Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands



Abstract
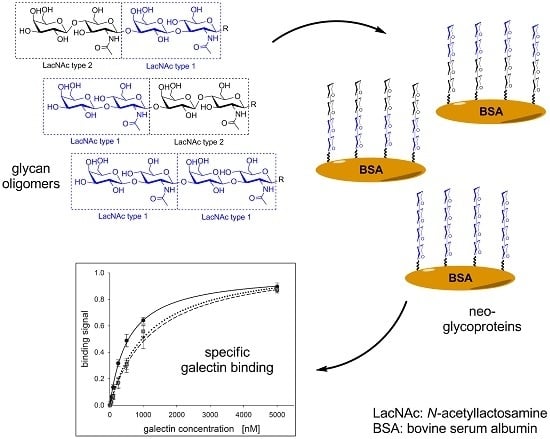
:
1. Introduction
2. Results and Discussion
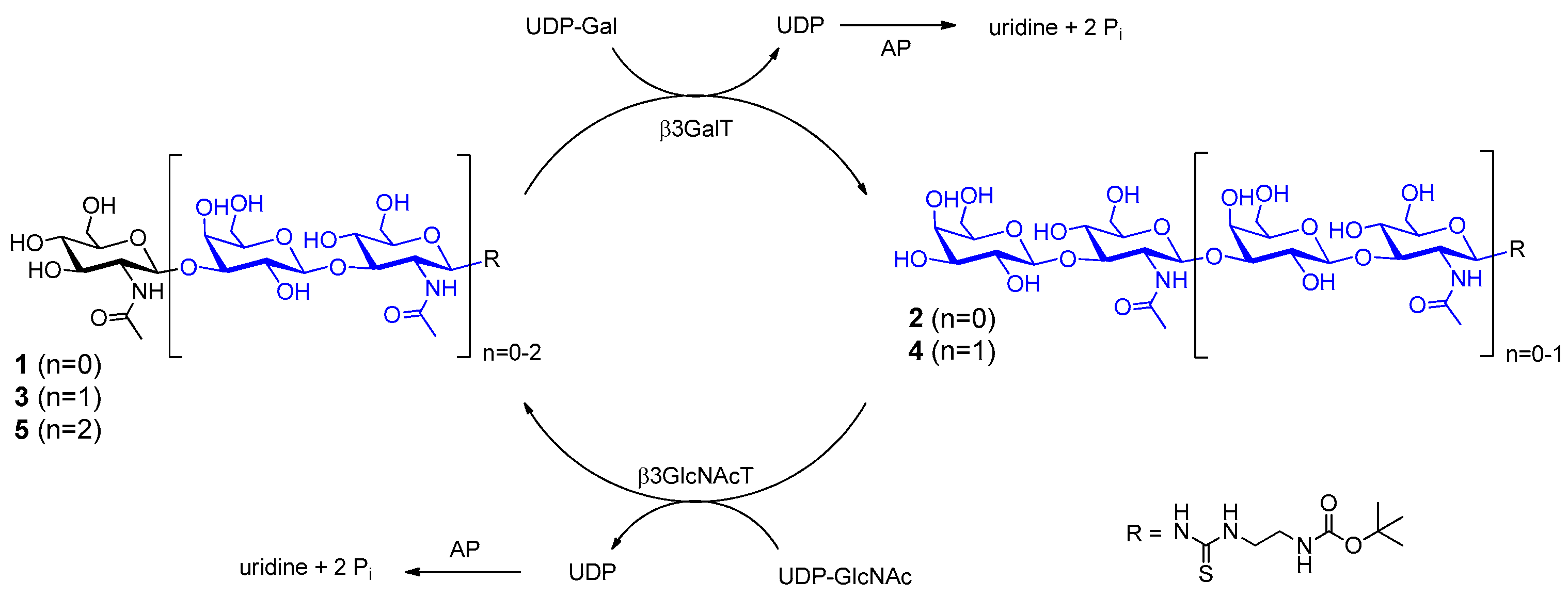
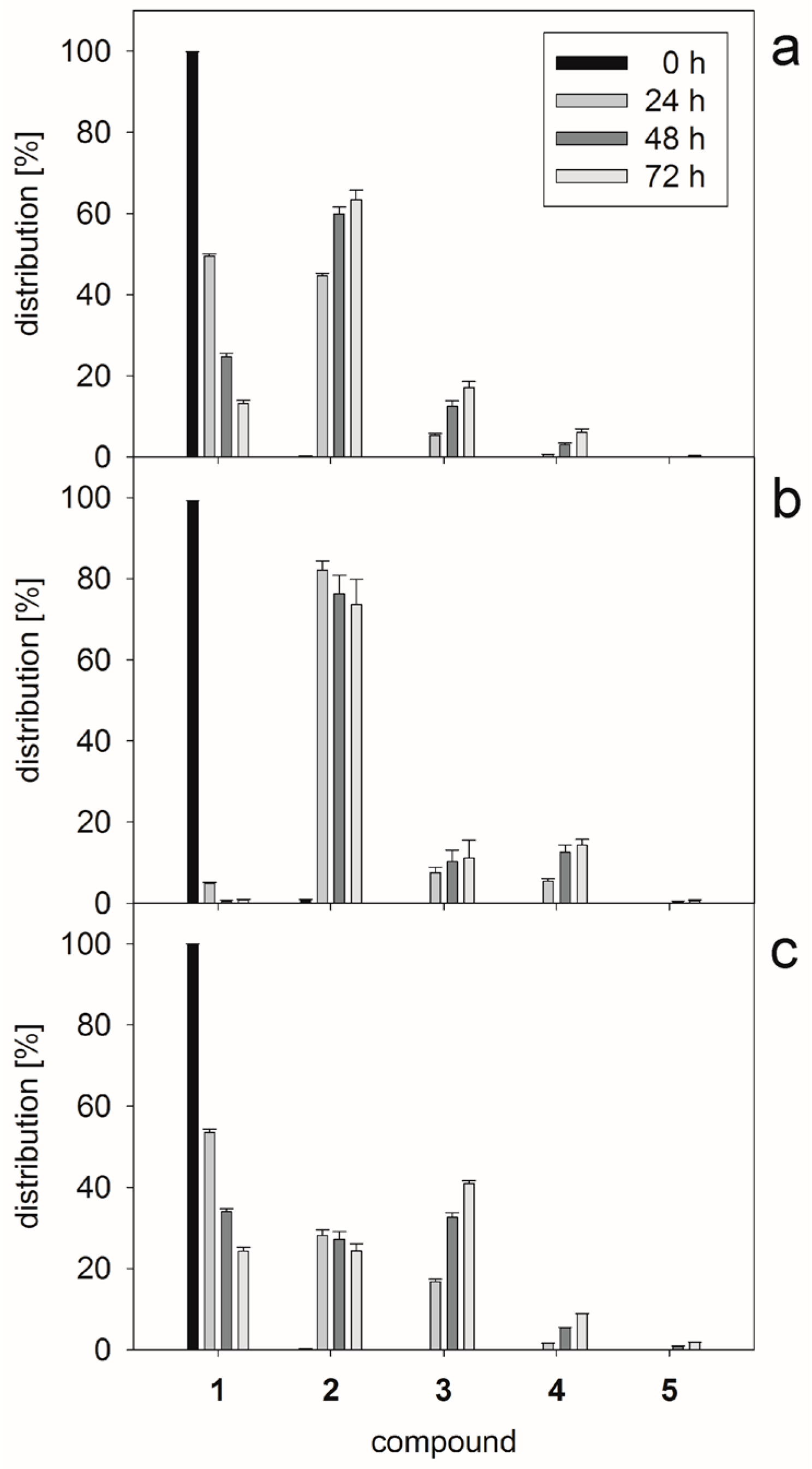
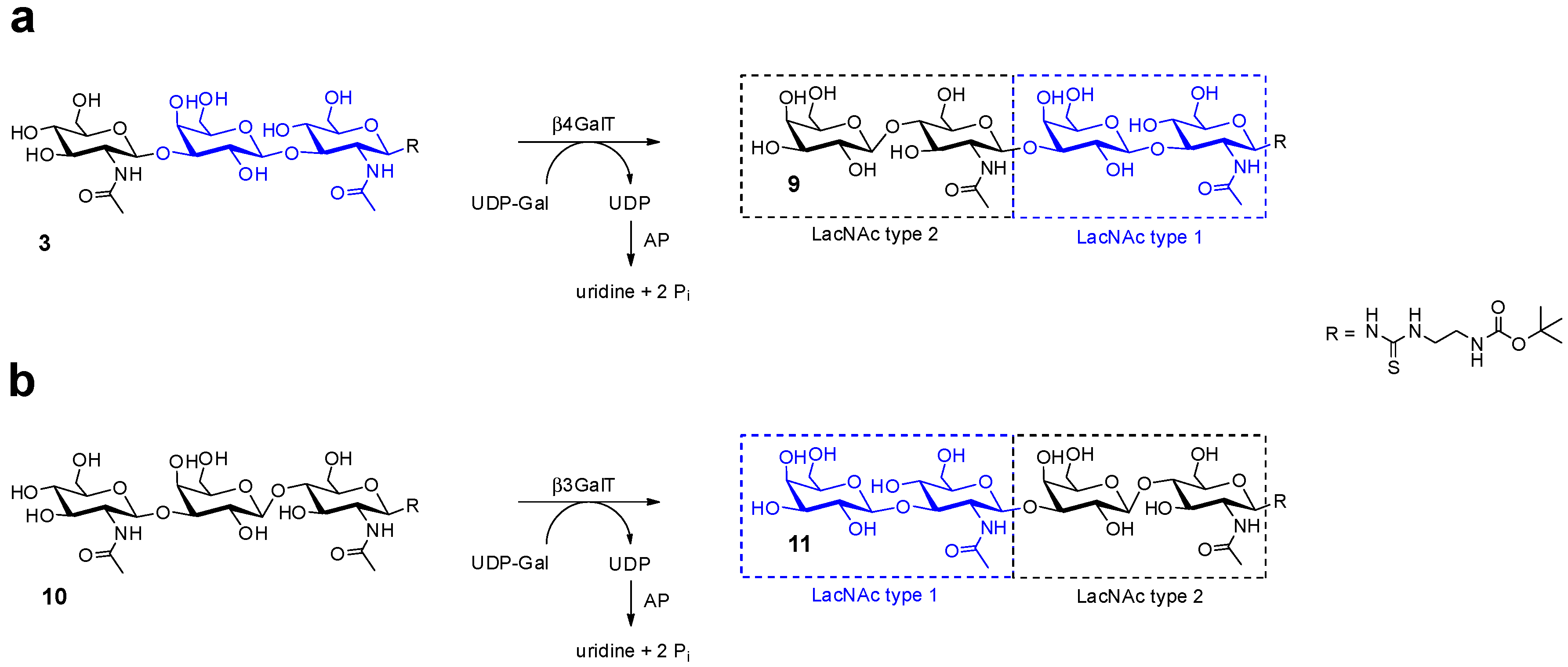
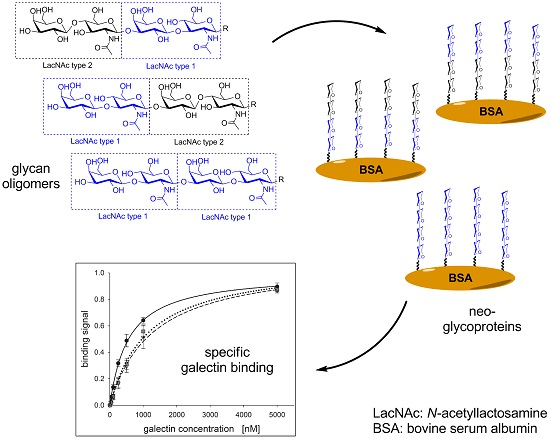
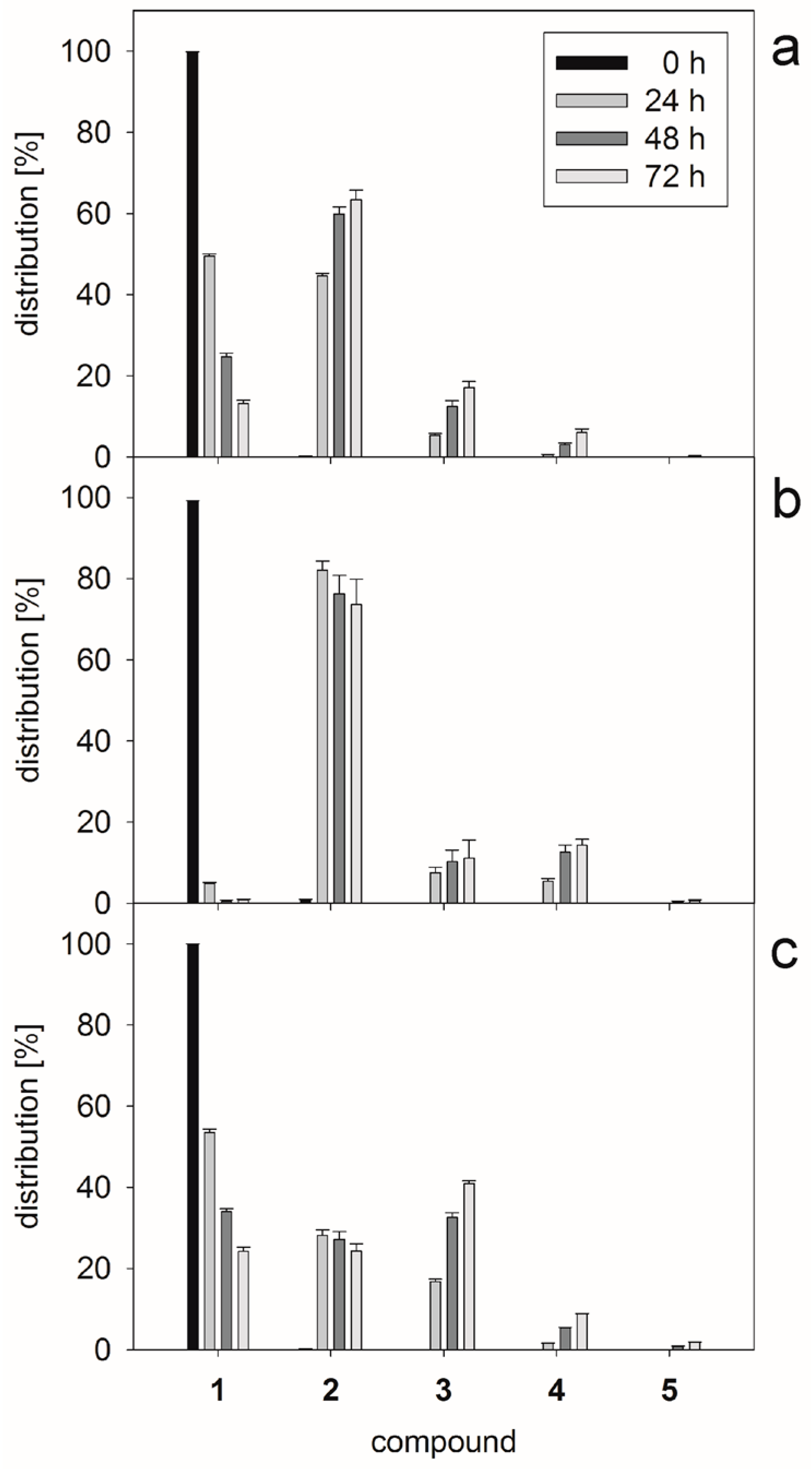
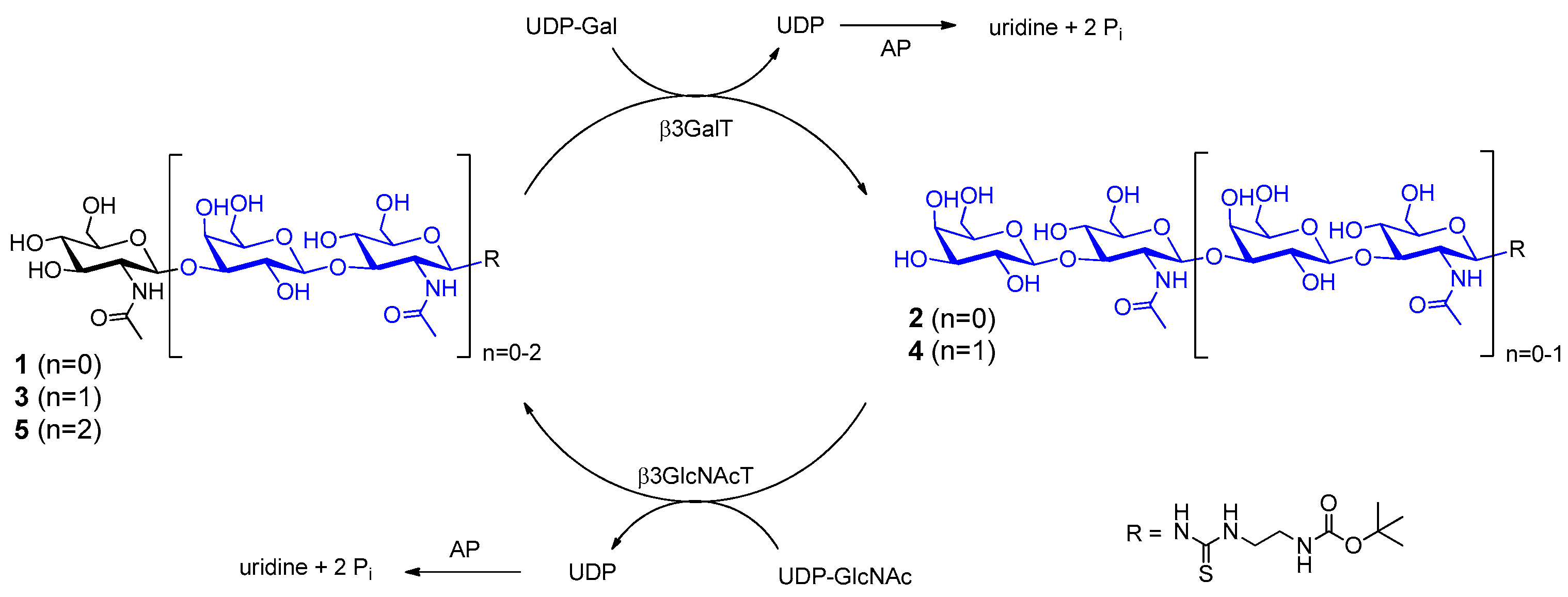
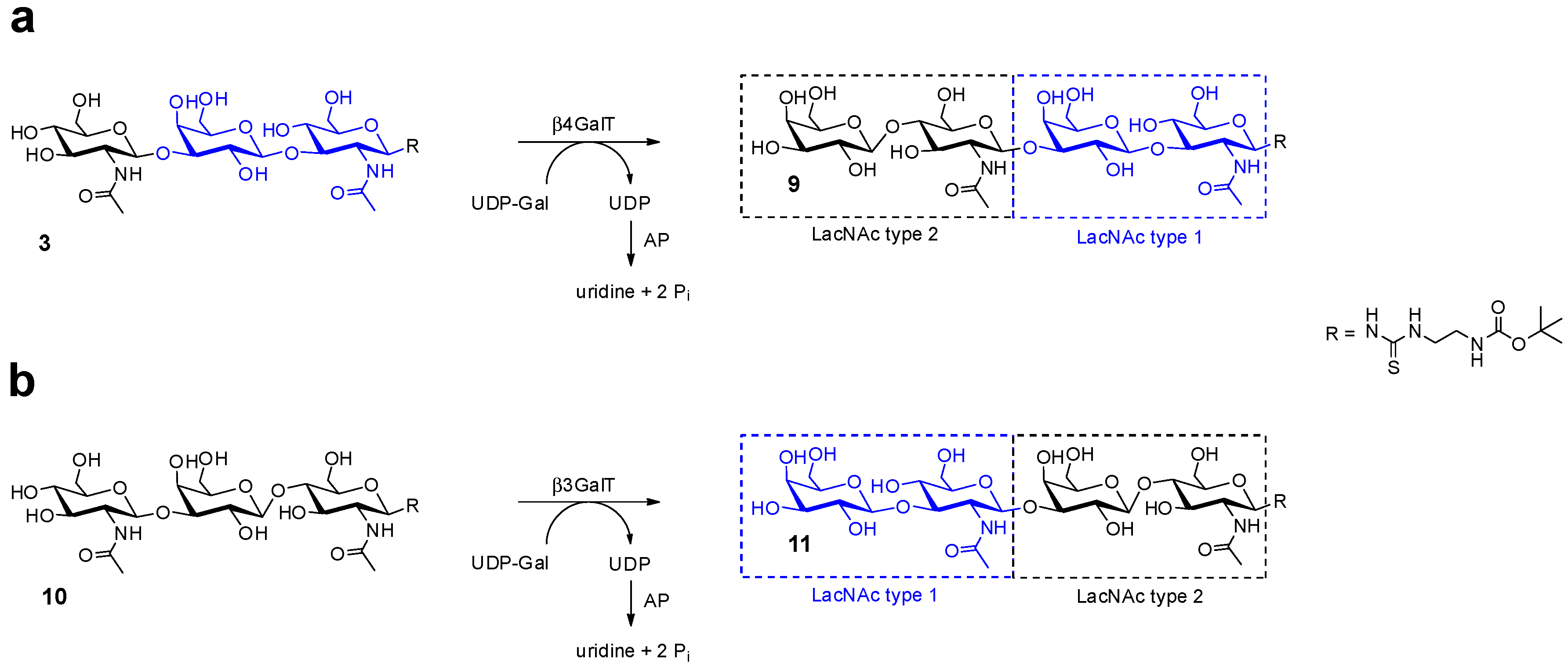
2.1. Glycan Synthesis
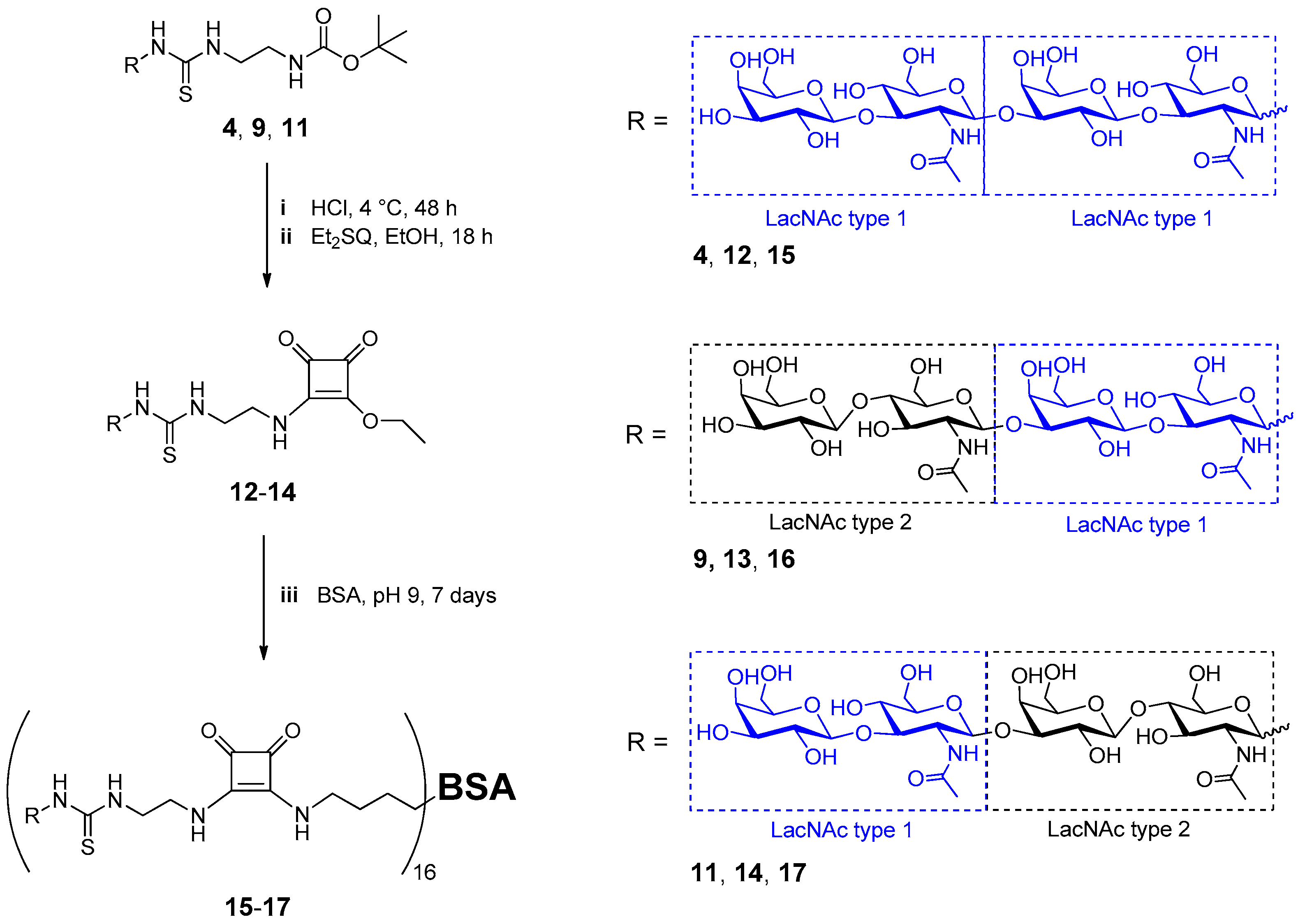
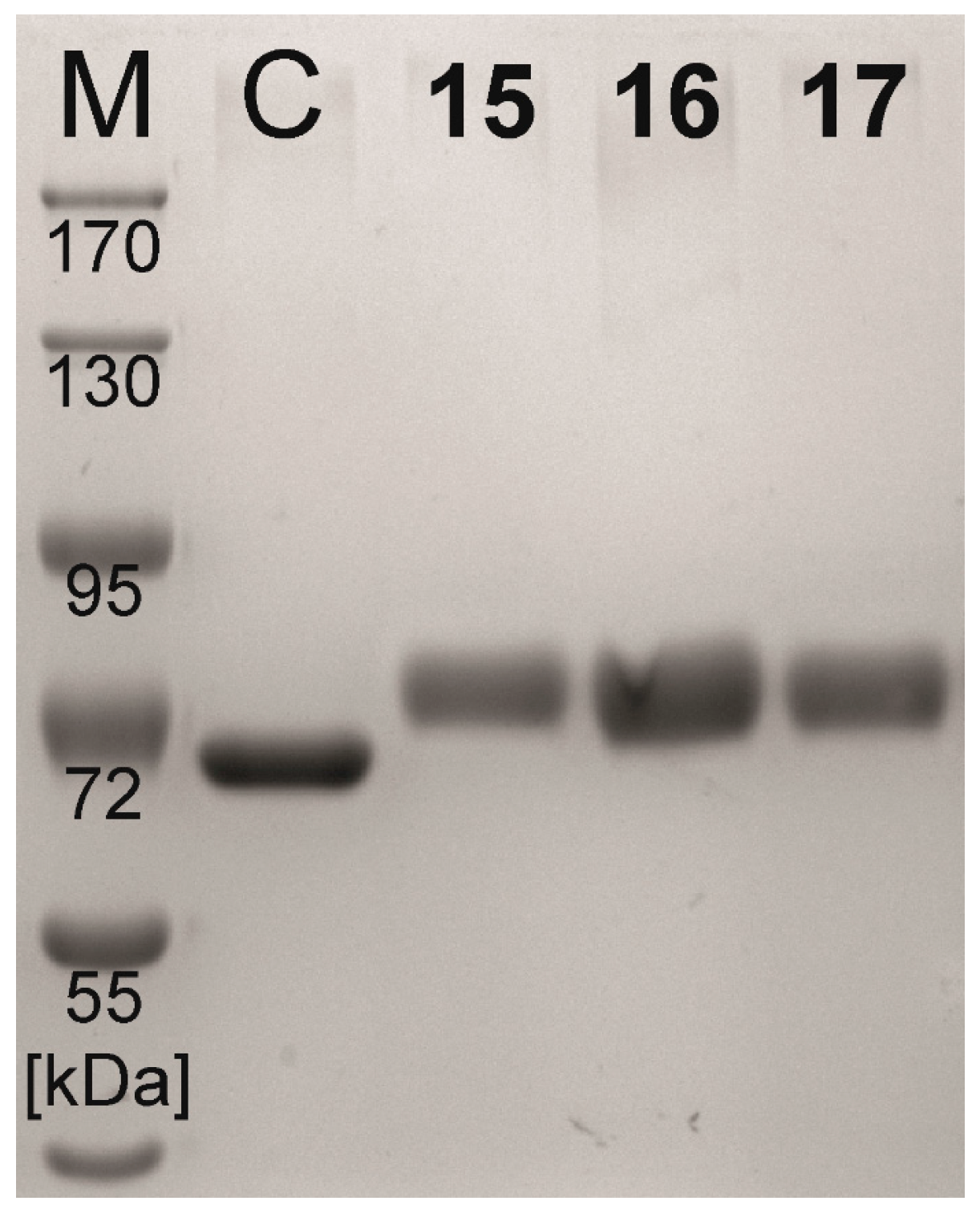
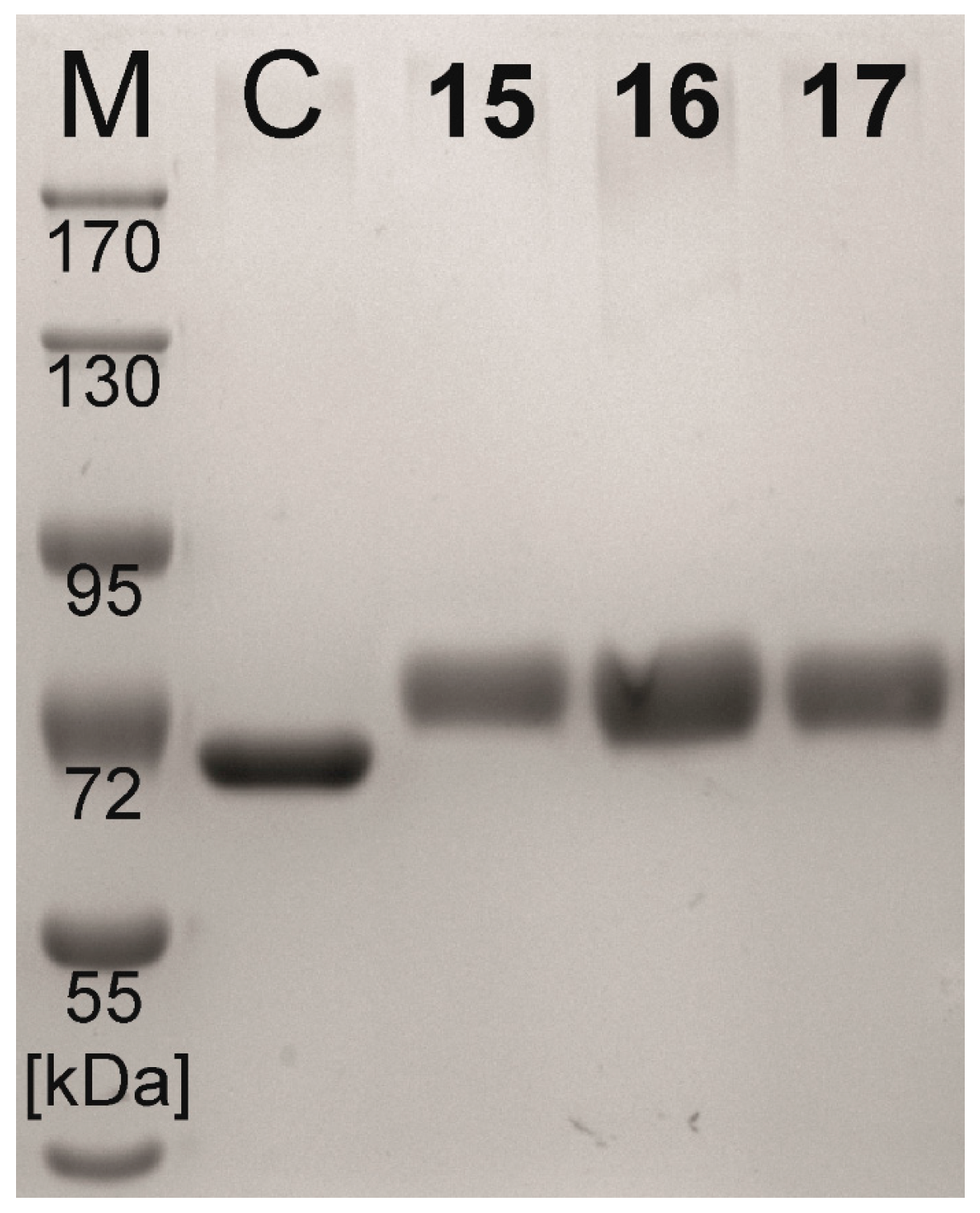
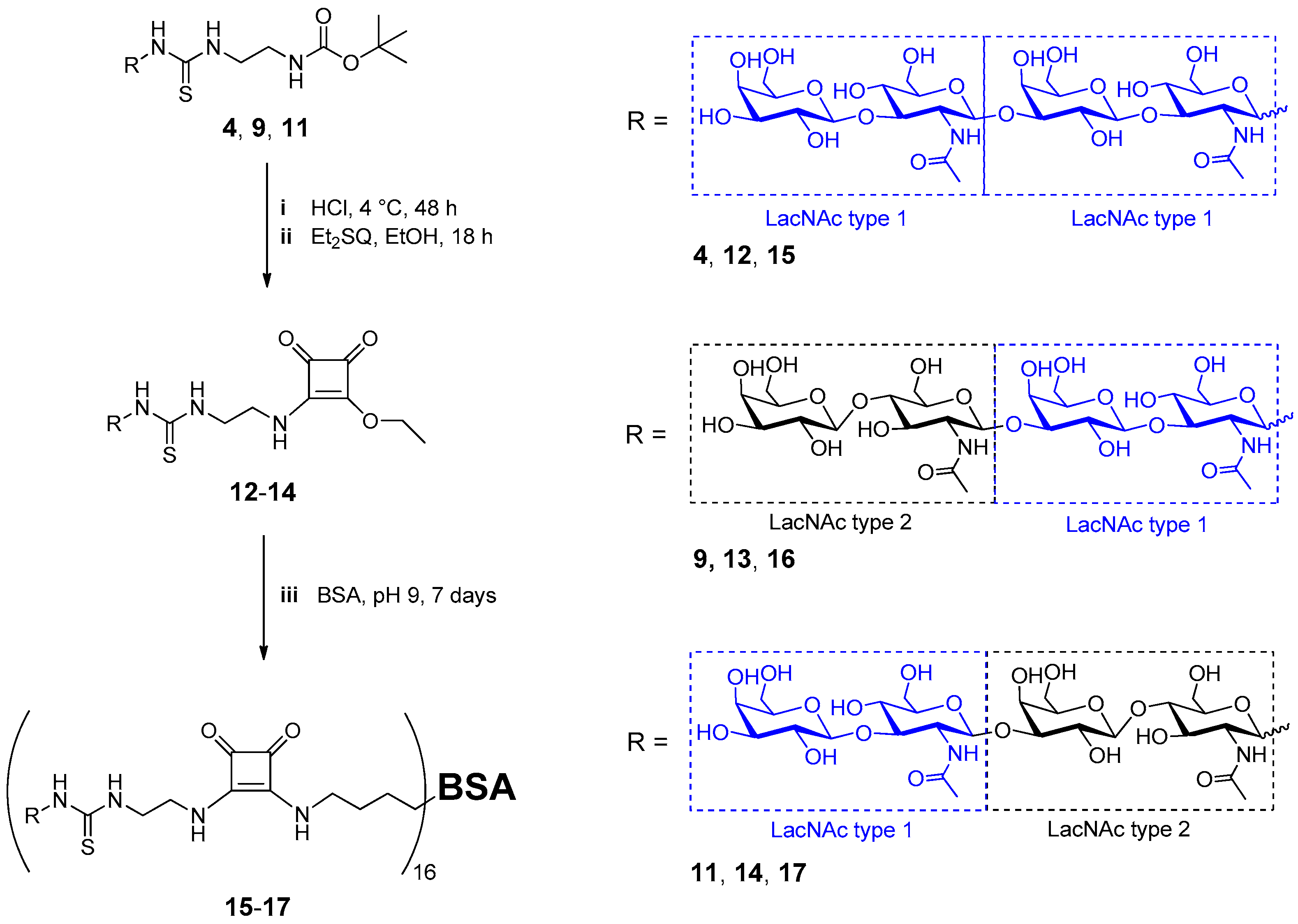
2.2. Neo-Glycoprotein Synthesis
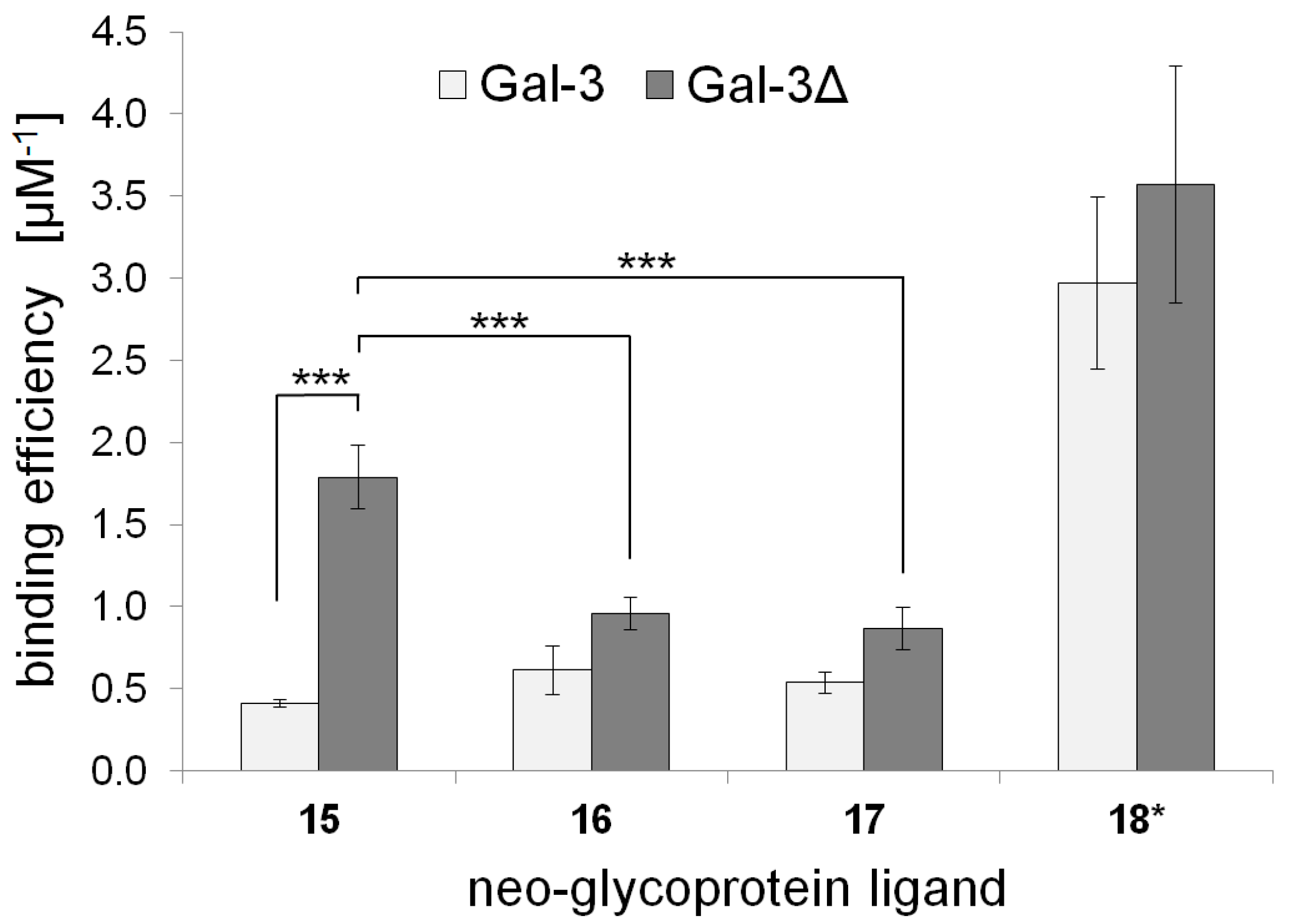
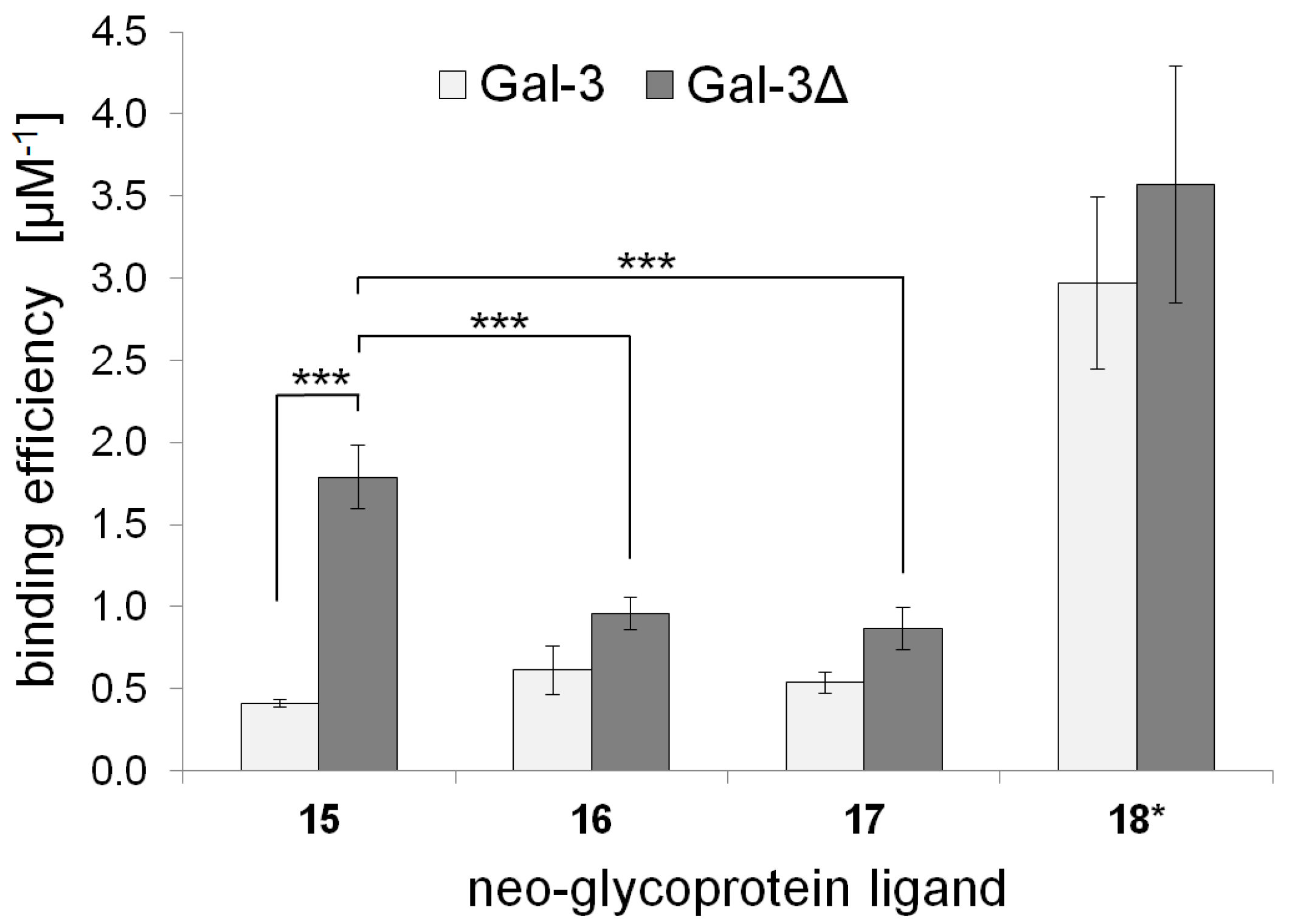
2.3. Galectin Binding Assays
3. Materials and Methods
3.1. Nucleotide Sugar Synthesis
3.2. Cloning of β3-Galactosyltransferase Fusion Protein
3.3. Production of Recombinant Enzymes and Human Galectins
3.4. Enzyme Activity Assays
3.5. One-Pot Synthesis of Poly-LacNAc Type 1 Oligomers
3.6. Sequential Synthesis of Poly-LacNAc Type 1 Oligomers
3.7. HPLC Analysis and Mass Spectrometry
3.8. Neo-Glycoprotein Synthesis
3.9. Galectin Binding Assays and Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Holgersson, J.; Löfling, J. Glycosyltransferases involved in type 1 chain and lewis antigen biosynthesis exhibit glycan and core chain specificity. Glycobiology 2006, 16, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Kerr, C.L.; Hanna, W.F.; Shaper, J.H.; Wright, W.W. Lewis X-containing glycans are specific and potent competitive inhibitors of the binding of zp3 to complementary sites on capacitated, acrosome-intact mouse sperm1. Biol. Reprod. 2004, 71, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.; Houles, C.; Sudakevitz, D.; Wimmerova, M.; Gautier, C.; Perez, S.; Wu, A.M.; Gilboa-Garber, N.; Imberty, A. Structural basis for oligosaccharide-mediated adhesion of pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Nat. Struct. Mol. Biol. 2002, 9, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Bode, L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology 2012, 22, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- German, J.B.; Freeman, S.L.; Lebrilla, C.B.; Mills, D.A. Human milk oligosaccharides: Evolution, structures and bioselectivity as substrates for intestinal bacteria. Nestle Nutr. Inst. Workshop Ser. Pediatr. Program 2008, 62, 205–218. [Google Scholar]
