Characterization of Proanthocyanidin Oligomers of Ephedra sinica

Abstract
:1. Introduction
2. Results and Discussion
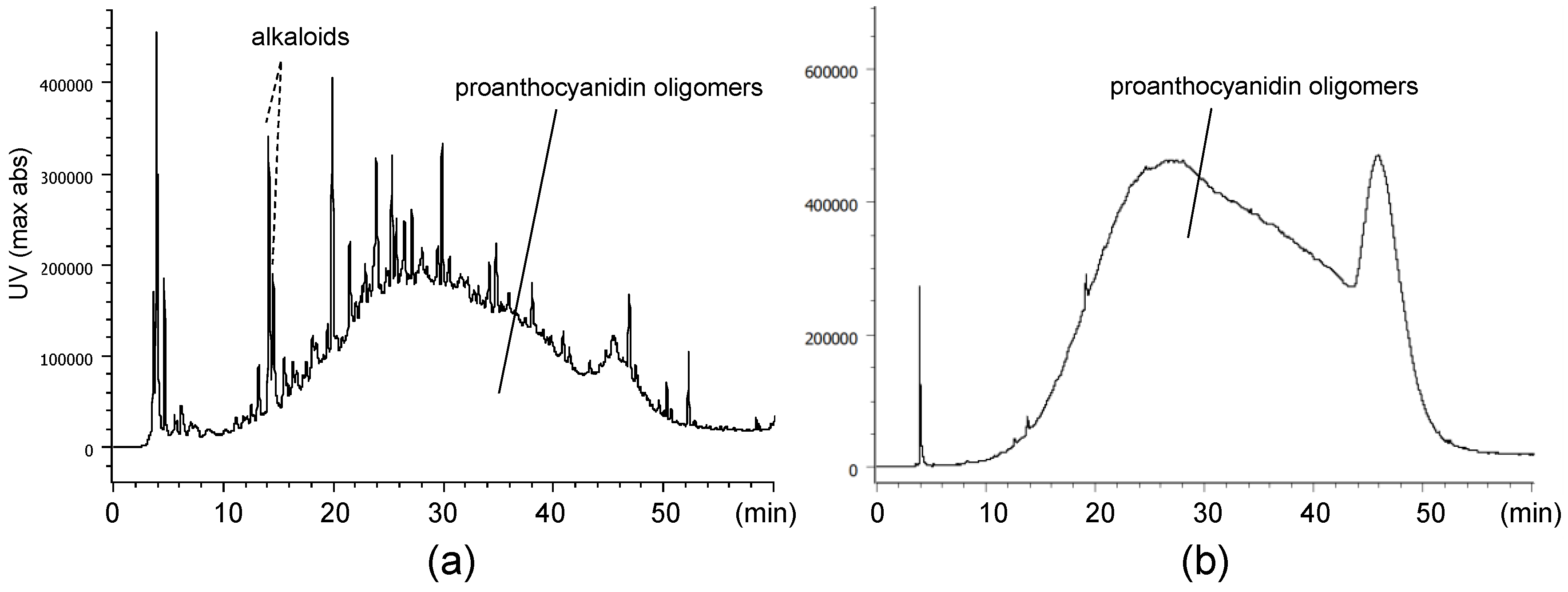
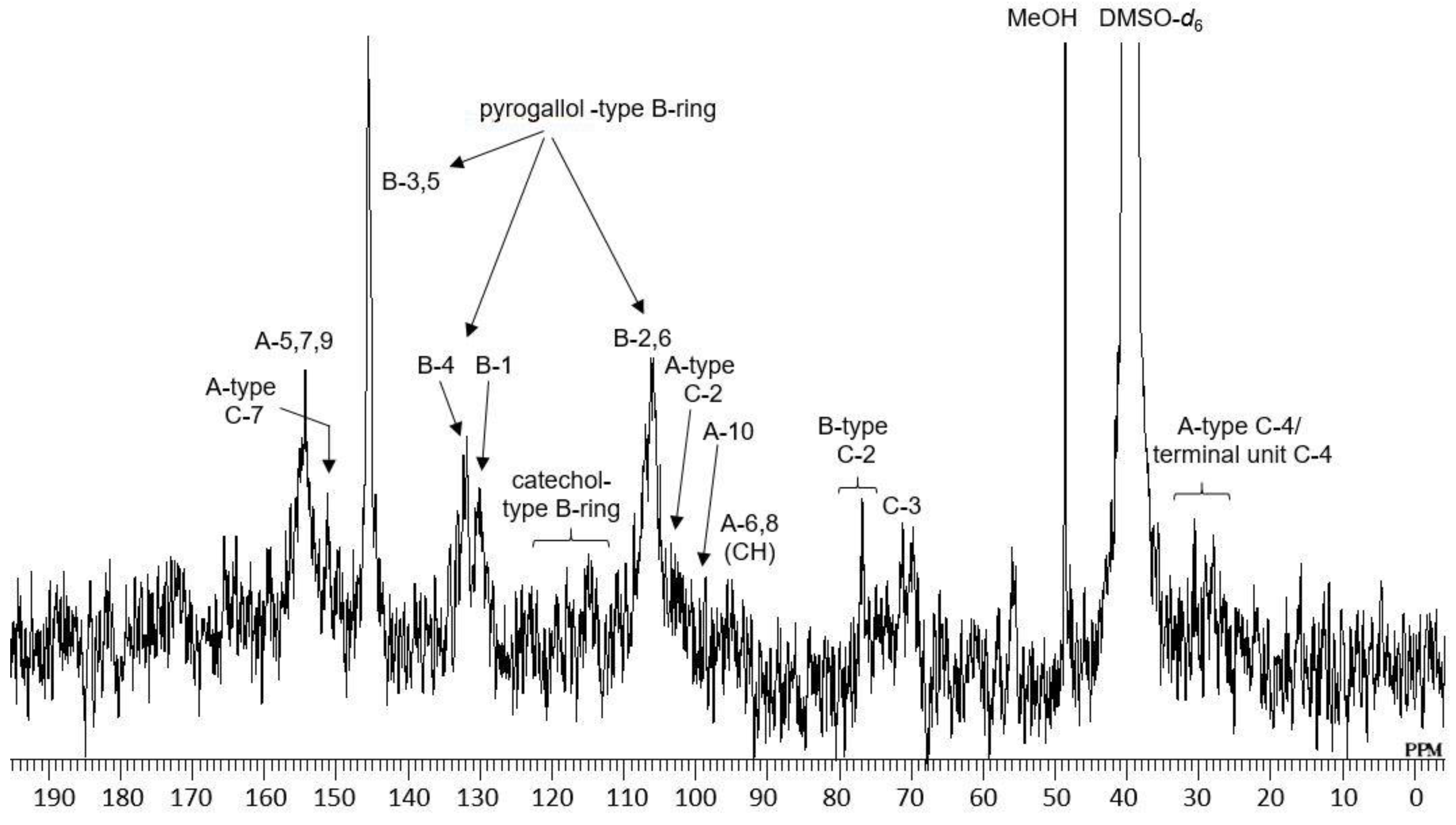
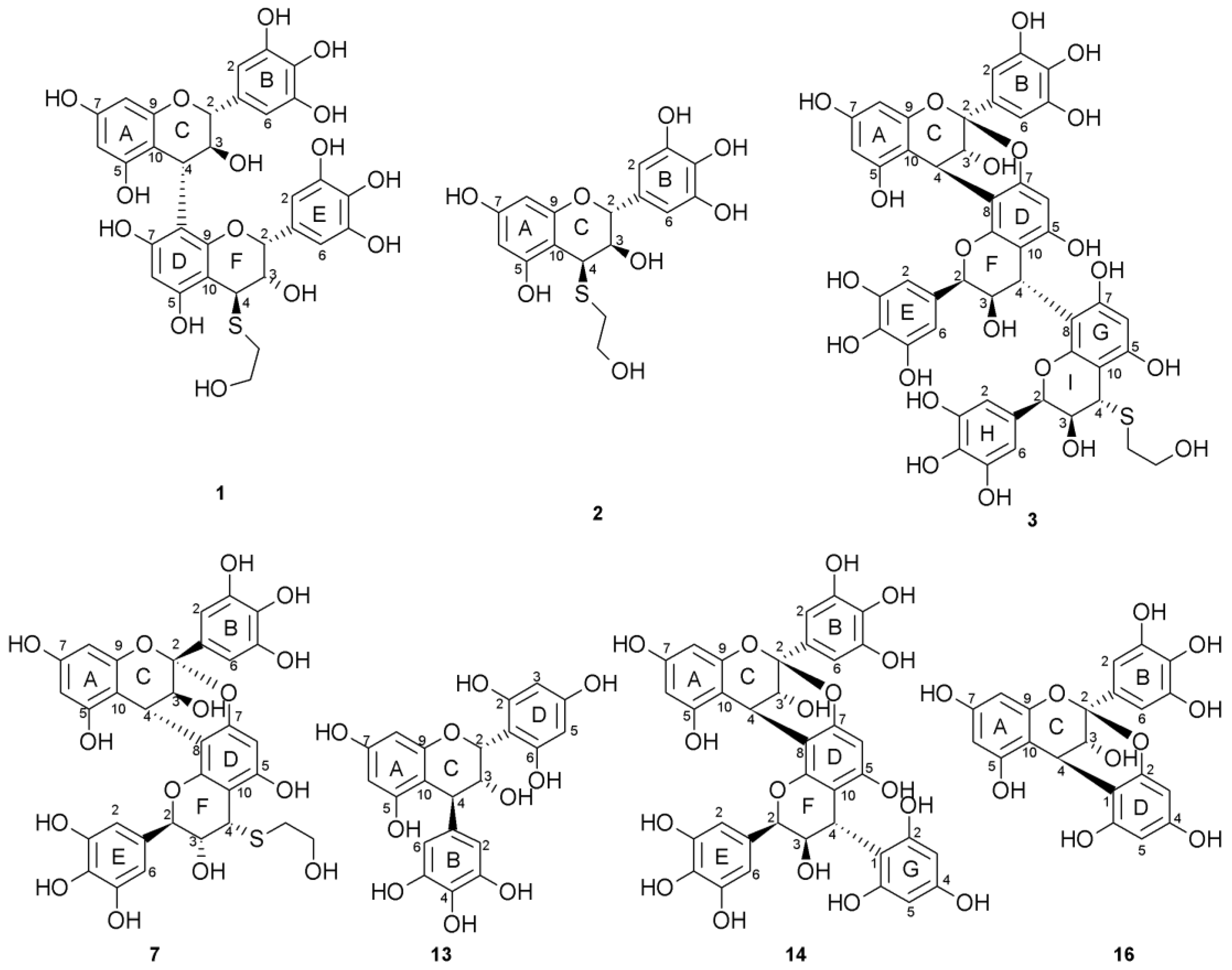
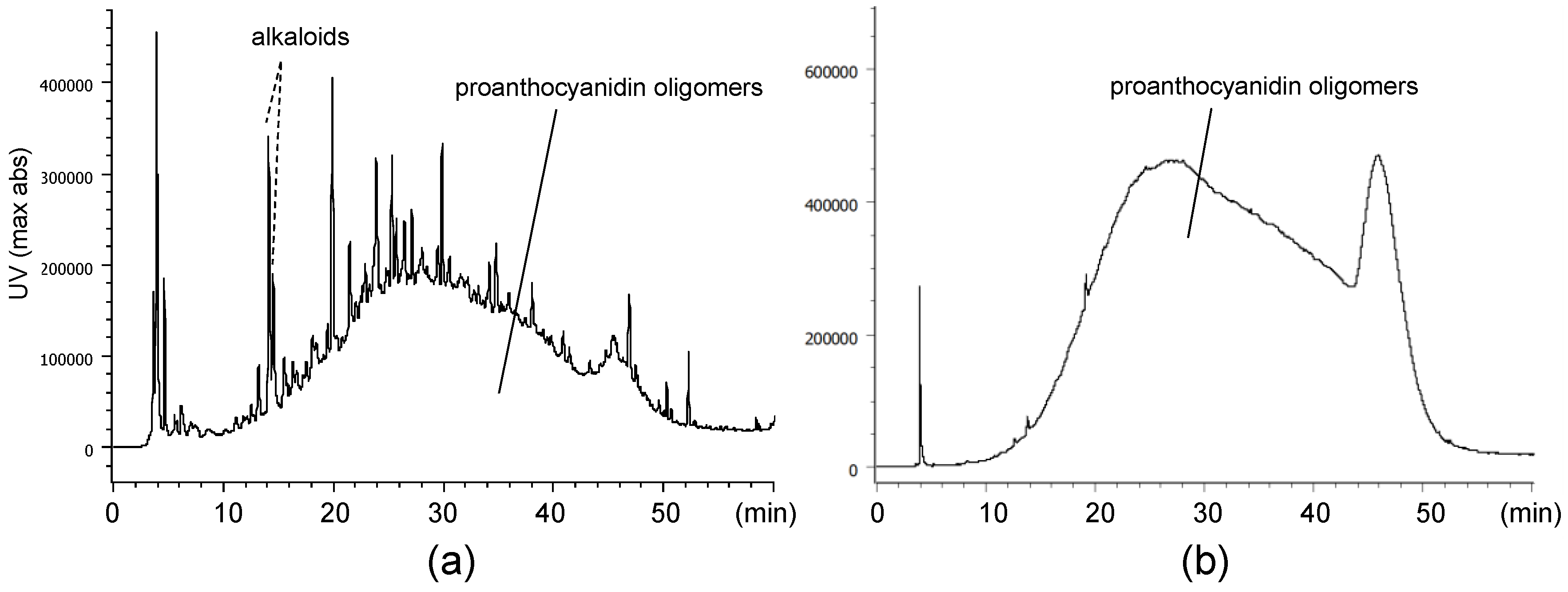
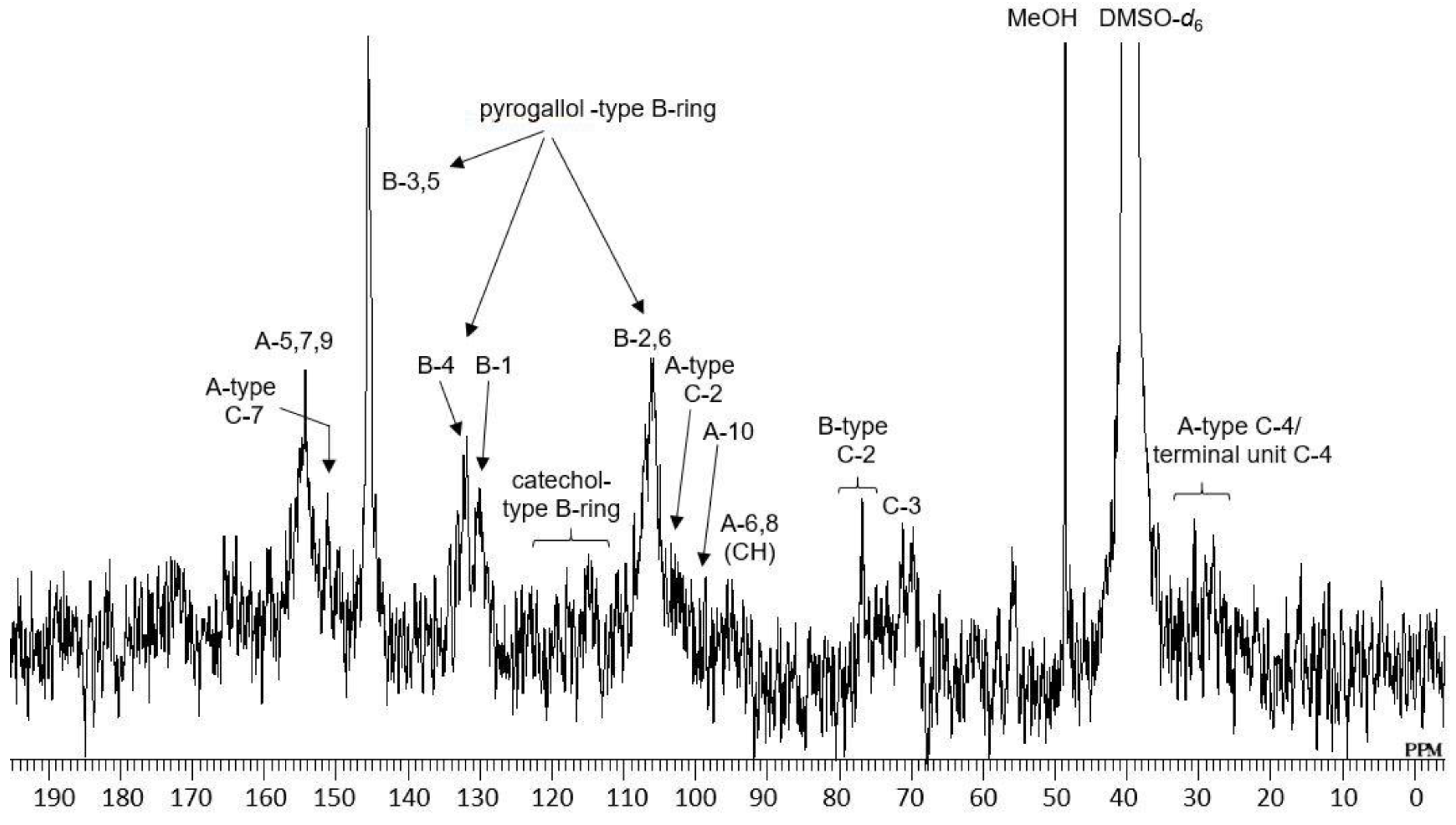
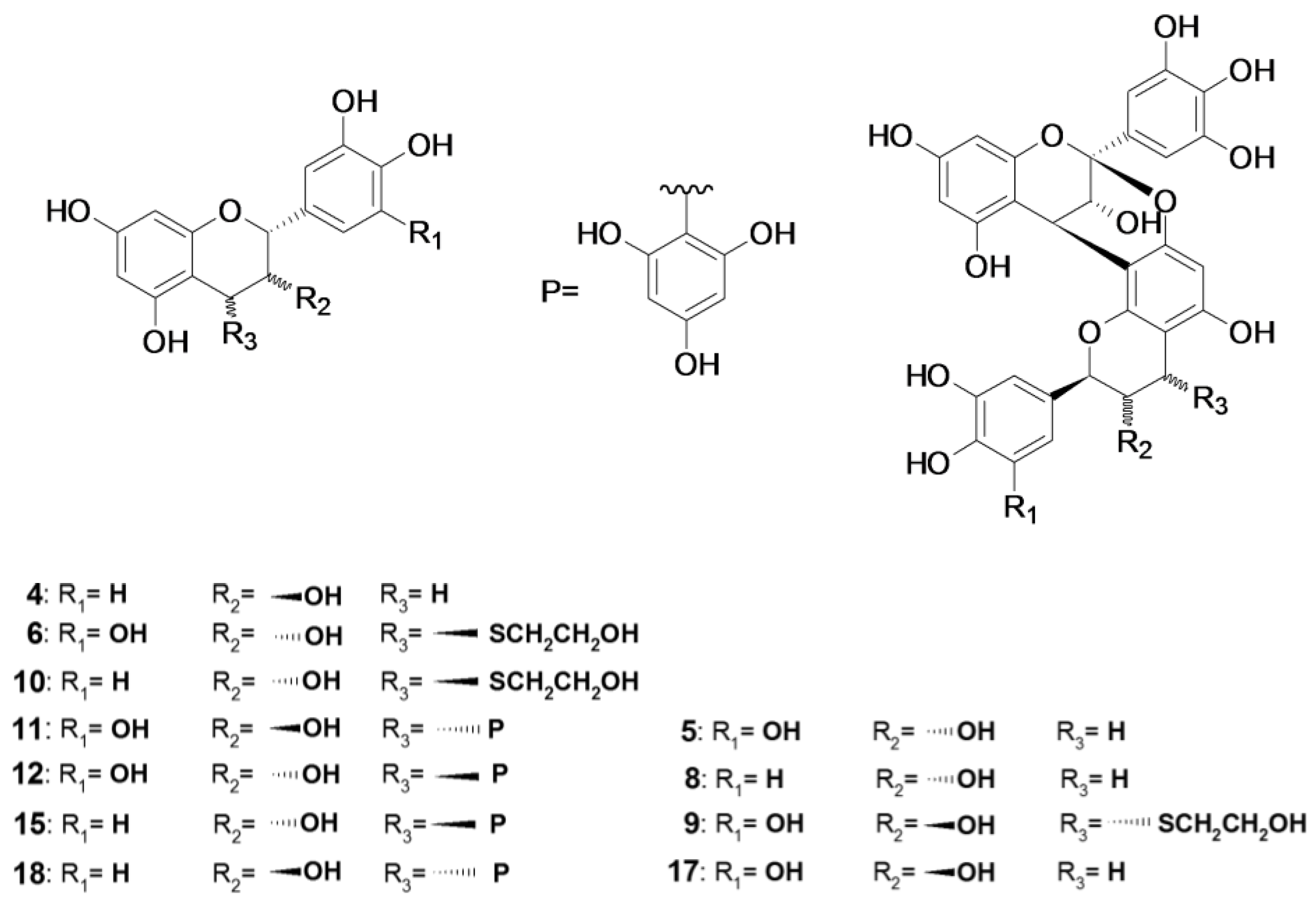
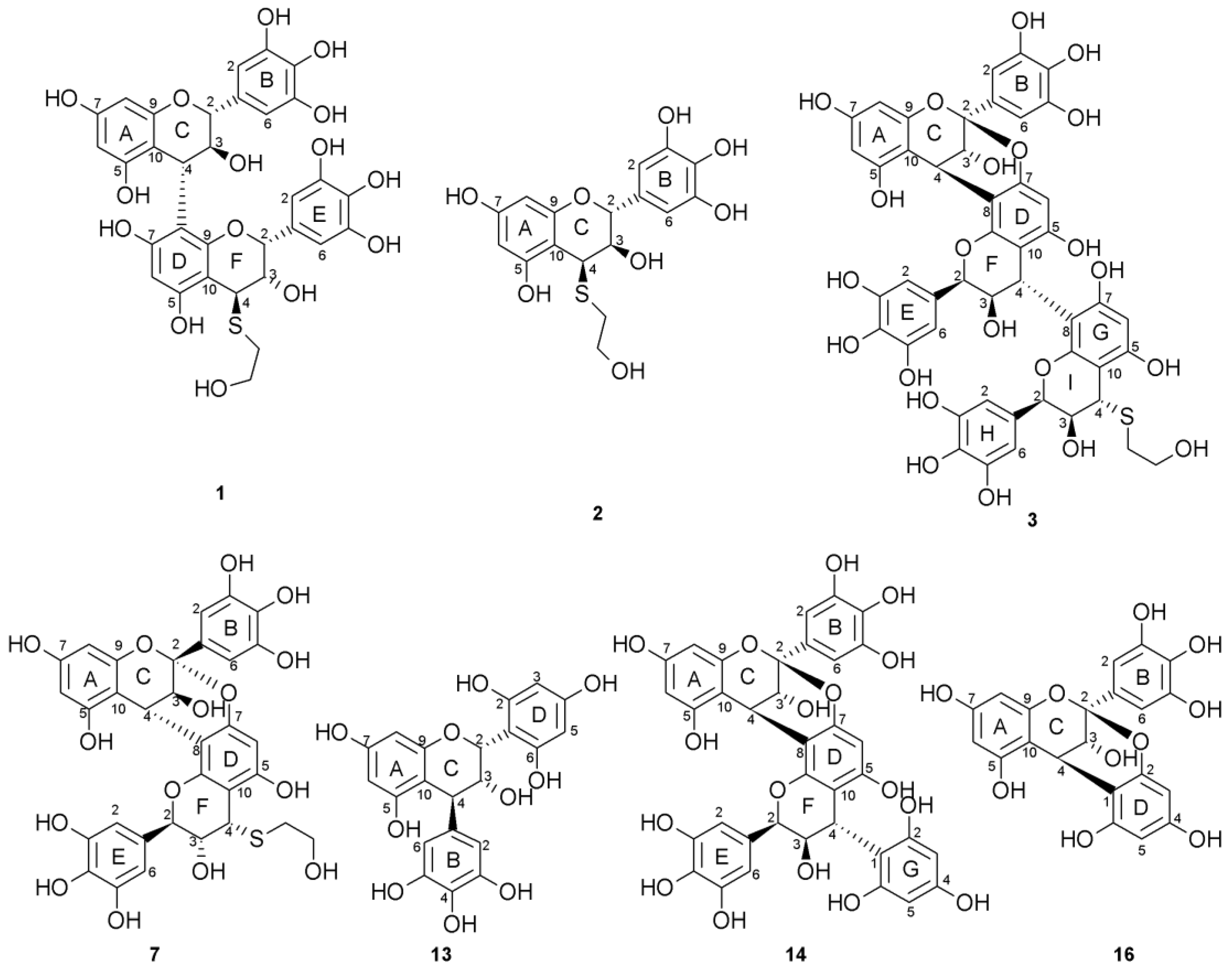
2.1. Composition of the Intact Proanthocyanidin Oligomer
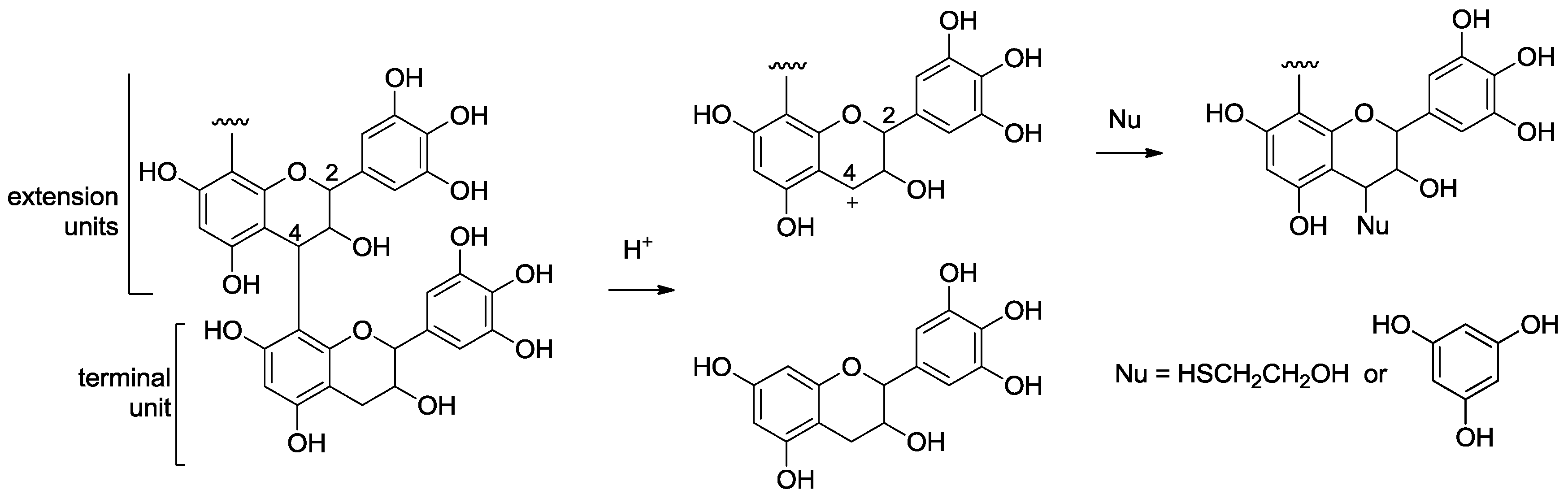
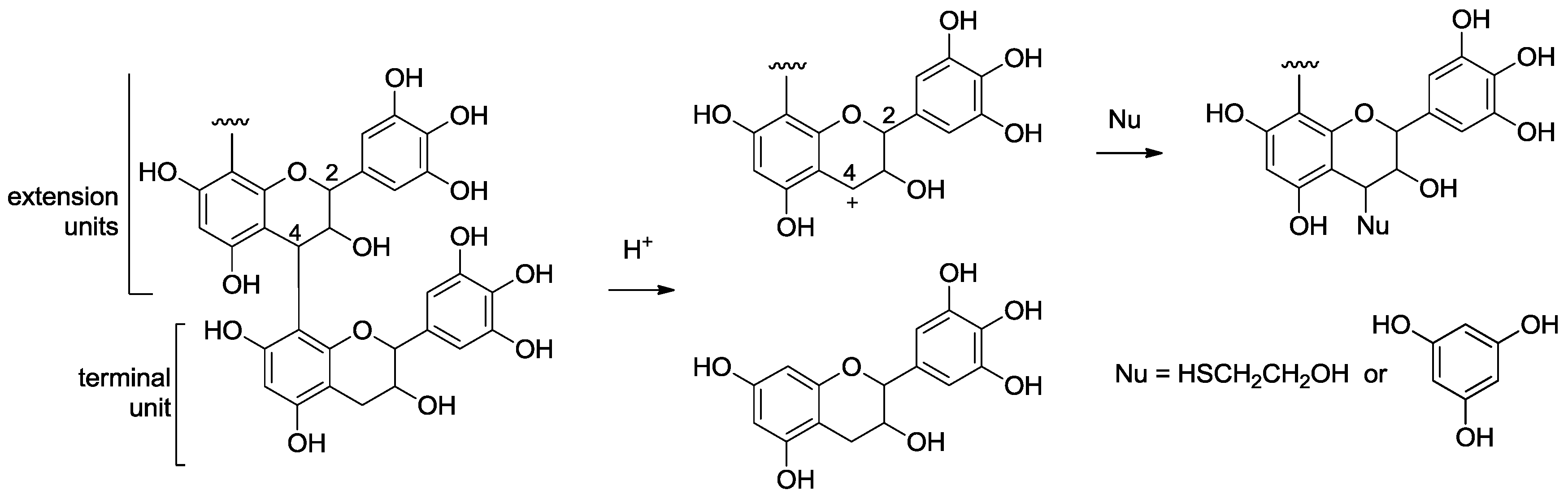
2.2. Acid-Catalyzed Degradation Products
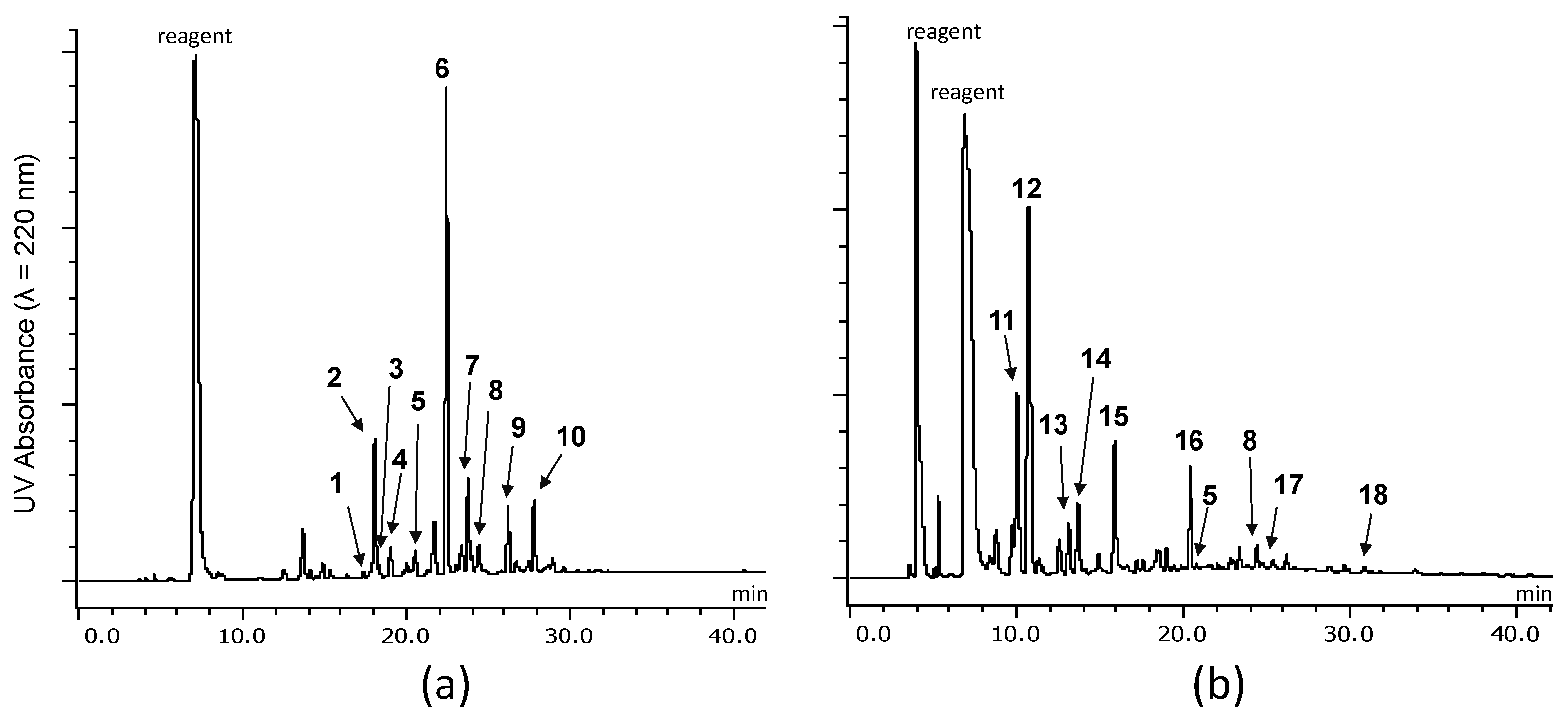
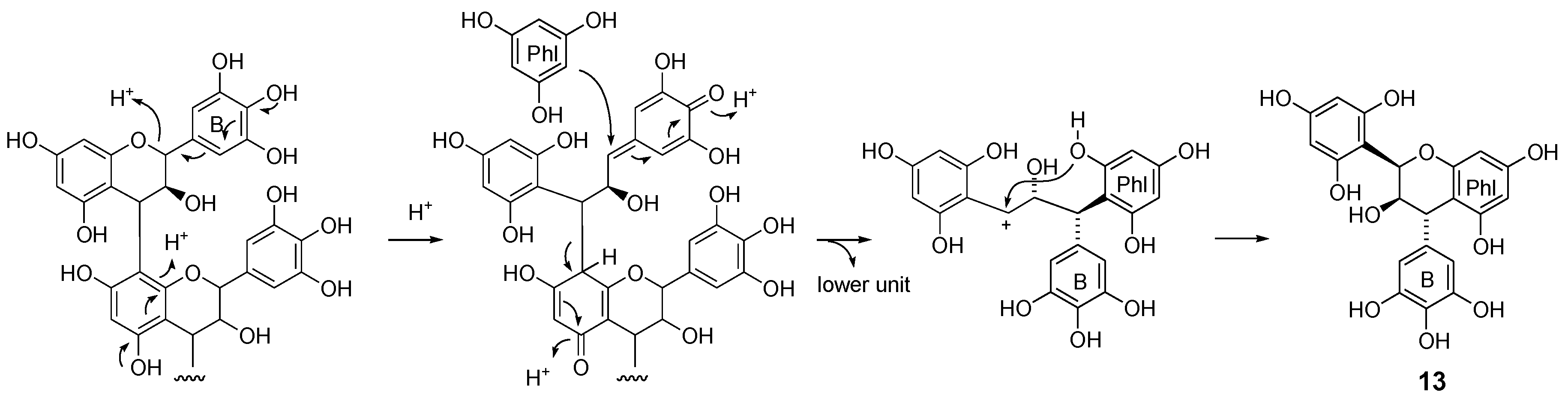
2.2.1. Identification of Known Products
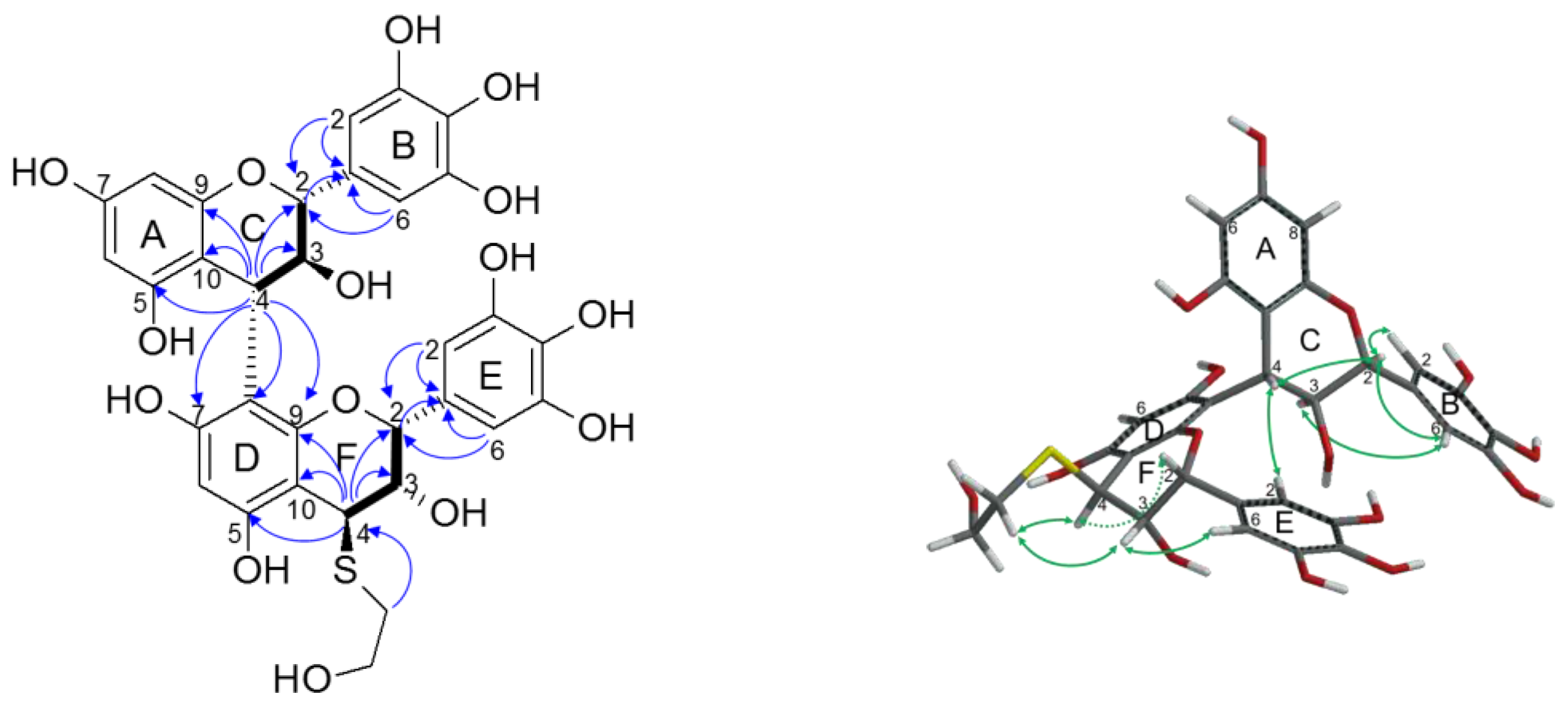
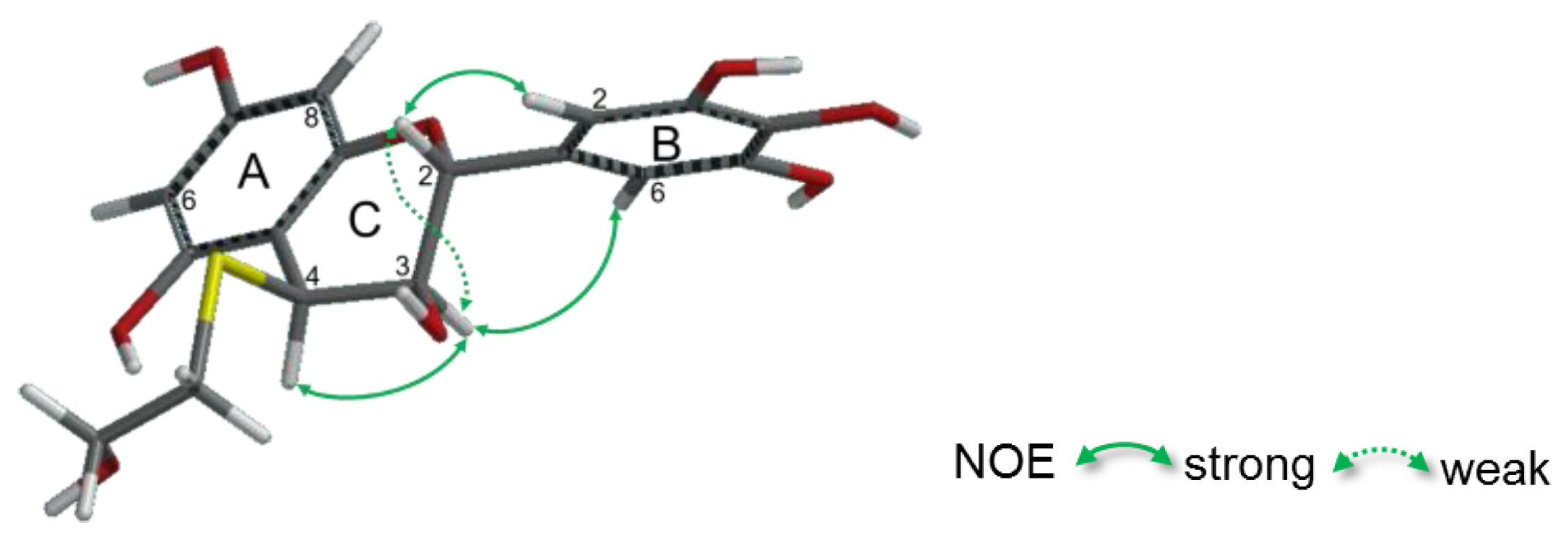
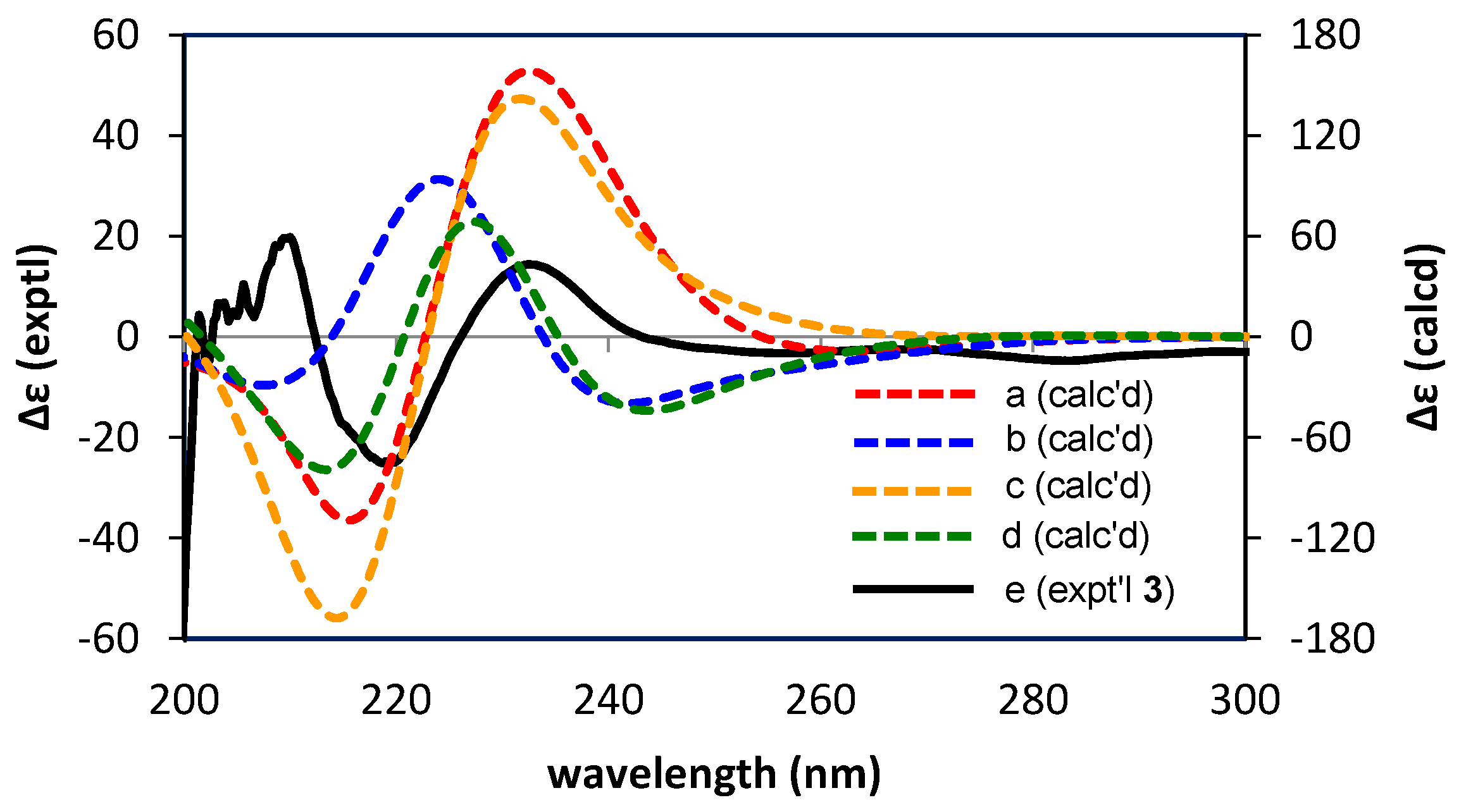
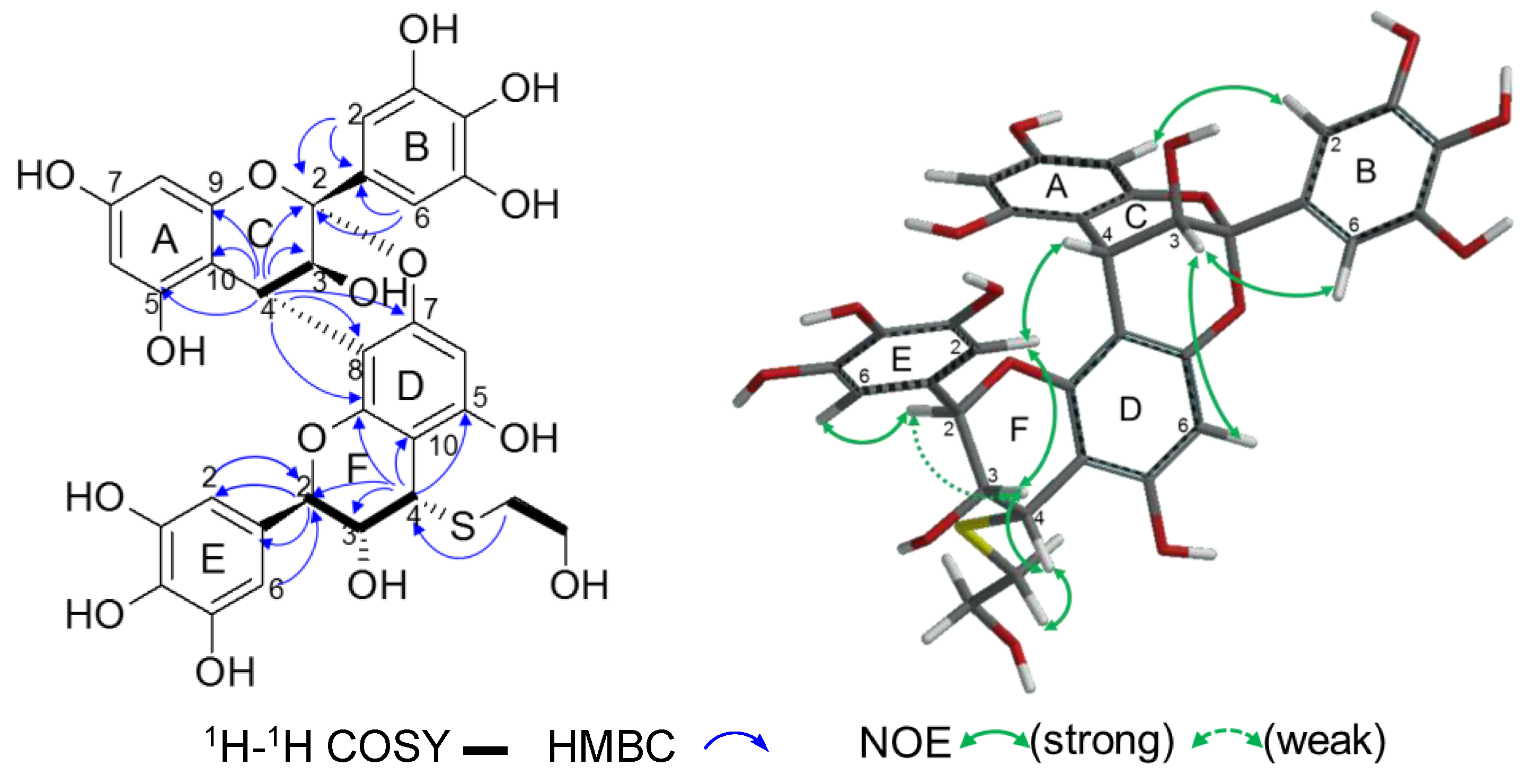
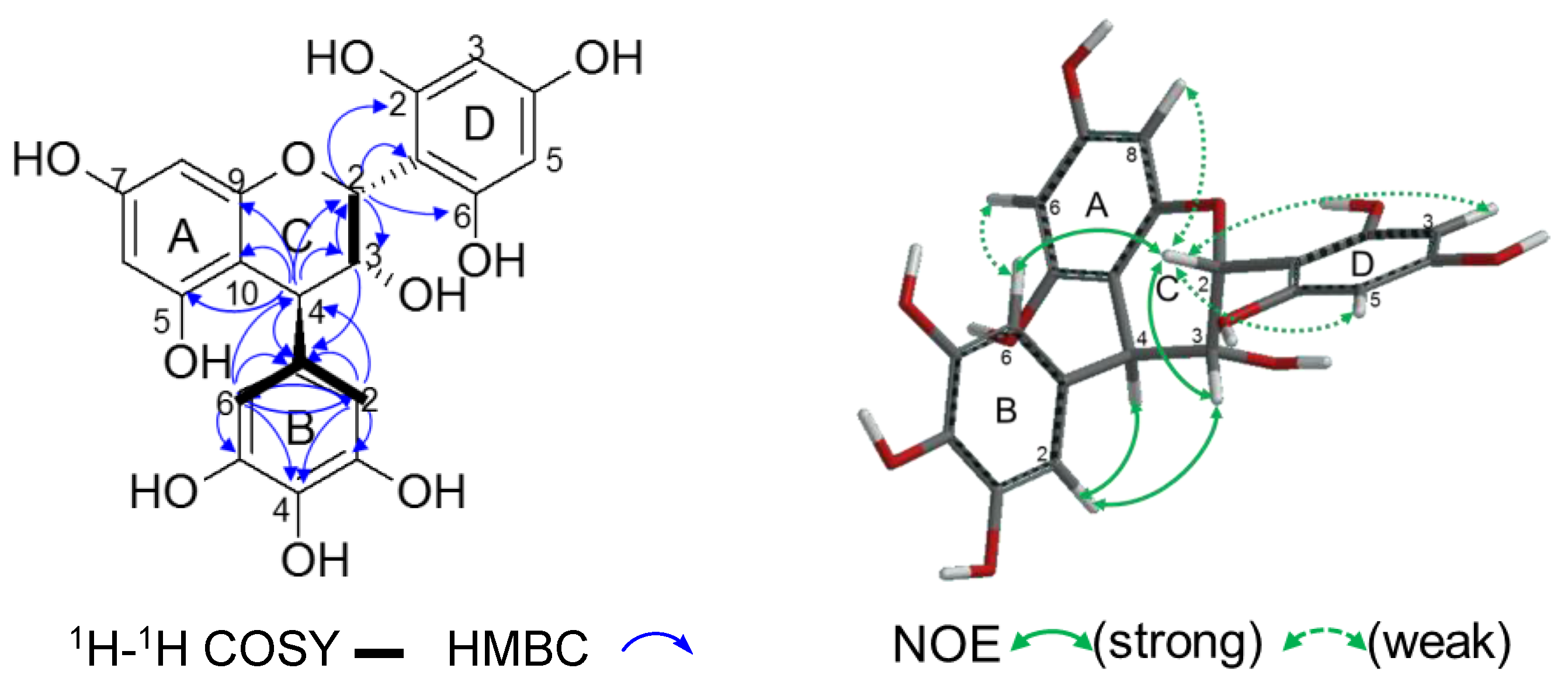
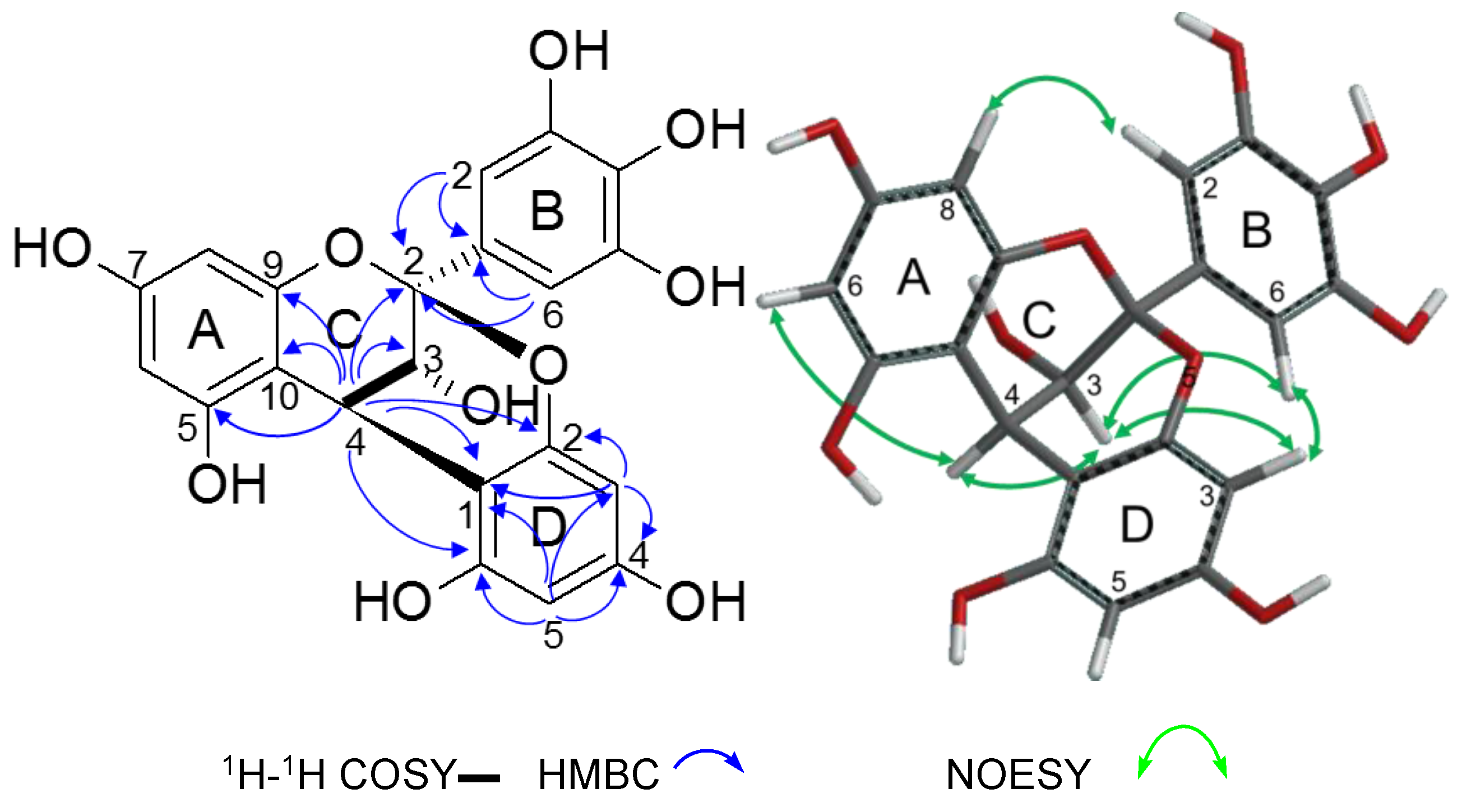
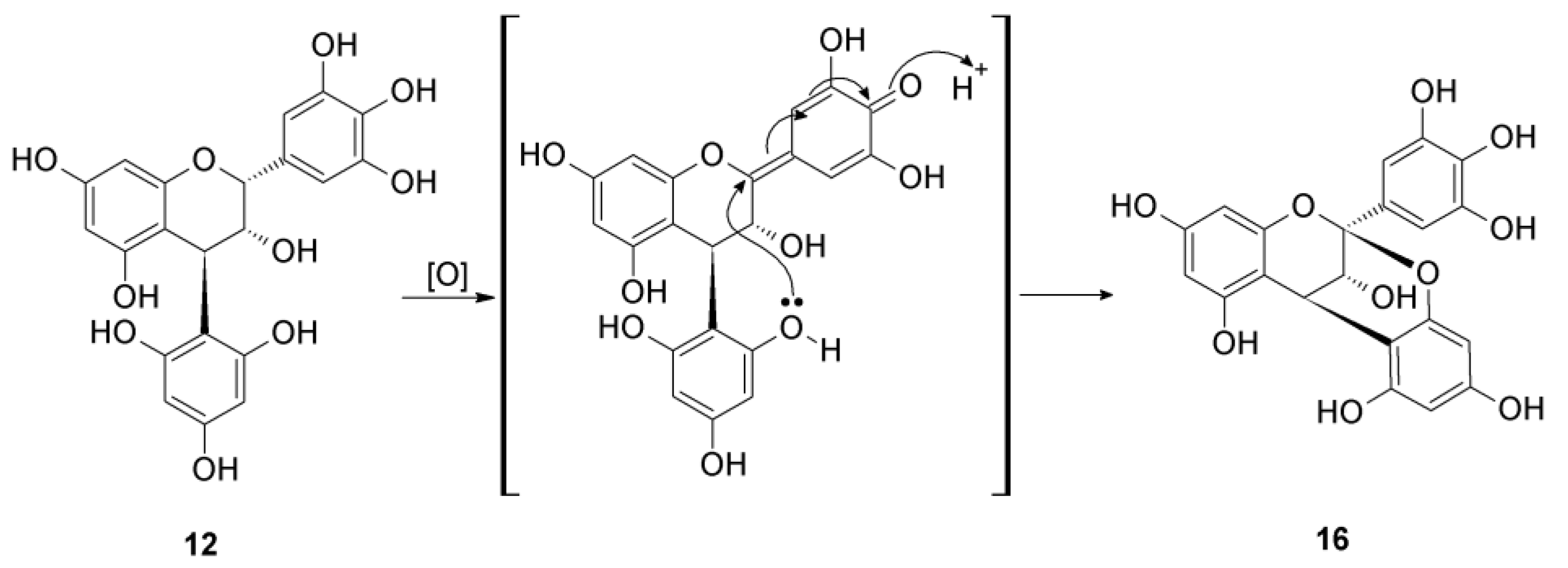
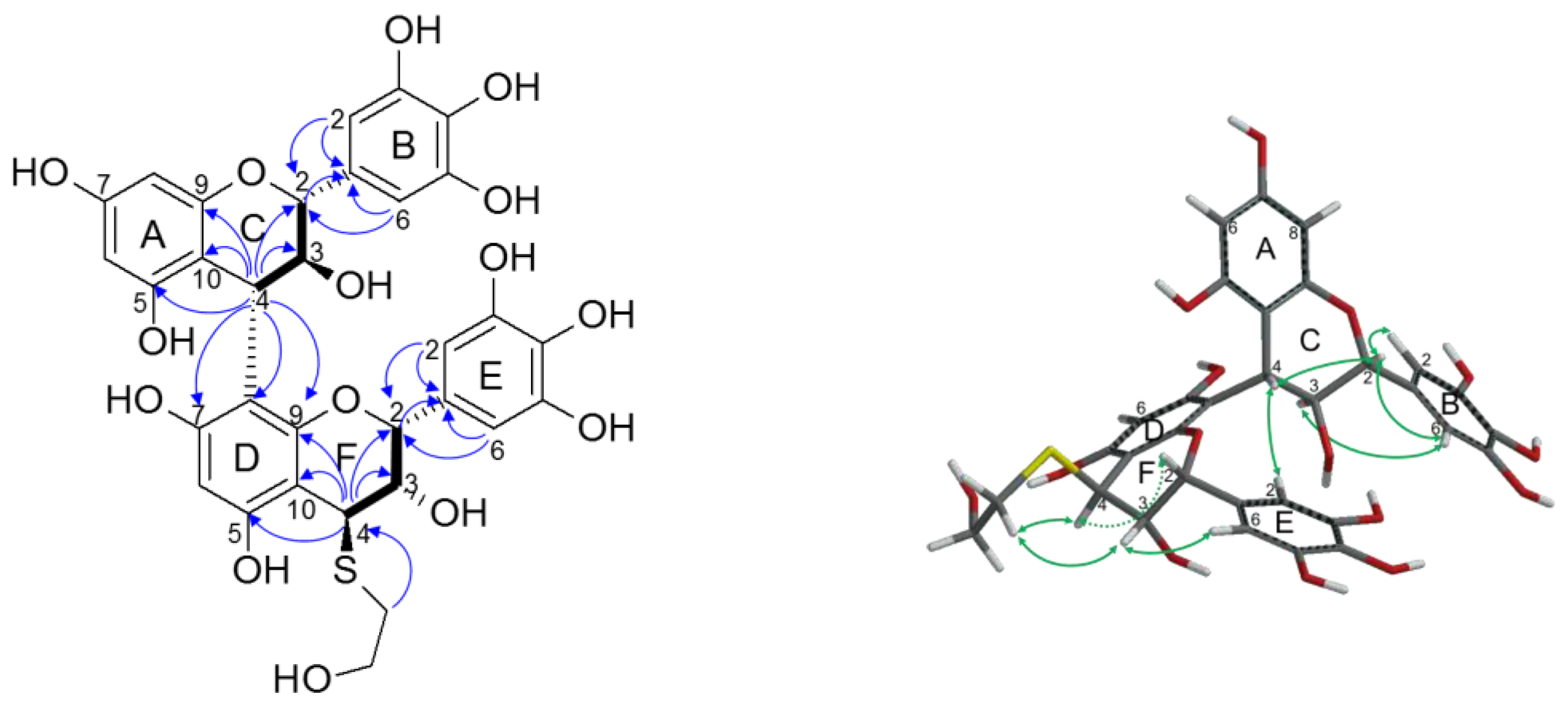
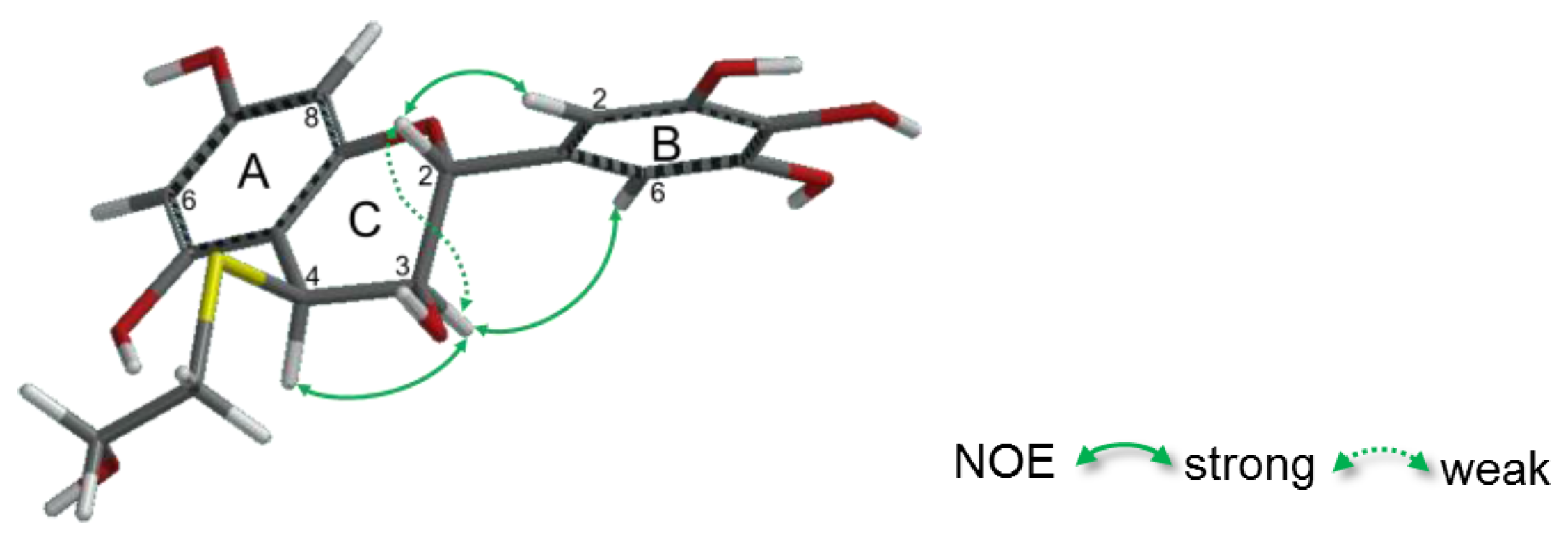
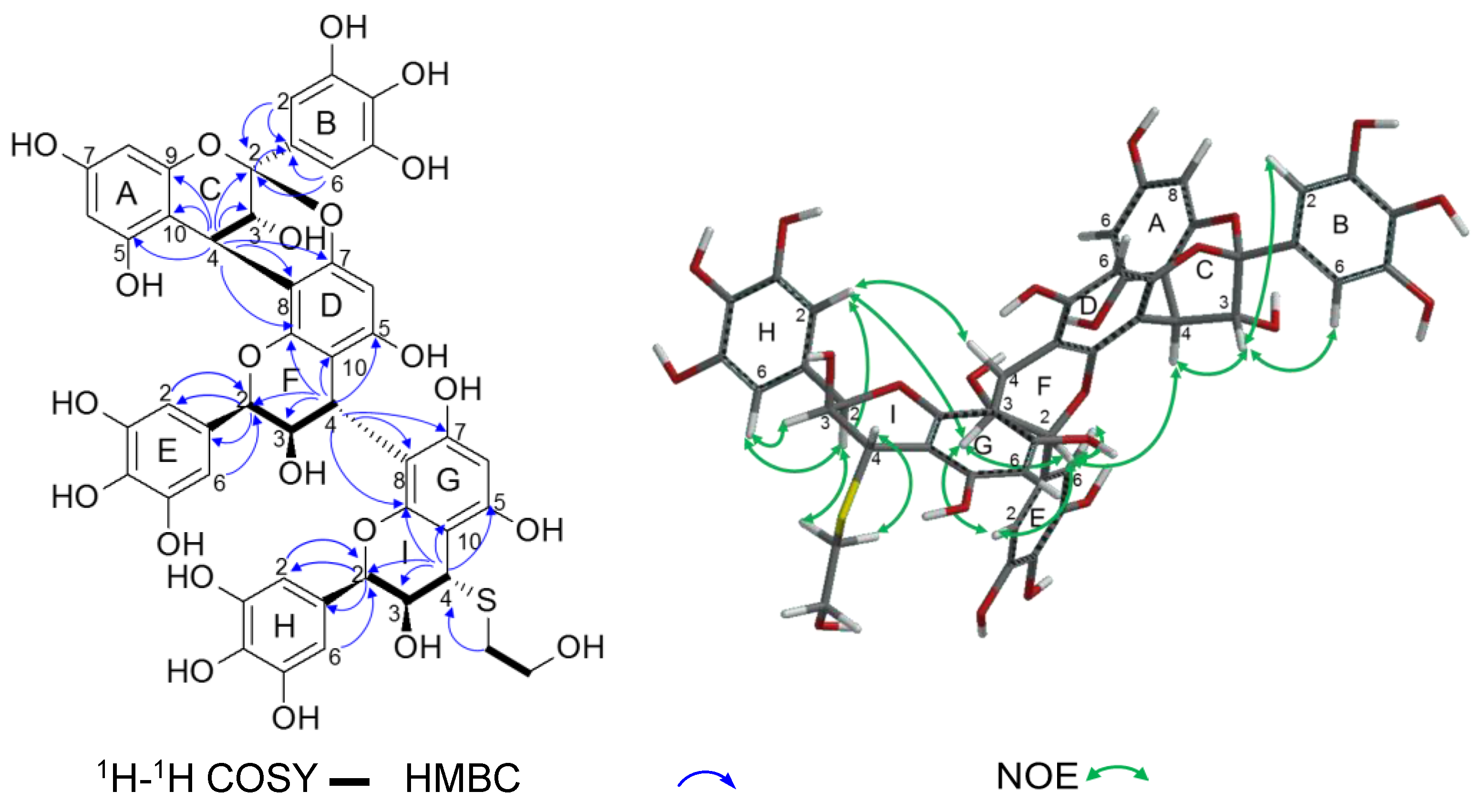
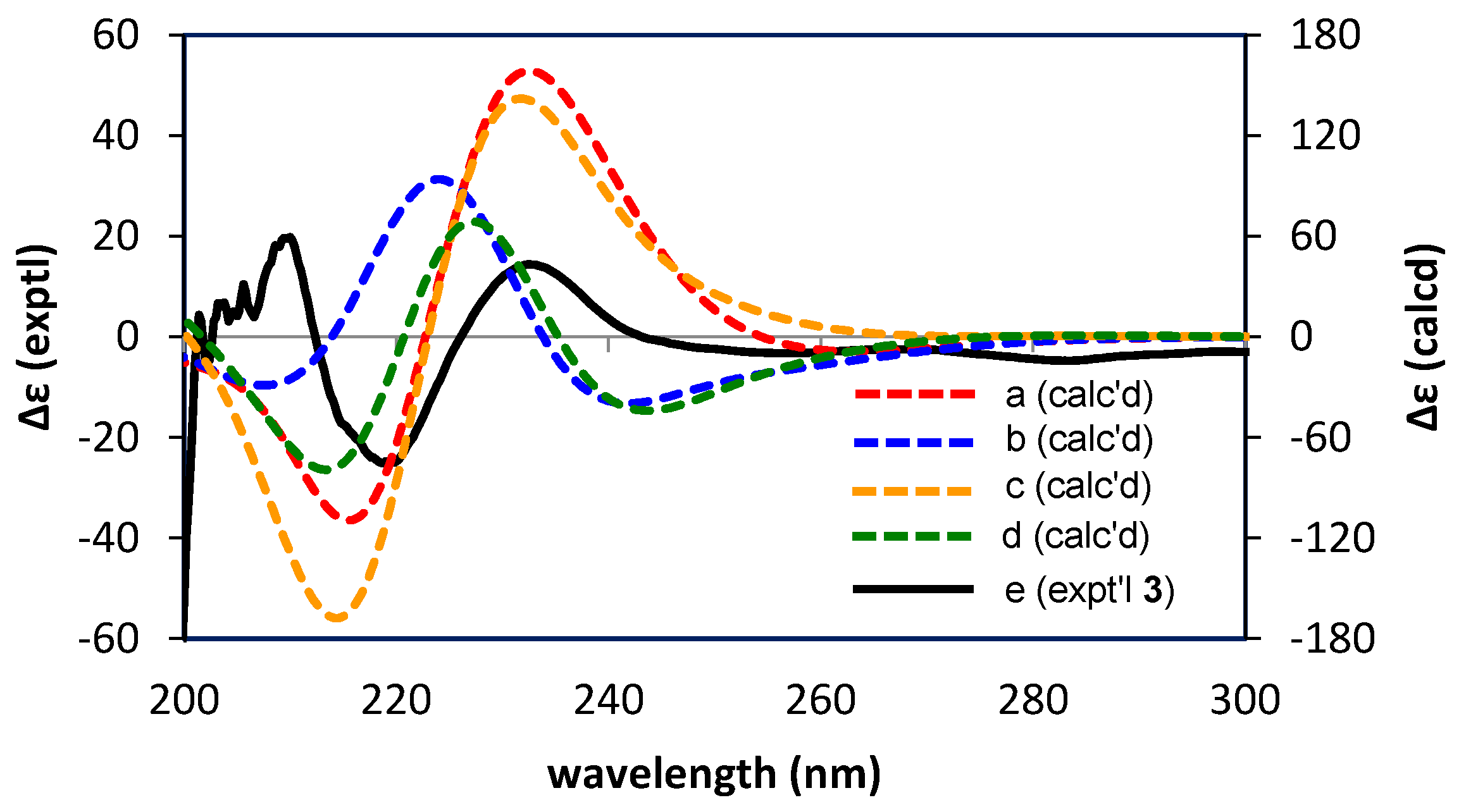
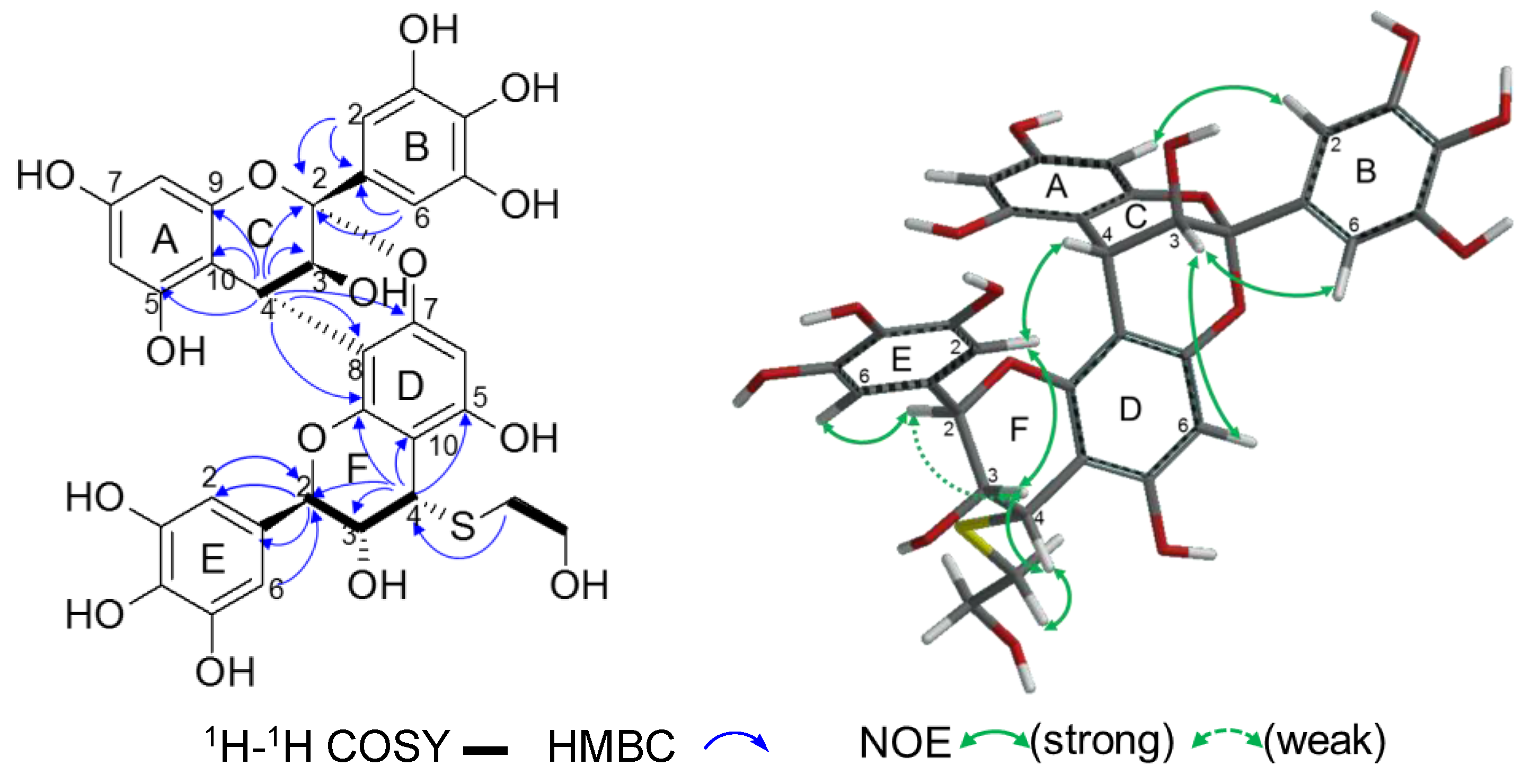
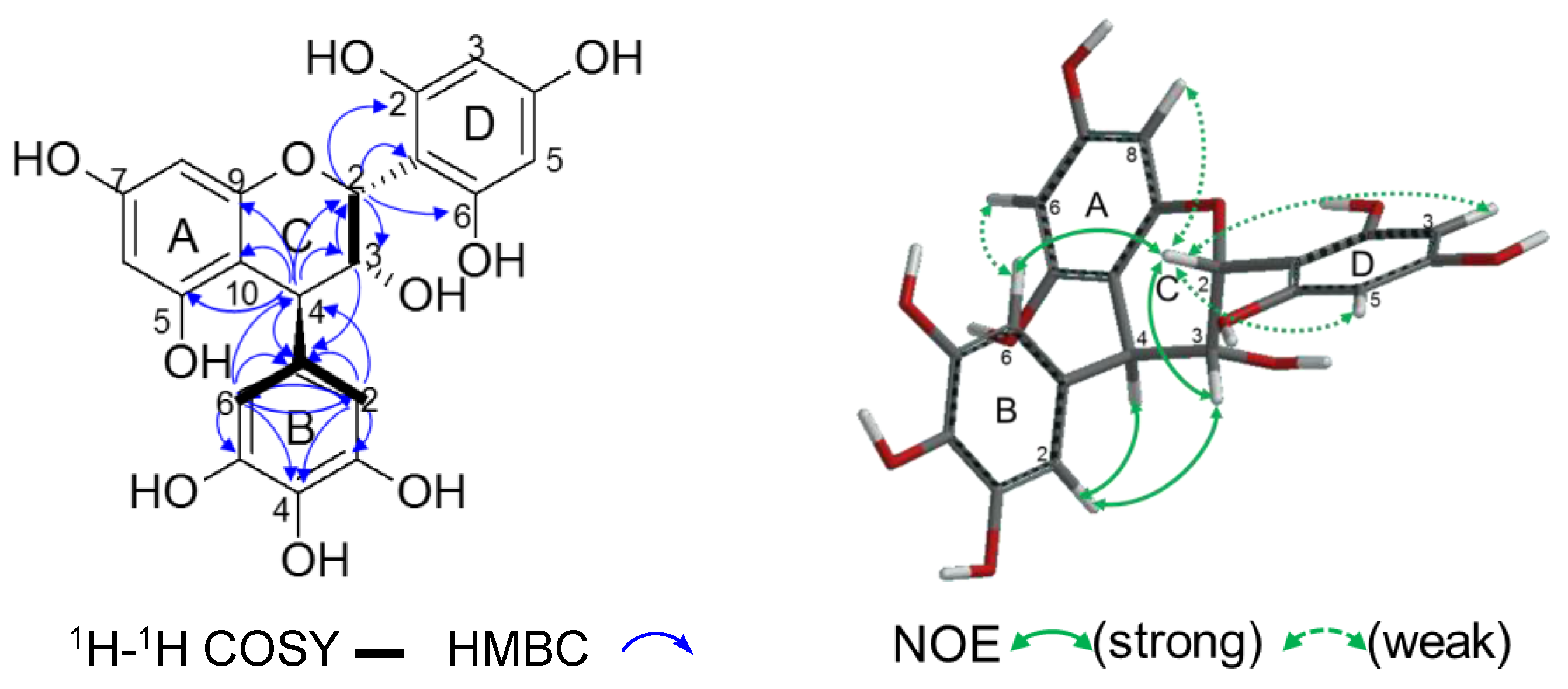
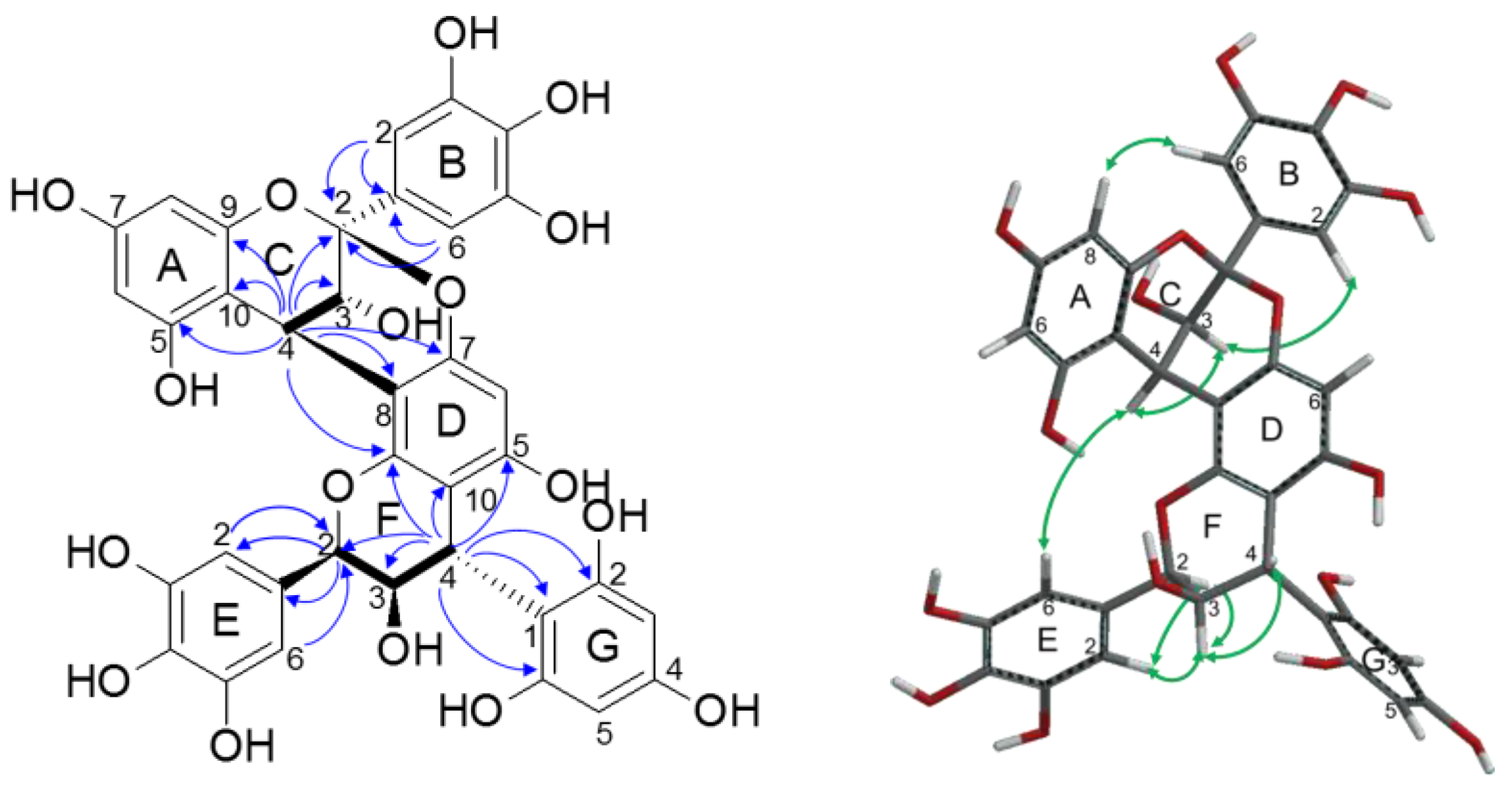
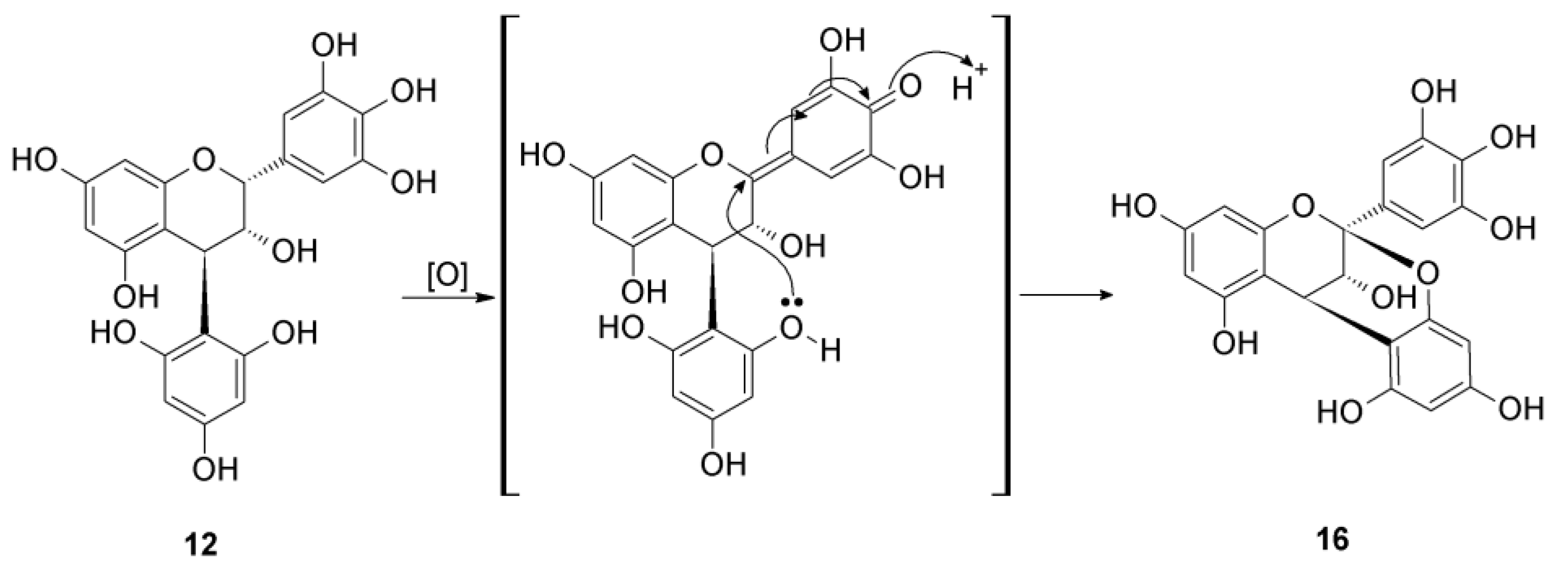
2.2.2. Structure Elucidation of New Degradation Products
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Thiolysis
3.5. Phloroglucinolysis
3.6. Calculations of ECD Spectra
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, Y. Chinese Materia Medica: Chemistry, Pharmacology and Applications; Harwood Academic Publisher: Amsterdam, The Netherlands, 1998; pp. 48–51. ISBN 90-5702-285-0. [Google Scholar]
- World Health Organization. WHO Monographs on Selected Medicinal Plants; World Health Organization: Geneva, Switzerland, 1999; Volume 1, pp. 145–153. ISBN 9241545178. [Google Scholar]
