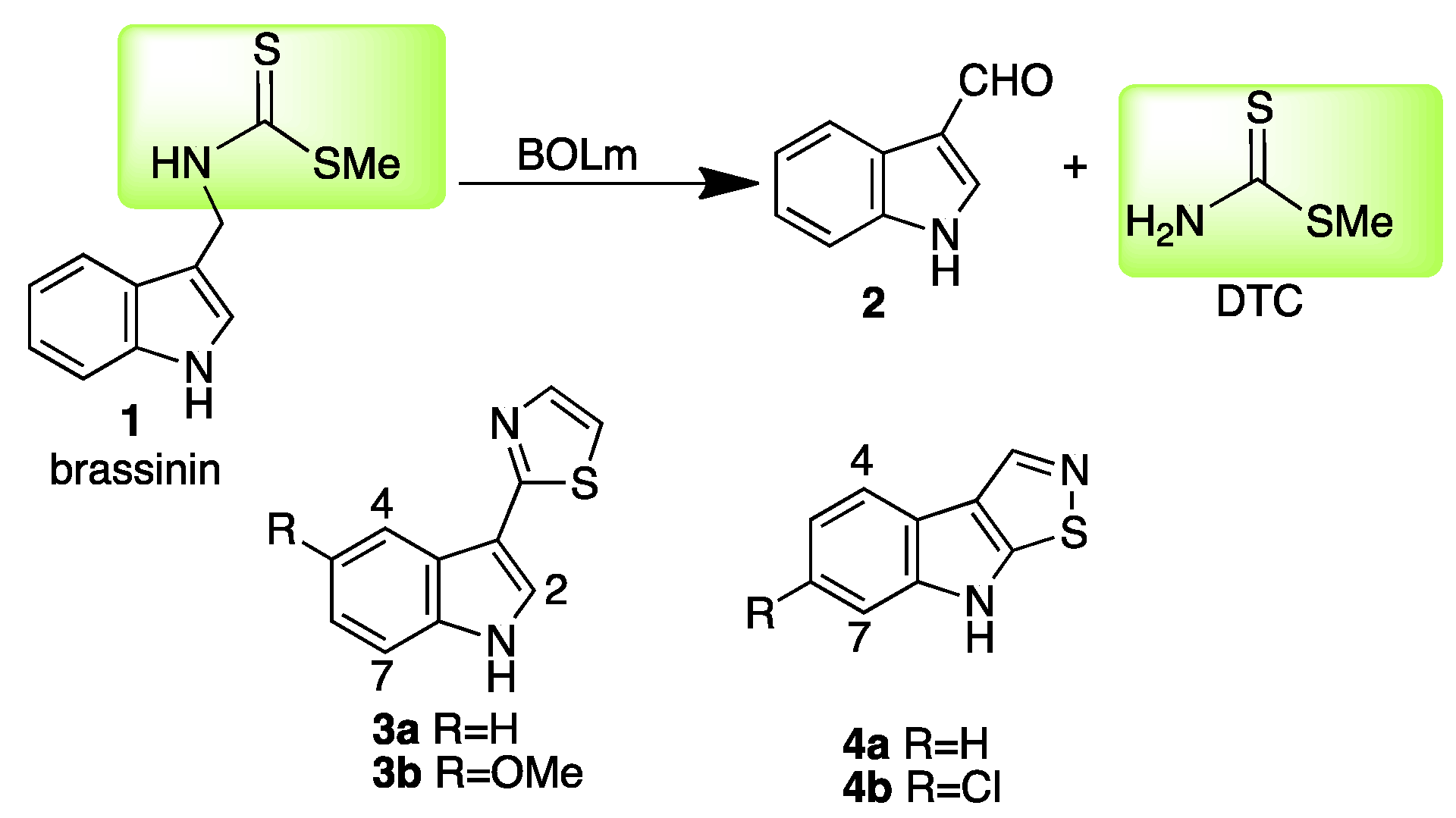
2.1.1. Quinolines
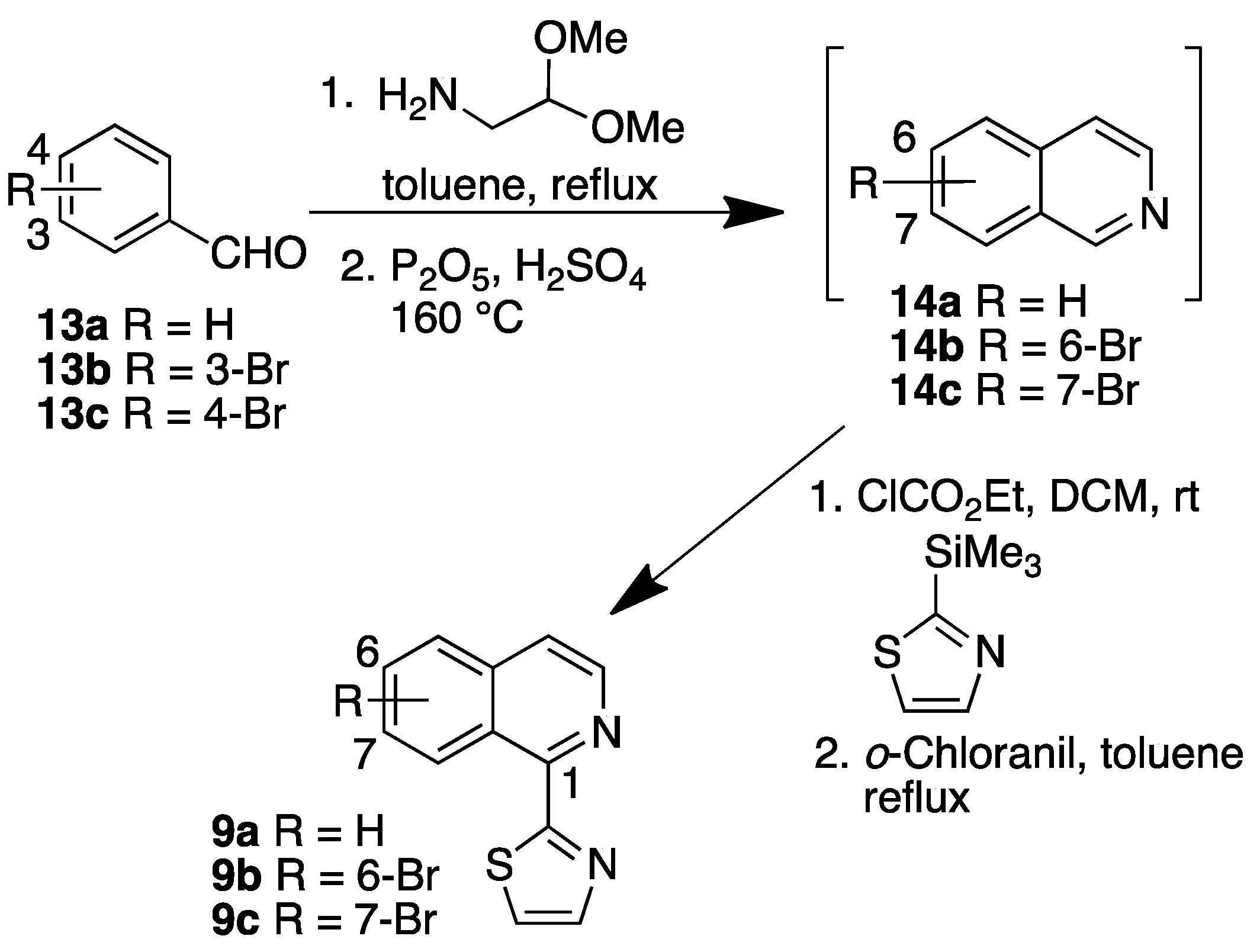
2.1.2. Isoquinolines
2.2. Antifungal Activity
2.3. Inhibition of Brassinin Oxidase Activity
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Phenylquinolines 5a–7g
General Procedure
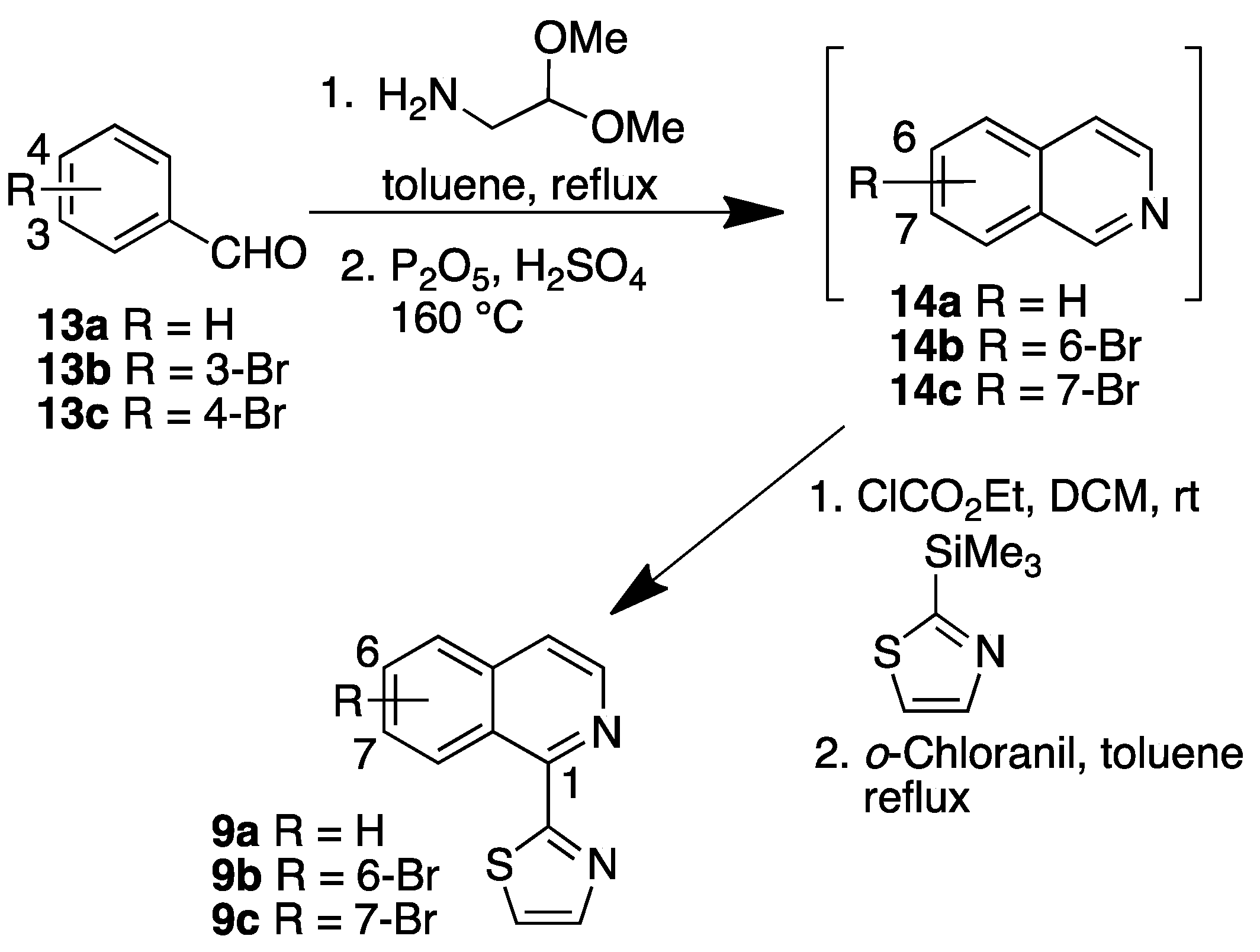
3.2.2. Synthesis Isoquinolines 9b, 9c and 10b
Isoquinolines 9b and 9c
3.3. Antifungal Activity
3.4. Fungal Cultures, Preparation of Cell-Free Protein Extracts, and BOLm Activity
Determination of BOLm Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pedras, M.S.C.; Minic, Z.; Jha, M. Brassinin oxidase, a fungal detoxifying enzyme to overcome a plant defense—Purification, characterization and inhibition. FEBS J. 2008, 275, 3691–3705. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Minic, Z.; Sarma-Mamillapalle, V.K. Brassinin oxidase mediated transformation of the phytoalexin brassinin: Structure of the elusive co-product, deuterium isotope effect and stereoselectivity. Bioorg. Med. Chem. 2011, 19, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Yaya, E.E.; Glawischnig, E. The phytoalexins from cultivated and wild crucifers: Chemistry and biology. Nat. Prod. Rep. 2011, 28, 1381–1405. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C. Protecting plants against fungal diseases. Can. Chem. News 2005, 57, 16–17. [Google Scholar]
- Pedras, M.S.C.; Minic, Z.; Sarma-Mamillapalle, V.K.; Suchy, M. Discovery of inhibitors of brassinin oxidase based on the scaffolds of the phytoalexins brassilexin and wasalexin. Bioorg. Med. Chem. 2010, 18, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Minic, Z.; Sarma-Mamillapalle, V.K. Synthetic inhibitors of the fungal detoxifying enzyme brassinin oxidase based on the phytoalexin camalexin scaffold. J. Agric. Food Chem. 2009, 57, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Abdoli, A. Pathogen inactivation of cruciferous phytoalexins: Detoxification reactions, enzymes and inhibitors. RSC Adv. 2017, 38, 23633–23646. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Jha, M. Toward the control of Leptosphaeria maculans: Design, syntheses, biological activity, and metabolism of potential detoxification inhibitors of the crucifer phytoalexin brassinin. Bioorg. Med. Chem. 2006, 14, 4958–4979. [Google Scholar] [CrossRef] [PubMed]
- Commercial Agriculture Fungicides are Classified According to Their Target Sites by the International Fungicide Resistance Action Committee (FRAC), cf. Available online: http://www.frac.info/docs/default-source/publications/frac-code-list/frac-code-list-2016.pdf?sfvrsn=2 (accessed on 22 April 2017).