- Petschacher, B.; Nidetzky, B. Biotechnological production of fucosylated human milk oligosaccharides: Prokaryotic fucosyltransferases and their use in biocatalytic cascades or whole cell conversion systems. J. Biotechnol. 2016, 235, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Thurin, M.; Kieber-Emmons, T. Sa-lea and tumor metastasis: The old prediction and recent findings. Hybrid. Hybridomics 2002, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Yu, S.Y.; Ito, H.; Kameyama, A.; Sato, T.; Lin, C.H.; Yu, L.C.; Narimatsu, H.; Khoo, K.H. Identification of further elongation and branching of dimeric type 1 chain on lactosylceramides from colonic adenocarcinoma by tandem mass spectrometry sequencing analyses. J. Biol. Chem. 2008, 283, 16455–16468. [Google Scholar] [CrossRef] [PubMed]
- Stroud, M.R.; Levery, S.B.; Nudelman, E.D.; Salyan, M.E.; Towell, J.A.; Roberts, C.E.; Watanabe, M.; Hakomori, S. Extended type 1 chain glycosphingolipids: Dimeric lea (III4V4Fuc2Lc6) as human tumor-associated antigen. J. Biol. Chem. 1991, 266, 8439–8446. [Google Scholar] [PubMed]
- Falk, K.-E.; Karlsson, K.-A.; Larson, G.; Thurin, J.; Blaszczyk, M.; Steplewski, Z.; Koprowski, H. Mass spectrometry of a human tumor glycolipid antigen being defined by mouse monoclonal antibody NS-19-9. Biochem. Biophys. Res. Commun. 1983, 110, 383–391. [Google Scholar] [CrossRef]
- Gong, E.; Hirohashi, S.; Shimosato, Y.; Watanabe, M.; Ino, Y.; Teshima, S.; Kodaira, S. Expression of carbohydrate antigen 19–9 and stage-specific embryonic antigen 1 in nontumorous and tumorous epithelia of the human colon and rectum. J. Natl. Cancer Inst. 1985, 75, 447–454. [Google Scholar] [PubMed]
- Magnani, J.L.; Nilsson, B.; Brockhaus, M.; Zopf, D.; Steplewski, Z.; Koprowski, H.; Ginsburg, V. A monoclonal antibody-defined antigen associated with gastrointestinal cancer is a ganglioside containing sialylated lacto-n-fucopentaose ii. J. Biol. Chem. 1982, 257, 14365–14369. [Google Scholar] [PubMed]