- Wang, L.; Kakiuchi, N.; Mikage, M. Studies of Ephedra plants in Asia. Part 6: Geographical changes of anatomical features and alkaloids content of Ephedra sinica. J. Nat. Med. 2009, 64, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheu, S.; Chiou, S.; Chang, H.; Chen, Y. A comparative study of commercial samples of Ephedrae herba. Planta Med. 1993, 59, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Westfall, T.; Westfall, D. Adrenergic agonists and antagonists. In Goodman & Gilman's the Pharmacological Basis of Therapeutics, 12th ed.; Brunton, L., Chabner, B., Knollman, B., Eds.; McGraw-Hill Medical: New York, NY, USA, 2011; pp. 277–333. ISBN 978-0-07-162442-8. [Google Scholar]
- Caveney, S.; Charlet, D.; Freitag, H.; Maier-Stolte, M.; Starratt, A. New observations on the secondary chemistry of world Ephedra (Ephedraceae). Am. J. Bot. 2001, 88, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, T.; Pieters, L.; Delstra, H.; Vlietinck, A. Condensed vegetable tannins: Biodiversity in structure and biological activities. Biochem. Syst. Ecol. 1999, 27, 445–459. [Google Scholar] [CrossRef]
- Magos, G.; Mateos, J.; Páez, E.; Fernández, G.; Lobato, C.; Márquez, C.; Enríquez, R. Hypotensive and vasorelaxant effects of the procyanidin fraction from Guazuma ulmifolia bark in normotensive and hypertensive rats. J. Ethnopharm. 2008, 117, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Terencio, M.; Paya, M. Isolation and hypotensive activity of a polymeric procyanidin fraction from Pistacia lentiscus L. Pharmazie 1992, 47, 466–467. [Google Scholar] [PubMed]
- Zeng, X.; Ma, Y.; Gu, H.; Gu, Y.; Huang, M. The effect of oligomeric proanthocyanidin on airway microenvironment in asthma. Eur. Respir. J. 2016, 48 (Suppl. S60). [Google Scholar] [CrossRef]
- Zhou, D.; Fang, S.; Zou, C.; Zhang, Q.; Gu, W. Proanthocyanidin from grape seed extract inhibits airway inflammation and remodeling in a murine model of chronic asthma. Nat. Prod. Commun. 2015, 10, 257–262. [Google Scholar] [PubMed]
- Zang, X.; Shang, M.; Xu, F.; Liang, J.; Wang, X.; Mikage, M.; Cai, S. A-Type proanthocyanidins from the stems of Ephedra sinica (Ephedraceae) and their antimicrobial activities. Molecules 2013, 18, 5172–5189. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Gavkosh, S.; Bigdeli, M.; Shams-Ghahfarokhi, M.; Razzaghi-Abyaneh, M. Inhibitory effects of Ephedra major host on Aspergillus parasiticus growth and aflatoxin production. Mycopathologia 2009, 168, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Okawa, M.; Kinjo, J.; Nohara, T.; Ono, M. DPPH (1,1-diphenyl-2-picrylhydrazyl) radical scavenging activity of flavonoids obtained from some medicinal plants. Biol. Pharm. Bull. 2001, 24, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Park, Y.J.; Yoon, S.; Lee, H. Ephedrannin A and B from roots of Ephedra sinica inhibit lipopolysaccharide-induced inflammatory mediators by suppressing nuclear factor-kB activation in RAW 264.7 macrophages. Int. Immunopharmacol. 2010, 10, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Son, K.; Chang, H.; Kang, S.; Kim, H. Inhibitory effects of plant extracts on adjuvant-induced arthritis. Arch. Pharm. Res. 1997, 20, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhu, G.; Xu, Z. Effect of different extracts from Ephedra on cell immunity. J. Nanjing Univ. Tradit. Chin. Med. 2001, 4, 15. [Google Scholar]
- Li, L.; Yu, C.; Ying, H.; Yu, J. Antiviral effects of modified Dingchuan decoction against respiratory syncytial virus infection in vitro and in an immunosuppressive mouse model. J. Ethnopharm. 2013, 147, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.; Lee, C.; Hong, D.; Kim, H.; Bae, K.; Ahn, B. Antiinvasive, antiangiogenic and antitumor activity of Ephedra sinica extract. Phytother. Res. 2003, 17, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Cheng, W.; Che, C.; Hsieh, D. Cytotoxicity assessment of Ma-huang (Ephedra) under different conditions of preparation. Toxicol. Sci. 2000, 56, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Wang, L.; Cui, Z.; Zhao, D.; Liu, Y. Dimeric proanthocyanidins from the roots of Ephedra sinica. Planta Med. 2008, 74, 1823–1825. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Fujioka, K.; Oura, H.; Tanaka, T.; Nonaka, G.; Nishioka, I. Decrease in uremic toxins, a newly found beneficial effect of Ephedrae Herba. Phytother. Res. 1995, 9, 382–384. [Google Scholar] [CrossRef]
- Wang, G.Z.; Hikokichi, O. Experimental study in treating chronic renal failure with dry extract and tannins of Herba Ephedra. Zhongguo Zhong Xi Yi Jie He Za Zhi 1994, 14, 485–488. [Google Scholar] [PubMed]
- King, F.; Clark-Lewis, J.; Forbes, W. The chemistry of extractives from hardwoods. Part XXV. (—)-epiAfzelechin, a new member of the catechin series. J. Chem. Soc. 1955, 2948–2956. [Google Scholar] [CrossRef]