- Lamberth, C.; Walter, H.; Kessabi, F.M.; Quaranta, L.; Beaudegnies, R.; Trah, S.; Jeanguenat, A.; Cederbaum, F. The significance of organosulfur compounds in crop protection: Current examples from fungicide research. Phosphorus Sulfur 2015, 190, 1225–1235. [Google Scholar] [CrossRef]
- Lamberth, C.; Kessabi, F.M.; Beaudegnies, R.; Quaranta, L.; Trah, S.; Berthon, G.; Cederbaum, F.; Knauf-Beiter, G.; Grasso, V.; Bieri, S.; et al. Synthesis and fungicidal activity of quinolin-6-yloxyacetamides, a novel class of tubulin polymerization inhibitors. Bioorg. Med. Chem. 2014, 22, 3922–3930. [Google Scholar] [CrossRef] [PubMed]
- Ayer, W.A.; Peter, A.C.; Ma, Y.; Miao, S. Synthesis of camalexin and related phytoalexins. Tetrahedron 1992, 48, 2919–2924. [Google Scholar] [CrossRef]
- Dondoni, A.; Dall’Occo, T.; Galliani, G.; Mastellari, A.; Medici, A. Addition of 2-silylazoles to heteroaryl cations. Synthesis of unsymetrical azadiaryls. Tetrahedron Lett. 1984, 25, 3637–3640. [Google Scholar] [CrossRef]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Synthesis of quinolines: A green perspective. ACS Sustain. Chem. Eng. 2016, 4, 4064–4078. [Google Scholar] [CrossRef]
- Chelucci, G.; Porcheddu, A. Synthesis of quinolines via a metal-catalyzed dehydrogenative N-heterocyclization. Chem. Rec. 2017, 17, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24476. [Google Scholar] [CrossRef]
- Cieslik, W.; Serda, M.; Kurczyk, A.; Musiol, R. Microwave assisted synthesis of monoazanaphthalene scaffolds. Curr. Org. Chem. 2013, 17, 491–503. [Google Scholar] [CrossRef]
- Li, W.Y.; Xiong, X.Q.; Zhao, D.M.; Shi, Y.F.; Yang, Z.H.; Yu, C.; Fan, P.W.; Cheng, M.S.; Shen, J.K. Quinoline-3-carboxamide derivatives as potential cholesteryl ester transfer protein inhibitors. Molecules 2012, 17, 5497–5507. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Ahmed, E.; Chen, X.; Cox, M.; Crew, A.P.; Dong, H.Q.; Jin, M.; Ma, L.; Panicker, B.; Siu, K.W.; et al. A highly effective one-pot synthesis of quinolines from o-nitroarylcarbaldehydes. Org. Biomol. Chem. 2007, 5, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Duckett, D.; Chen, W.; Habel, J.; Ling, Y.Y.; LoGrasso, P.; Kamenecka, T.M. 3,5-Disubstituted quinolines as novel c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6378–6382. [Google Scholar] [CrossRef] [PubMed]