- Ugorski, M.; Laskowska, A. Sialyl lewis(a): A tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells. Acta Biochim. Pol. 2002, 49, 303–311. [Google Scholar] [PubMed]
- Pettijohn, D.E.; Pfenninger, O.; Brown, J.; Duke, R.; Olsson, L. Tumorigenic human squamous lung cancer cells have defined cell surface carbohydrates that are absent from nontumorigenic cells. Proc. Natl. Acad. Sci. USA 1988, 85, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Ugorski, M.; Påhlsson, P.; Dus, D.; Nilsson, B.; Skouv, J.; Radzikowski, C. The sialosyl lewisa ganglioside is present in tumorigenic human urothelial cell lines. Int. J. Cancer 1990, 45, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Henze, M.; Schmidtke, S.; Hoffmann, N.; Steffens, H.; Pietruszka, J.; Elling, L. Combination of glycosyltransferases and a glycosynthase in sequential and one-pot reactions for the synthesis of type 1 and type 2 N-acetyllactosamine oligomers. ChemCatChem 2015, 7, 3131–3139. [Google Scholar] [CrossRef]
- Liu, X.-W.; Xia, C.; Li, L.; Guan, W.-Y.; Pettit, N.; Zhang, H.-C.; Chen, M.; Wang, P.G. Characterization and synthetic application of a novel β1,3-galactosyltransferase from escherichia coli O55:H7. Bioorg. Med. Chem. 2009, 17, 4910–4915. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Thon, V.; Li, Y.; Yu, H.; Ding, L.; Lau, K.; Qu, J.; Hie, L.; Chen, X. One-pot three-enzyme synthesis of udp-glcnac derivatives. Chem. Commun. 2011, 47, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Wahl, C.; Hirtz, D.; Elling, L. Multiplexed capillary electrophoresis as analytical tool for fast optimization of multi-enzyme cascade reactions – synthesis of nucleotide sugars. Biotechnol. J. 2016, 11, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Barondes, S.H.; Cooper, D.N.; Gitt, M.A.; Leffler, H. Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 1994, 269, 20807–20810. [Google Scholar] [PubMed]
- Fred Brewer, C. Binding and cross-linking properties of galectins. Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 255–262. [Google Scholar] [CrossRef]
- Leffler, H.; Carlsson, S.; Hedlund, M.; Qian, Y.; Poirier, F. Introduction to galectins. Glycoconj. J. 2004, 19, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Klyosov, A.A.; Witczak, Z.J.; Platt, D. Galectins and their functions in plain language. In Galectins; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 9–31. [Google Scholar]