- Hikino, H.; Takahashi, M.; Konno, C. Structure of ephedrannin A, a hypotensive principle of Ephedra roots. Tetrahedron Lett. 1982, 23, 673–676. [Google Scholar] [CrossRef]
- Hikino, H.; Shimoyama, N.; Kasahara, Y.; Takahashi, M.; Konno, C. Structures of mahuannin A and B, hypotensive principles of Ephedra roots. Heterocycles 1982, 19, 1381–1384. [Google Scholar] [CrossRef]
- Kasahara, H.; Hikino, H. Structure of mahuannin D, a hypotensive principle of Ephedra roots. Heterocycles 1983, 20, 1953–1956. [Google Scholar] [CrossRef]
- Nonaka, G.; Morimoto, S.; Kinjo, J.; Nohara, T.; Nishioka, I. Tannins and related compounds L. Structures of proanthocyanidin A-1 and related compounds. Chem. Pharm. Bull. 1987, 35, 149–155. [Google Scholar] [CrossRef]
- Kennedy, J.; Jones, G. Analysis of proanthocyanidin cleavage products following acid-catalysis in the presence of excess phloroglucinol. J. Agric. Food Chem. 2001, 49, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, A.; Shoji, T.; Shibusawa, Y. Separation of proanthocyanidins by degree of polymerization by means of size-exclusion chromatography and related techniques. J. Biochem. Biophys. Methods 2003, 56, 311–322. [Google Scholar] [CrossRef]
- Kolodziej, H. 1H NMR spectral studies of procyanidin derivatives: Diagnostic 1H NMR parameters applicable to the structural elucidation of oligomeric procyanidins. In Plant Polyphenols-Synthesis, Properties, Significance; Hemingway, R., Laks, P.E., Eds.; Plenum Press: New York, NY, USA, 1992; pp. 295–319. ISBN 978-1-4615-3476-1. [Google Scholar]
- Porter, L.; Newman, R.; Foo, L.; Wong, H.; Hemingway, R. Polymeric proanthocyanidins. 13C-NMR studies of procyanidins. J. Chem. Soc. Perkin Trans. 1 1982, 1217–1221. [Google Scholar] [CrossRef]
- Foo, L.; Newman, R.; Waghorn, G.; McNabb, W.; Ulyatt, M. Proanthocyanidins from Lotus corniculatus. Phytochemistry 1996, 41, 617–624. [Google Scholar] [CrossRef]
- Chai, W.; Shi, Y.; Feng, H.; Qiu, L.; Zhou, H.; Deng, Z.; Yan, C.; Chen, X. NMR, HPLC-ESI-MS, and MALDI-TOF MS analysis of condensed tannins from Delonix regia (Bojer ex Hook.) Raf. and their bioactivities. J. Agric. Food Chem. 2012, 60, 5013–5022. [Google Scholar] [CrossRef] [PubMed]