- Czakó, B.; Kürti, L.; Mammoto, A.; Ingber, D.E.; Corey, E.J. Discovery of potent and practical antiangiogenic agents inspired by cortistatin A. J. Am. Chem. Soc. 2009, 131, 9014–9019. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.J.; Schade, M.A.; Cheemala, M.N.; Wirth, S.; Guevara, S.A.; Cahiez, G.; Knochel, P. Cobalt-catalyzed cross-coupling reactions of heterocyclic chlorides with arylmagnesium halides and of polyfunctionalized arylcopper reagents with aryl bromides, chlorides, fluorides and tosylates. Synthesis 2006, 21, 3547–3574. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Abdoli, A. Metabolism of the phytoalexins camalexins, their bioisosteres and analogues in the plant pathogenic fungus Alternaria brassicicola. Bioorg. Med. Chem. 2013, 21, 4541–4549. [Google Scholar] [CrossRef] [PubMed]
- Movassaghi, M.; Hill, M.D. Synthesis of substituted pyridine derivatives via the ruthenium-catalyzed cycloisomerization of 3-azadienynes. J. Am. Chem. Soc. 2006, 128, 4592–4593. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Huang, H.; Li, Y.; Chen, H.; Jiang, H. Palladium-catalyzed sequential formation of C-C bonds: Efficient assembly of 2-substituted and 2,3-disubstituted quinolines. Angew. Chem. Int. Ed. Engl. 2012, 51, 7292–7296. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Rong, L.; Shi, C.; Zhuang, Q.; Wang, X.; Tu, S.; Hu, H. Low-valent titanium reagent-promoted intramolecular reductive coupling reactions of ketomalononitriles: A facile synthesis of benzo[4,5]indene, acridine and quinoline derivatives. Synthesis 2005, 5, 717–724. [Google Scholar] [CrossRef]
- Huo, Z.; Gridnev, I.D.; Yamamoto, Y. A method for the synthesis of substituted quinolines via electrophilic cyclization of 1-azido-2-(2-propynyl)benzene. J. Org. Chem. 2010, 75, 1266–1270. [Google Scholar] [CrossRef] [PubMed]
- Meléndez Gómez, C.M.; Kouznetsov, V.V.; Sortino, M.A.; Álvarez, S.L.; Zacchino, S.A. In vitro antifungal activity of polyfunctionalized 2-(hetero)arylquinolines prepared through imino Diels-Alder reactions. Bioorg. Med. Chem. 2008, 16, 7908–7920. [Google Scholar] [CrossRef] [PubMed]
- Colomb, J.; Billard, T. Palladium-catalyzed desulfitative arylation of 3-haloquinolines with arylsulfinates. Tetrahedron Lett. 2013, 54, 1471–1474. [Google Scholar] [CrossRef]