- Gabius, H.-J.; Siebert, H.-C.; André, S.; Jiménez-Barbero, J.; Rüdiger, H. Chemical biology of the sugar code. ChemBioChem 2004, 5, 740–764. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.-J. Glycans: Bioactive signals decoded by lectins. Biochem. Soc. Trans. 2008, 36, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Compagno, D.; Gentilini, L.D.; Jaworski, F.M.; Perez, I.G.; Contrufo, G.; Laderach, D.J. Glycans and galectins in prostate cancer biology, angiogenesis and metastasis. Glycobiology 2014, 24, 899–906. [Google Scholar] [CrossRef] [PubMed]
- D’Haene, N.; Maris, C.; Rorive, S.; Decaestecker, C.; Le Mercier, M.; Salmon, I. Galectins and neovascularization in central nervous system tumors. Glycobiology 2014, 24, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Thijssen, V.L. Galectins in tumor angiogenesis. Ann. Transl. Med. 2014, 2, 90. [Google Scholar] [PubMed]
- Le Mercier, M.; Fortin, S.; Mathieu, V.; Kiss, R.; Lefranc, F. Galectins and gliomas. Brain Pathol. 2010, 20, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Oka, N.; Raz, A. On the role of galectin-3 in cancer apoptosis. Apoptosis 2005, 10, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; van Kooyk, Y.; Cobb, B.A. Glycobiology of immune responses. Ann. N. Y. Acad. Sci. 2012, 1253, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.I.; Liu, F.-T.; Panjwani, N. Galectin-3 is an important mediator of vegf- and bfgf-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Elola, M.T.; Blidner, A.G.; Ferragut, F.; Bracalente, C.; Rabinovich, G.A. Assembly, organization and regulation of cell-surface receptors by lectin-glycan complexes. Biochem. J. 2015, 469, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Iacobini, C.; Pesce, C.M.; Menini, S. Galectin-3: An emerging all-out player in metabolic disorders and their complications. Glycobiology 2015, 25, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Guevremont, M.; Martel-Pelletier, J.; Boileau, C.; Liu, F.T.; Richard, M.; Fernandes, J.C.; Pelletier, J.P.; Reboul, P. Galectin-3 surface expression on human adult chondrocytes: A potential substrate for collagenase-3. Ann. Rheum. Dis. 2004, 63, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Green, B.; Evans, S.; James, O.; Warfield, P. Modulation of the biological functions of galectin-3 by matrix metalloproteinases. Biochim. Biophys. Acta Gen. Subj. 1998, 1379, 97–106. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Raz, T.; Tait, L.; Hogan, V.; Fridman, R.; Raz, A. Galectin-3 cleavage: A novel surrogate marker for matrix metalloproteinase activity in growing breast cancers. Cancer Res. 2007, 67, 11760–11768. [Google Scholar] [CrossRef] [PubMed]
- John, C.M.; Leffler, H.; Kahl-Knutsson, B.; Svensson, I.; Jarvis, G.A. Truncated galectin-3 inhibits tumor growth and metastasis in orthotopic nude mouse model of human breast cancer. Clin. Cancer Res. 2003, 9, 2374–2383. [Google Scholar] [PubMed]
- Mirandola, L.; Nguyen, D.D.; Rahman, R.L.; Grizzi, F.; Yuefei, Y.; Figueroa, J.A.; Jenkins, M.R.; Cobos, E.; Chiriva-Internati, M. Anti-galectin-3 therapy: A new chance for multiple myeloma and ovarian cancer? Int. Rev. Immunol. 2014, 33, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Yu, Y.; Chui, K.; Jenkins, M.R.; Cobos, E.; John, C.M.; Chiriva-Internati, M. Galectin-3c inhibits tumor growth and increases the anticancer activity of bortezomib in a murine model of human multiple myeloma. PLoS ONE 2011, 6, e21811. [Google Scholar] [CrossRef] [PubMed]
- Engels, L.; Elling, L. Wbgl: A novel bacterial alpha1,2-fucosyltransferase for the synthesis of 2′-fucosyllactose. Glycobiology 2014, 24, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Sauerzapfe, B.; Namdjou, D.J.; Schumacher, T.; Linden, N.; Křenek, K.; Křen, V.; Elling, L. Characterization of recombinant fusion constructs of human β1,4-galactosyltransferase 1 and the lipase pre-propeptide from staphylococcus hyicus. J. Mol. Catal. B Enzym. 2008, 50, 128–140. [Google Scholar] [CrossRef]