- Kusano, R.; Ogawa, S.; Matsuo, Y.; Tanaka, T.; Yazaki, Y.; Kouno, I. α-amylase and lipase inhibitory activity and structural characterization of acacia bark proanthocyanidins. J. Nat. Prod. 2011, 74, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Ortin, A.; Molero, N.; Marín, F.; Ruiz-García, Y.; Gómez-Plaza, E. Reactivity of pure and commercial grape skin tannins with cell wall material. Eur. Food Res. Technol. 2015, 240, 645–654. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, R.; Kouno, I.; Nonaka, G. Chemical evidence for the de-astringency (insolubilization of tannins) of persimmon fruit. J. Chem. Soc. Perkin Trans. 1 1994, 3013–3022. [Google Scholar] [CrossRef]
- Tanaka, T.; Kondou, K.; Kouno, I. Oxidation and epimerization of epigallocatechin in banana fruits. Phytochemistry 2000, 53, 311–316. [Google Scholar] [CrossRef]
- Steynberg, J.; Brandt, E.; Hoffman, M.; Ferreira, D. Conformations of proanthocyanidins. In Plant Polyphenols-Synthesis, Properties, Significance; Hemingway, R., Laks, P.E., Eds.; Plenum Press: New York, NY, USA, 1992; pp. 501–519. ISBN 978-1-4615-3476-1. [Google Scholar]
- Slade, D.; Ferreira, D.; Marais, J.P.J. Circular dichroism, a powerful tool for the assessment of absolute configuration of flavonoids. Phytochemistry 2005, 66, 2177–2215. [Google Scholar] [CrossRef] [PubMed]
- Esatbeyoglu, T.; Jaschok-Kentner, B.; Wray, V.; Winterhalter, P. Structure elucidation of proanthocyanidin oligomers by low-temperature 1H NMR spectroscopy. J. Agric. Food Chem. 2011, 59, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.W.; Phansaklar, R.S.; Lankin, D.C.; McAlpine, J.B.; Leme-Kraus, A.A.; Vidal, C.M.; Gan, L.S.; Bedran-Russo, A.; Chen, S.N.; Pauli, G.F. Absolute configuration of native oligomeric proanthocyanidins with dentin biomodification potency. J. Org. Chem. 2017, 82, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.; Klyne, W.; Scopes, P.; Fletcher, A.; Porter, L.; Haslam, E. Plant proanthocyanidins. Part 6. Chiroptical studies. Part 95. Circular dichroism of procyanidins. J. Chem. Soc. Perkin Trans. 1 1979, 2375–2377. [Google Scholar] [CrossRef]