- Wang, Y.; Xin, X.; Liang, Y.; Lin, Y.; Zhang, R.; Dong, D. A facile and efficient one-pot synthesis of substituted quinolines from α-arylamino ketones under vilsmeier conditions. Eur. J. Org. Chem. 2009, 24, 4165–4169. [Google Scholar] [CrossRef]
- Saunthwal, R.K.; Patel, M.; Verma, A.K. Regioselective synthesis of C-3-functionalized quinolines via Hetero-Diels-Alder cycloaddition of azadienes with terminal alkynes. J. Org. Chem. 2016, 81, 6563–6572. [Google Scholar] [CrossRef] [PubMed]
- Monrad, R.N.; Madsen, R. Ruthenium-catalysed synthesis of 2- and 3-substituted quinolines from anilines and 1,3-diols. Org. Biomol. Chem. 2011, 9, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Thapa, S.; Gurung, S.K.; Pike, R.A.S.; Giri, R. General copper-catalyzed coupling of alkyl-, aryl-, and alkynylaluminum reagents with organohalides. J. Org. Chem. 2016, 81, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Mongin, F.; Mojovic, L.; Guillamet, B.; Trécourt, F.; Quéguiner, G. Cross-coupling reactions of phenylmagnesium halides with fluoroazines and fluorodiazines. J. Org. Chem. 2002, 67, 8991–8994. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; An, Y.; Peng, J.; Wu, W.; Jiang, H. Palladium-catalyzed allylic C-H oxidative annulation for assembly of functionalized 2-substituted quinoline derivatives. J. Org. Chem. 2016, 81, 12189–12196. [Google Scholar] [CrossRef] [PubMed]
- Larivée, A.; Mousseau, J.J.; Charette, A.B. Palladium-catalyzed direct C–H arylation of N-iminopyridinium ylides: Application to the synthesis of (±)-anabasine. J. Am. Chem. Soc. 2008, 130, 52–54. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
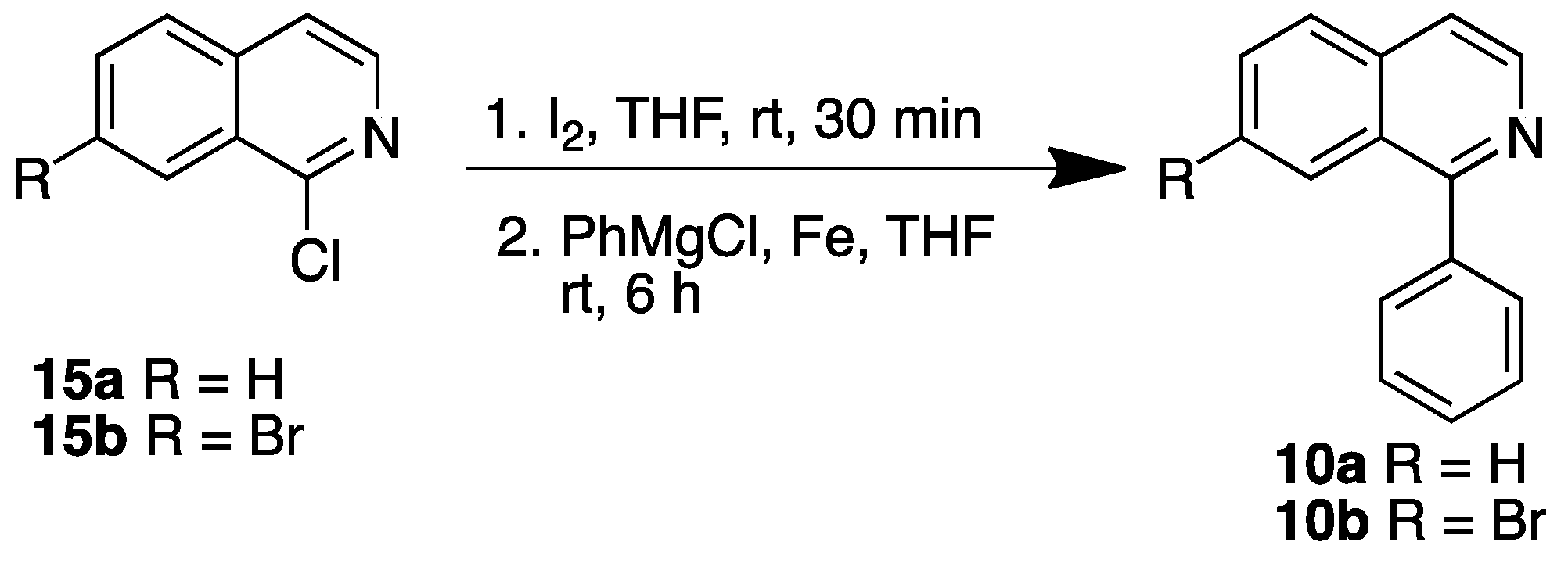

{kind=link}
{kind=link}
{kind=link}
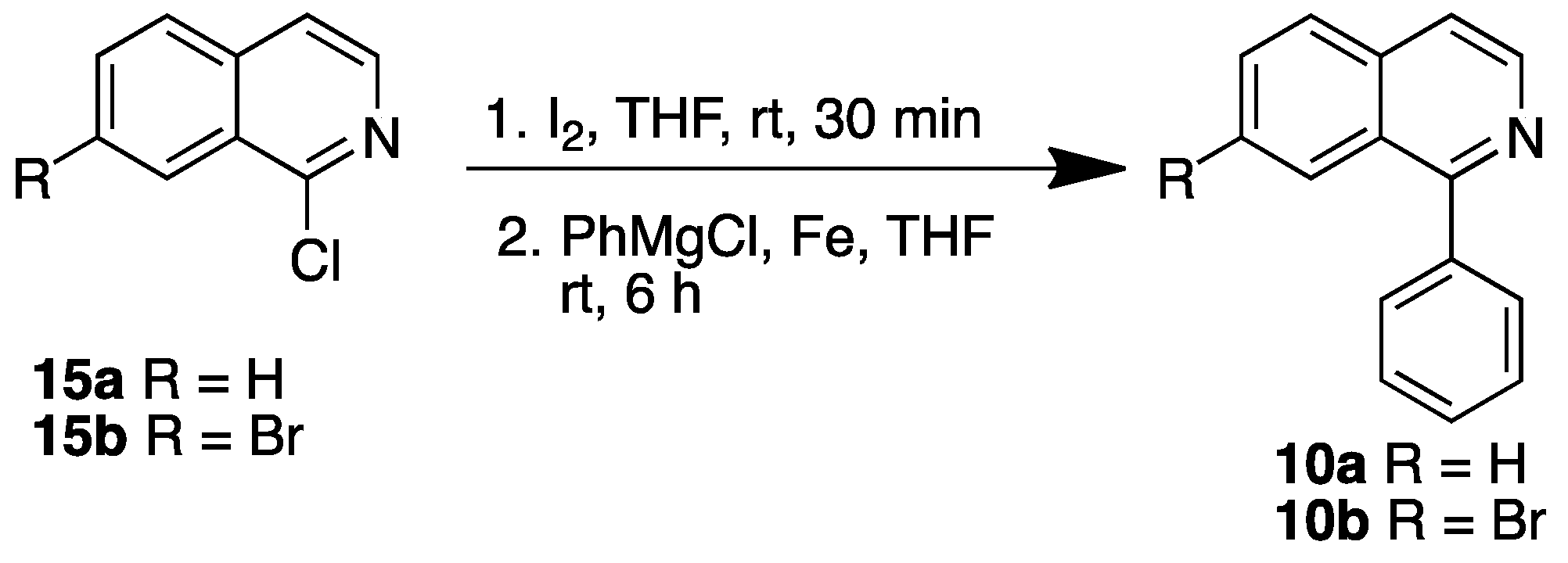{kind=link}
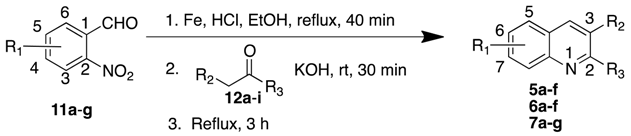
| Starting Material | R1 (C#) | 12 | R2 | R3 | Product | %Yield (Reported Yield) |
|---|---|---|---|---|---|---|
| 11a | H | 12a | H | Ph | 5a a | 83 (99) |
| 11b | 6-Cl | 12a | H | Ph | 5b b | 84 |
| 11c | 5-Cl | 12a | H | Ph | 5c b | 63 |
| 11d | 5-Br | 12a | H | Ph | 5d b | 64 |
| 11e | 5-OH | 12a | H | Ph | 5e b | 42 |
| 11f | 5-OMe | 12a | H | Ph | 5f b | 60 |
| 11a | H | 12b | Ph | H | 6a a | 63 (87) |
| 11b | 6-Cl | 12b | Ph | H | 6b b | 80 |
| 11c | 5-Cl | 12b | Ph | H | 6c b | 64 |
| 11d | 5-Br | 12b | Ph | H | 6d b | 36 |
| 11e | 5-OH | 12b | Ph | H | 6e c | 39 |