- Logan, S.M.; Altman, E.; Mykytczuk, O.; Brisson, J.-R.; Chandan, V.; Michael, F.S.; Masson, A.; Leclerc, S.; Hiratsuka, K.; Smirnova, N.; et al. Novel biosynthetic functions of lipopolysaccharide rfaj homologs from helicobacter pylori. Glycobiology 2005, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Sauerzapfe, B.; Křenek, K.; Schmiedel, J.; Wakarchuk, W.W.; Pelantová, H.; Křen, V.; Elling, L. Chemo-enzymatic synthesis of poly-N-acetyllactosamine (poly-LacNAc) structures and their characterization for cgl2-galectin-mediated binding of ecm glycoproteins to biomaterial surfaces. Glycoconj. J. 2009, 26, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Rech, C.; Rosencrantz, R.R.; Křenek, K.; Pelantová, H.; Bojarová, P.; Römer, C.E.; Hanisch, F.-G.; Křen, V.; Elling, L. Combinatorial one-pot synthesis of poly-N-acetyllactosamine oligosaccharides with leloir-glycosyltransferases. Adv. Synth. Catal. 2011, 353, 2492–2500. [Google Scholar] [CrossRef]
- Henze, M.; You, D.-J.; Kamerke, C.; Hoffmann, N.; Angkawidjaja, C.; Ernst, S.; Pietruszka, J.; Kanaya, S.; Elling, L. Rational design of a glycosynthase by the crystal structure of β-galactosidase from bacillus circulans (Bgac) and its use for the synthesis of N-acetyllactosamine type 1 glycan structures. J. Biotechnol. 2014, 191, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kamerke, C.; Pattky, M.; Huhn, C.; Elling, L. Synthesis of nucleotide-activated disaccharides with recombinant β3-galactosidase c from bacillus circulans. J. Mol. Catal. B Enzym. 2013, 89, 73–81. [Google Scholar] [CrossRef]
- Böcker, S.; Laaf, D.; Elling, L. Galectin binding to neo-glycoproteins: Lacdinac conjugated bsa as ligand for human galectin-3. Biomolecules 2015, 5, 1671–1696. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.J. Maximising multivalency effects in protein-carbohydrate interactions. Org. Biomol. Chem. 2009, 7, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.J.; Arnusch, C.J.; Breukink, E. Membrane permeabilization by multivalent anti-microbial peptides. Protein Pept. Lett. 2009, 16, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.J.; Saksena, R.; Kovac, P. Preparation of glycoconjugates by dialkyl squarate chemistry revisited. Carbohydr. Res. 2008, 343, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Laaf, D.; Bojarová, P.; Mikulová, B.; Pelantová, H.; Křen, V.; Elling, L. Two-step enzymatic synthesis of β-d-N-acetylgalactosamine-(1→4)-d-N-acetylglucosamine (LacdiNAc) chitooligomers for deciphering galectin binding behavior. Adv. Synth. Catal. 2017, 359, 2101–2108. [Google Scholar] [CrossRef]