- Yu, R.J.; Liu, H.B.; Yu, Y.; Liang, L.; Xu, R.; Liang, C.; Tang, J.S.; Yao, X.S. Anticancer activities of proanthocyanidins from the plant Urceola huaitingii and their synergistic effects in combination with chemotherapeutics. Fitoterapia 2016, 112, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.; Kolodziej, H.; Hemingway, R.; Steynberg, J.; Young, D.; Ferreira, D. Oligomeric flavonoids. Part 15a. Base-catalyzed pyran rearrangements of procyanidin B-2, and evidence for the oxidative transformation of B- to A-type procyanidins. Tetrahedron 1990, 46, 5733–5740. [Google Scholar] [CrossRef]
- Koerner, J.; Hsu, V.; Lee, J.K. Determination of proanthocyanidin A2 content in phenolic polymer isolates by reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Cronjé, A.; Burger, J.; Brandt, E.; Kolodziej, H.; Ferreira, D. Assessment of 3,4-trans and 3,4-cis relative configurations in the A-series of 4,8-linked proanthocyanidins. Tetrahedron Lett. 1990, 31, 3789–3792. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
Sample Availability: Samples of compounds 1–18 are all available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 7 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δH | ( J in Hz) | δC | δH | ( J in Hz) | δC | δH | ( J in Hz) | δC | δH | ( J in Hz) | δC | ||||
| C | 2 | 4.34 | d (9.5) | 83.19 | 4.77 | d(9.7) | 78.77 | 99.91 | 100.07 | |||||||
| 3 | 4.50 | dd (7.8, 9.5) | 73.26 | 4.07 | dd(9.7, 4.4) | 71.59 | 4.20 | d (3.4) | 67.31 | 4.15 | d (3.6) | 66.82 | ||||
| 4 | 4.68 | d (7.8) | 38.11 | 4.36 | d(4.4) | 44.90 | 4.38 | d (3.4) | 28.68 | 4.17 | d (3.6) | 28.47 | ||||
| A | 5 | 158.27 | 157.51 | 156.59 | 156.04 | |||||||||||
| 6 | 5.82 | s | 97.04 | 6.00 | d(2.3) | 96.67 | 5.85 | d (2.4) | 97.58 | 5.89 | d (2.3) | 97.62 | ||||
| 7 | 157.15 | 159.14 | 157.69 | 157.88 | ||||||||||||
| 8 | 5.82 | d (1.7) | 95.82 | 5.78 | d(2.3) | 94.96 | 6.05 | s | 96.00 | 6.02 | d (2.3) | 96.12 | ||||
| 9 | 156.93 | 155.88 | 151.65 | 153.76 | ||||||||||||
| 10 | 106.09 | 102.14 | 103.92 | 103.38 | ||||||||||||
| B | 1 | 131.56 | 130.36 | 131.50 | 130.96 | |||||||||||
| 2 | 6.57 | s | 107.94 | 6.50 | s | 108.19 | 6.77 | s | 107.23 | 6.74 | s | 107.23 | ||||
| 3 | 146.16 | 146.08 | 145.63 | 145.64 | ||||||||||||
| 4 | 133.28 | 133.49 | 133.74 | 133.81 | ||||||||||||
| 5 | 146.16 | 146.08 | 145.63 | 145.64 | ||||||||||||