| 11f | 5-OMe | 12b | Ph | H | 6f b | 53 |
| 11g | 5-Ph | 12c | H | H | 7a b | 62 |
| 11g | 5-Ph | 12d | Me | H | 7b b | 52 |
| 11g | 5-Ph | 12e | H | Me | 7c c | 52 |
| 11g | 5-Ph | 12f | Et | H | 7d c | 82 |
| 11g | 5-Ph | 12g | i-Pro | H | 7e c | 69 |
| 11g | 5-Ph | 12h | Ph | H | 7f b | quantitative |
| 11g | 5-Ph | 12i | H | Ph | 7g c | 62 |
| Compound (#) | Inhibition ± SD (%) a | ||
|---|---|---|---|
| 0.50 mM | 0.20 mM | 0.10 mM | |
| Brassinin (1) | 50 ± 3 i,g | 27 ± 0 l | 14 ± 3 j |
| Camalexin (3a) | 100 ± 0 c | 41 ± 2 i,j | 24 ± 3 i |
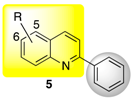
| 2-Phenylquinoline (5a) | 54 ± 2 h | 31 ± 4 k,l | 16 ± 5 j |
| 5-Chloro-2-phenylquinoline (5b) | 39 ± 2 l | 18 ± 4 m,n | 9 ± 2 k |
| 6-Chloro-2-phenylquinoline (5c) | 26 ± 4 m | 18 ± 5 m,n | 5 ± 2 l |
| 6-Bromo-2-phenylquinoline (5d) | 27 ± 4 m | 14 ± 4 n,o | 0 m |
| 6-Hydroxy-2-phenylquinoline (5e) | 68 ± 5 e,f | 42 ± 4 h,i,j | 32 ± 4 f,g,h |
| 6-Methoxy-2-phenylquinoline (5f) | 36 ± 4 l | 20 ± 4 m | 8 ± 2 k |
| 3-Phenylquinoline (6a) | 100 ± 0 c | 77 ± 4 d | 39 ± 7 f |
| 5-Chloro-3-phenylquinoline (6b) | 17 ± 3 n | 11 ± 2 o | 8 ± 4 k,l |
| 6-Chloro-3-phenylquinoline (6c) | 28 ± 3 m | 17 ± 3 m,n | 5 ± 2 l |
| 6-Bromo-3-phenylquinoline (6d) | 45 ± 2 k | 34 ± 2 k | 25 ± 0 i |
| 6-Hydroxy-3-phenylquinoline (6e) | 73 ± 5 e | 45 ± 4 h,i | 30 ± 4 g,h |
| 6-Methoxy-3-phenylquinoline (6f) | 100 ± 0 c | 68 ± 5 e | 55 ± 5 c |
| 6-Methyl-3-phenylquinoline (6g) | 69 ± 3 e,f | 61 ± 2 f | 47 ± 0 e |
| 6-Phenylquinoline (7a) | 64 ± 0 g | 55 ± 0 g | 30 ± 0 h |
| 2-Methyl-6-phenylquinoline (7b) | 68 ± 2 f | 62 ± 3 f | 48 ± 3 d,e |
| 3-Methyl-6-phenylquinoline (7c) | 74 ± 2 e | 67 ± 4 e | 53 ± 0 c |
| 3-Ethyl-6-phenylquinoline (7d) | 67 ± 3 f | 61 ± 3 f | 48 ± 3 d,e |
| 3-Isopropyl-6-phenylquinoline (7e) | 72 ± 3 e | 66 ± 3 e | 56 ± 3 c |
| 2,6-Diphenylquinoline (7f) | 49 ± 4 i,j,k | 40 ± 0 j | 33 ± 0 f,g |
| 3,6-Diphenylquinoline (7g) | 0 o | 0 p | 0 m |
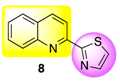
| 2-(2-Thiazolyl)quinoline (8) | 54 ± 3 h | 46 ± 4 h | 8 ± 3 k,l |
| 1-(2-Thiazolyl)isoquinoline (9a) | 100 ± 0 c | 100 ± 0 c | 52 ± 4 c,d |
| 6-Bromo-1-(2-thiazolyl)isoquinoline (9b) | 75 ± 6 d,e | 54 ± 3 g | 15 ± 4 j |
| 7-Bromo-1-(2-thiazolyl)isoquinoline (9c) | 82 ± 5 d | 63 ± 3 e,f | 25 ± 6 i |
| 1-Phenylisoquinoline (10a) | 54 ± 5 h,i | 32 ± 4 k | 30 ± 3 h |