- Xu, P.; Kelly, M.; Vann, W.F.; Qadri, F.; Ryan, E.T.; Kovac, P. Conjugate vaccines from bacterial antigens by squaric acid chemistry: A closer look. ChemBioChem 2017, 18, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Böcker, S.; Elling, L. Binding characteristics of galectin-3 fusion proteins. Glycobiology 2017, 27, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Kupper, C.E.; Bocker, S.; Liu, H.; Adamzyk, C.; van de Kamp, J.; Recker, T.; Lethaus, B.; Jahnen-Dechent, W.; Neuss, S.; Muller-Newen, G.; et al. Fluorescent snap-tag galectin fusion proteins as novel tools in glycobiology. Curr. Pharm. Des. 2013, 19, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Arthur, C.M.; Mehta, P.; Slanina, K.A.; Blixt, O.; Leffler, H.; Smith, D.F.; Cummings, R.D. Galectin-1, -2 and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J. Biol. Chem. 2008, 283, 10109–10123. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.; Tan, H.L.; Ding, V.; Castangia, R.; Belgacem, O.; Liau, B.; Hartley-Tassell, L.; Haslam, S.M.; Dell, A.; Choo, A. Characterization of h type 1 and type 1 N-acetyllactosamine glycan epitopes on ovarian cancer specifically recognized by the anti-glycan monoclonal antibody mab-a4. J. Biol. Chem. 2017, 292, 6163–6176. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.H. Anti-cancer vaccines—A one-hit wonder? Yale J. Biol. Med. 2014, 87, 481–489. [Google Scholar] [PubMed]
- Šimonová, A.; Kupper, C.E.; Böcker, S.; Müller, A.; Hofbauerová, K.; Pelantová, H.; Elling, L.; Křen, V.; Bojarová, P. Chemo-enzymatic synthesis of LacdiNAc dimers of varying length as novel galectin ligands. J. Mol. Catal. B Enzym. 2014, 101, 47–55. [Google Scholar] [CrossRef]
Sample Availability: Samples of the LacNAc type 1 containing compounds are available from the authors. |
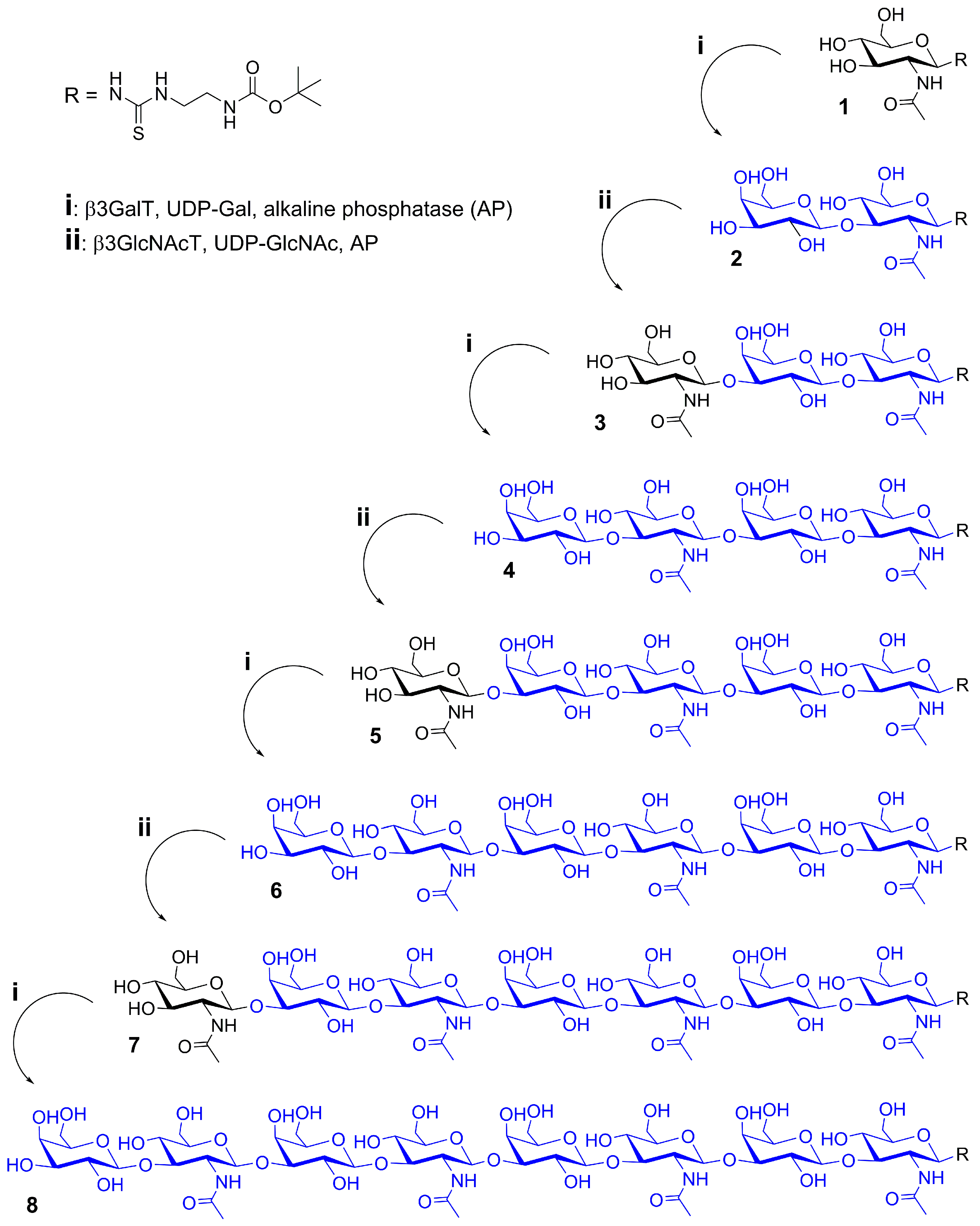

{kind=link}
{kind=link}
{kind=link}
{kind=link}
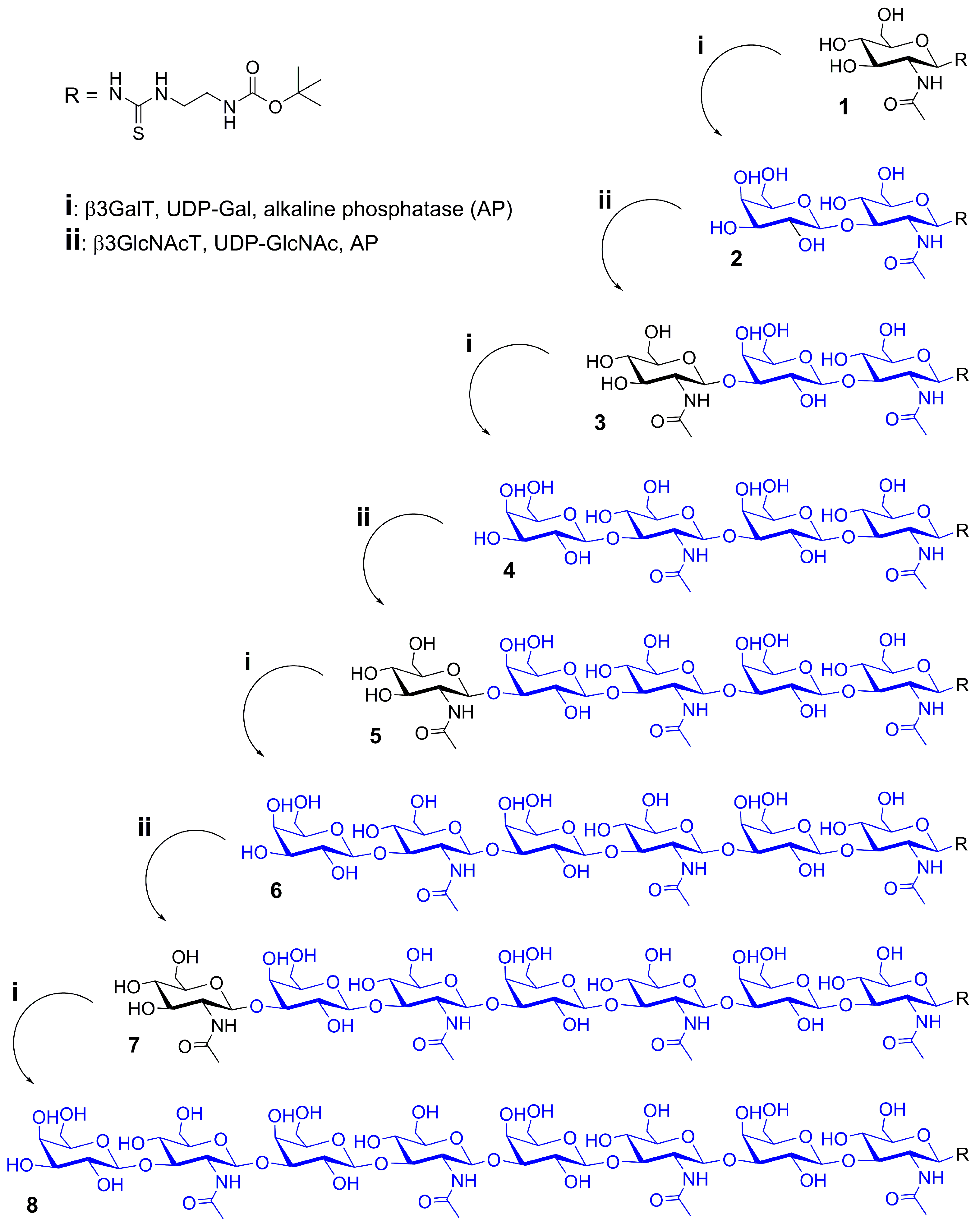{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Amount (µmol) | Stepwise Enzymatic Conversion (%) | Stepwise Molar Yield (%) | Calculated m/z | Observed m/z |
|---|---|---|---|---|---|
| 2 | 9.97 | 100.0 | 98.3 | 583.2 a | 583.1 a |