| 6 | 6.57 | s | 107.94 | 6.50 | s | 108.19 | 6.77 | s | 107.23 | 6.74 | s | 107.23 | ||||
| F | 2 | 5.31 | br s | 75.05 | 5.31 | br s | 78.02 | 4.99 | d (9.8) | 79.57 | ||||||
| 3 | 4.04 | d (1.1) | 71.95 | 3.95 | d (2.2) | 72.84 | 4.21 | dd (9.8, 4.3) | 71.02 | |||||||
| 4 | 4.05 | d (1.1) | 43.67 | 4.79 | d (2.2) | 36.21 | 4.39 | d (4.3) | 44.43 | |||||||
| D | 5 | 156.49 | 155.79 | 155.74 | ||||||||||||
| 6 | 6.04 | s | 97.69 | 6.05 | s | 96.00 | 6.13 | s | 96.92 | |||||||
| 7 | 156.49 | 153.78 | 153.02 | |||||||||||||
| 8 | 108.15 | 106.13 | 105.98 | |||||||||||||
| 9 | 154.34 | 151.31 | 149.62 | |||||||||||||
| 10 | 98.91 | 104.48 | 104.33 | |||||||||||||
| E | 1 | 130.97 | 130.62 | 129.08 | ||||||||||||
| 2 | 6.67 | s | 106.11 | 6.56 | s | 106.42 | 6.64 | s | 108.00 | |||||||
| 3 | 145.94 | 146.27 | 146.32 | |||||||||||||
| 4 | 132.60 | 133.08 | 134.05 | |||||||||||||
| 5 | 145.94 | 146.27 | 146.32 | |||||||||||||
| 6 | 6.67 | s | 106.11 | 6.56 | s | 106.42 | 6.64 | s | 108.00 | |||||||
| I | 2 | 5.29 | br s | 75.06 | ||||||||||||
| 3 | 4.14 | d (2.3) | 71.18 | |||||||||||||
| 4 | 4.12 | d (2.3) | 43.83 | |||||||||||||
| G | 5 | 156.77 | ||||||||||||||
| 6 | 5.96 | s | 97.45 | |||||||||||||
| 7 | 157.05 | |||||||||||||||
| 8 | 106.91 | |||||||||||||||
| 9 | 153.90 | |||||||||||||||
| 10 | 99.96 | |||||||||||||||
| H | 1 | 130.93 | ||||||||||||||
| 2 | 6.70 | s | 106.42 | |||||||||||||
| 3 | 146.14 | |||||||||||||||
| 4 | 132.74 | |||||||||||||||
| 5 | 146.14 | |||||||||||||||
| 6 | 6.70 | s | 106.42 | |||||||||||||
| CH2OH- | 3.73-3.89 | m | 62.65 | 3.68-3.88 | m | 62.68 | 3.73-3.94 | m | 62.72 | 3.73-3.84 | m | 62.47 | ||||
| SCH2- | 2.75-2.96 | m | 34.95 | 2.75-3.11 | m | 37.22 | 2.78-2.99 | m | 35.14 | 2.77-3.09 | m | 37.54 | ||||
| 13 | 14 | 16 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δ H | (J in Hz) | δC | δH | (J in Hz) | δC | δH | (J in Hz) | δC | |||
| C | 2 | 5.53 | br s | 70.22 | 99.97 | 100.21 | ||||||
| 3 | 4.05 | dd (2.4,1.0) | 74.27 | 4.22 | d (3.5) | 67.26 | 4.15 | d (3.6) | 66.99 | |||
| 4 | 4.06 | br s | 45.70 | 4.38 | d (3.5) | 28.66 | 4.25 | d (3.6) | 28.74 | |||
| A | 5 | 158.59 | 156.60 | 155.32 | ||||||||
| 6 | 5.95 | d (2.4) | 96.59 | 5.84 | d (2.4) | 97.58 | 6.04 | d (1.5) | 97.21 | |||