| 7-Bromo-1-phenylisoquinoline (10b) | 47 ± 3 j,k | 31 ± 4 k,l | 12 ± 2 j |
| Potential Inhibitor (#) a | % Inhibition b | Potential Inhibitor (#) a | % Inhibition b | ||
|---|---|---|---|---|---|
| 0.10 mM | 0.30 mM | 0.10 mM | 0.30 mM | ||
 |  | ||||
| 30 ± 4 c | 53 ± 4 d | n.i. | n.i. | ||
 | 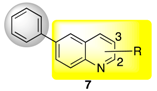 | ||||
| 5a R = H | n.i. | n.i. | 7a R = H | 16 ± 2 d,e | 36 ± 2 e,f |
| 5b R = 5-Cl | n.i. | n.i. | 7b R = 2-Me | n.i. | n.i. |
| 5c R = 6-Cl | n.i. | n.i. | 7c R = 3-Me | 19 ± 4 d | 33 ± 6 e,f |
| 5d R = 6-Br | n.i. | n.i. | 7d R = 3-Et | 30 ± 4 c | 64 ± 5 c |
| 5e R = 6-OH | 11 ± 3 e | 22 ± 1 h | 7e R = 3-CH(CH3)2 | 11 ± 3 e | 33 ± 5 e,f |
| 5f R = 6-OMe | n.i. | n.i. | 7f R = 2-Ph | 10 ± 2 e | 19 ± 1 h |
| 7g R = 3-Ph | 12 ± 1 e | 25 ± 1 g,h | |||
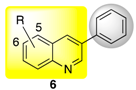 | 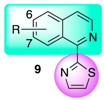 | ||||
| 6a R = H | 14 ± 2 d,e | 30 ± 4 f,g | 9a R = H | n.i. | n.i. |
| 6b R = 5-Cl | n.i. | n.i. | 9b R = 6-Br | n.i. | n.i. |
| 6c R = 6-Cl | n.i. | n.i. | 9c R = 7-Br | n.i. | n.i. |
| 6d R = 6-Br | n.i. | n.i. |  | ||
| 6e R = 6-OH | 20 ± 4 d | 34 ± 2 e,f | |||
| 6f R = 6-OMe | 19 ± 1 d | 40 ± 3 e | |||
| 6g R = 6-Me | 10 ± 3 e | 32 ± 3 f | |||
| 10a R = H | n.i. | n.i. | |||
| 10b R = 7-Br | n.i. | n.i. | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedras, M.S.C.; Abdoli, A.; Sarma-Mamillapalle, V.K. Inhibitors of the Detoxifying Enzyme of the Phytoalexin Brassinin Based on Quinoline and Isoquinoline Scaffolds. Molecules 2017, 22, 1345. https://doi.org/10.3390/molecules22081345
Pedras MSC, Abdoli A, Sarma-Mamillapalle VK. Inhibitors of the Detoxifying Enzyme of the Phytoalexin Brassinin Based on Quinoline and Isoquinoline Scaffolds. Molecules. 2017; 22(8):1345. https://doi.org/10.3390/molecules22081345
Chicago/Turabian StylePedras, M. Soledade C., Abbas Abdoli, and Vijay K. Sarma-Mamillapalle. 2017. "Inhibitors of the Detoxifying Enzyme of the Phytoalexin Brassinin Based on Quinoline and Isoquinoline Scaffolds" Molecules 22, no. 8: 1345. https://doi.org/10.3390/molecules22081345
APA StylePedras, M. S. C., Abdoli, A., & Sarma-Mamillapalle, V. K. (2017). Inhibitors of the Detoxifying Enzyme of the Phytoalexin Brassinin Based on Quinoline and Isoquinoline Scaffolds. Molecules, 22(8), 1345. https://doi.org/10.3390/molecules22081345