| 3 | 9.82 | 100.0 | 98.5 | 786.3 a | 786.1 a |
| 4 | 9.61 | 100.0 | 97.9 | 948.4 a | 948.2 a |
| 5 | 9.10 | 100.0 | 94.7 | 575.3 b | 575.3 b |
| 6 | 2.29 | 25.2 | 24.2 | 656.3 b | 656.5 b |
| 7 | 2.22 | 100.0 | 96.8 | 757.8 b | 758.0 b |
| 8 | 0.43 | 20.9 | 19.5 | 838.8 b | 839.1 b |
| Compound | Amount (µmol) | Stepwise Enzymatic Conversion (%) | Stepwise Molar Yield (%) | Calculated m/z | Observed m/z |
|---|---|---|---|---|---|
| 9 | 4.0 | 100 | 95.9 | 948.4 a | 948.3 a |
| 11 | 4.0 | 100 | 97.5 | 948.4 a | 948.5 a |
| Compound | Coupling Efficiency (%) | Conjugated Glycans (TNBSA Assay) (mol/mol BSA) | Molecular Mass (Calculated) (kDa) | Molecular Mass (SDS-PAGE) (kDa) |
|---|---|---|---|---|
| BSA | - | 0.0 ± 0.21 | 66.4 | 65.4 |
| 15 | 73.7 | 16.6 ± 0.20 | 81.8 | 78.8 |
| 16 | 73.3 | 16.3 ± 0.10 | 81.5 | 78.3 |
| 17 | 72.4 | 16.5 ± 0.26 | 81.7 | 78.6 |
| Neo-Glycoprotein | Galectin Binding Efficiency (µM−1) | Maximal Binding Signal (Bmax) | Apparent Kd Value (µM) | |||
|---|---|---|---|---|---|---|
| Gal-3 | Gal-3Δ | Gal-3 | Gal-3Δ | Gal-3 | Gal-3Δ | |
| 15 | 0.41 ± 0.02 | 1.79 ± 0.20 | 1.05 ± 0.02 | 1.00 ± 0.04 | 2.56 ± 0.09 | 0.56 ± 0.04 |
| 16 | 0.61 ± 0.07 | 0.96 ± 0.13 | 1.11 ± 0.10 | 1.08 ± 0.03 | 1.81 ± 0.36 | 1.13 ± 0.12 |
| 17 | 0.54 ± 0.15 | 0.87 ± 0.10 | 1.15 ± 0.10 | 1.10 ± 0.04 | 2.14 ± 0.41 | 1.27 ± 0.10 |
| 18 * | 2.97 ± 0.52 | 3.57 ± 0.72 | 1.01 ± 0.09 | 1.00 ± 0.06 | 0.34 ± 0.09 | 0.28 ± 0.04 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischöder, T.; Laaf, D.; Dey, C.; Elling, L. Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands. Molecules 2017, 22, 1320. https://doi.org/10.3390/molecules22081320
Fischöder T, Laaf D, Dey C, Elling L. Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands. Molecules. 2017; 22(8):1320. https://doi.org/10.3390/molecules22081320
Chicago/Turabian StyleFischöder, Thomas, Dominic Laaf, Carina Dey, and Lothar Elling. 2017. "Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands" Molecules 22, no. 8: 1320. https://doi.org/10.3390/molecules22081320
APA StyleFischöder, T., Laaf, D., Dey, C., & Elling, L. (2017). Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands. Molecules, 22(8), 1320. https://doi.org/10.3390/molecules22081320