| 7 | 158.05 | 157.65 | 157.69 | |||||||||
| 8 | 5.94 | d (2.4) | 95.24 | 6.04 | d (2.4) | 96.17 | 6.07 | br s | 96.21 | |||
| 9 | 157.76 | 153.79 | 153.96 | |||||||||
| 10 | 102.49 | 104.06 | 104.26 | |||||||||
| B | 1 | 135.98 | 131.53 | 131.23 | ||||||||
| 2 | 6.19 | s | 108.50 | 6.78 | s | 107.29 | 6.53 | s | 107.32 | |||
| 3 | 146.47 | 145.62 | 145.62 | |||||||||
| 4 | 132.19 | 133.73 | 133.76 | |||||||||
| 5 | 146.47 | 145.62 | 145.62 | |||||||||
| 6 | 6.19 | s | 108.50 | 6.78 | s | 107.29 | 6.53 | s | 107.32 | |||
| F | 2 | 5.32 | br s | 78.12 | ||||||||
| 3 | 3.95 | d (2.3) | 72.61 | |||||||||
| 4 | 4.63 | d (2.3) | 36.24 | |||||||||
| D | 1 | 104.59 | 106.96 | |||||||||
| 2 | 158.59 | 153.81 | ||||||||||
| 3 | 5.79 | s | 95.95 | 5.96 | d (1.5) | 96.01 | ||||||
| 4 | 159.52 | 158.18 | ||||||||||
| 5 | 5.79 | s | 95.95 | 156.16 | 6.03 | d (1.5) | 97.19 | |||||
| 6 | 158.59 | 6.02 | s | 95.63 | 154.23 | |||||||
| 7 | 151.54 | |||||||||||
| 8 | 105.79 | |||||||||||
| 9 | 157.65 | |||||||||||
| 10 | 104.58 | |||||||||||
| E | 1 | 130.96 | ||||||||||
| 2 | 6.58 | s | 106.42 | |||||||||
| 3 | 146.27 | |||||||||||
| 4 | 133.02 | |||||||||||
| 5 | 146.27 | |||||||||||
| 6 | 6.58 | s | 106.42 | |||||||||
| G | 1 | 106.90 | ||||||||||
| 2 | 151.31 | |||||||||||
| 3 | 6.05 | s | 95.96 | |||||||||
| 4 | 155.43 | |||||||||||
| 5 | 6.05 | s | 95.96 | |||||||||
| 6 | 151.31 | |||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orejola, J.; Matsuo, Y.; Saito, Y.; Tanaka, T. Characterization of Proanthocyanidin Oligomers of Ephedra sinica. Molecules 2017, 22, 1308. https://doi.org/10.3390/molecules22081308
Orejola J, Matsuo Y, Saito Y, Tanaka T. Characterization of Proanthocyanidin Oligomers of Ephedra sinica. Molecules. 2017; 22(8):1308. https://doi.org/10.3390/molecules22081308
Chicago/Turabian StyleOrejola, Joanna, Yosuke Matsuo, Yoshinori Saito, and Takashi Tanaka. 2017. "Characterization of Proanthocyanidin Oligomers of Ephedra sinica" Molecules 22, no. 8: 1308. https://doi.org/10.3390/molecules22081308
APA StyleOrejola, J., Matsuo, Y., Saito, Y., & Tanaka, T. (2017). Characterization of Proanthocyanidin Oligomers of Ephedra sinica. Molecules, 22(8), 1308. https://doi.org/10.3390/molecules22081308