Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
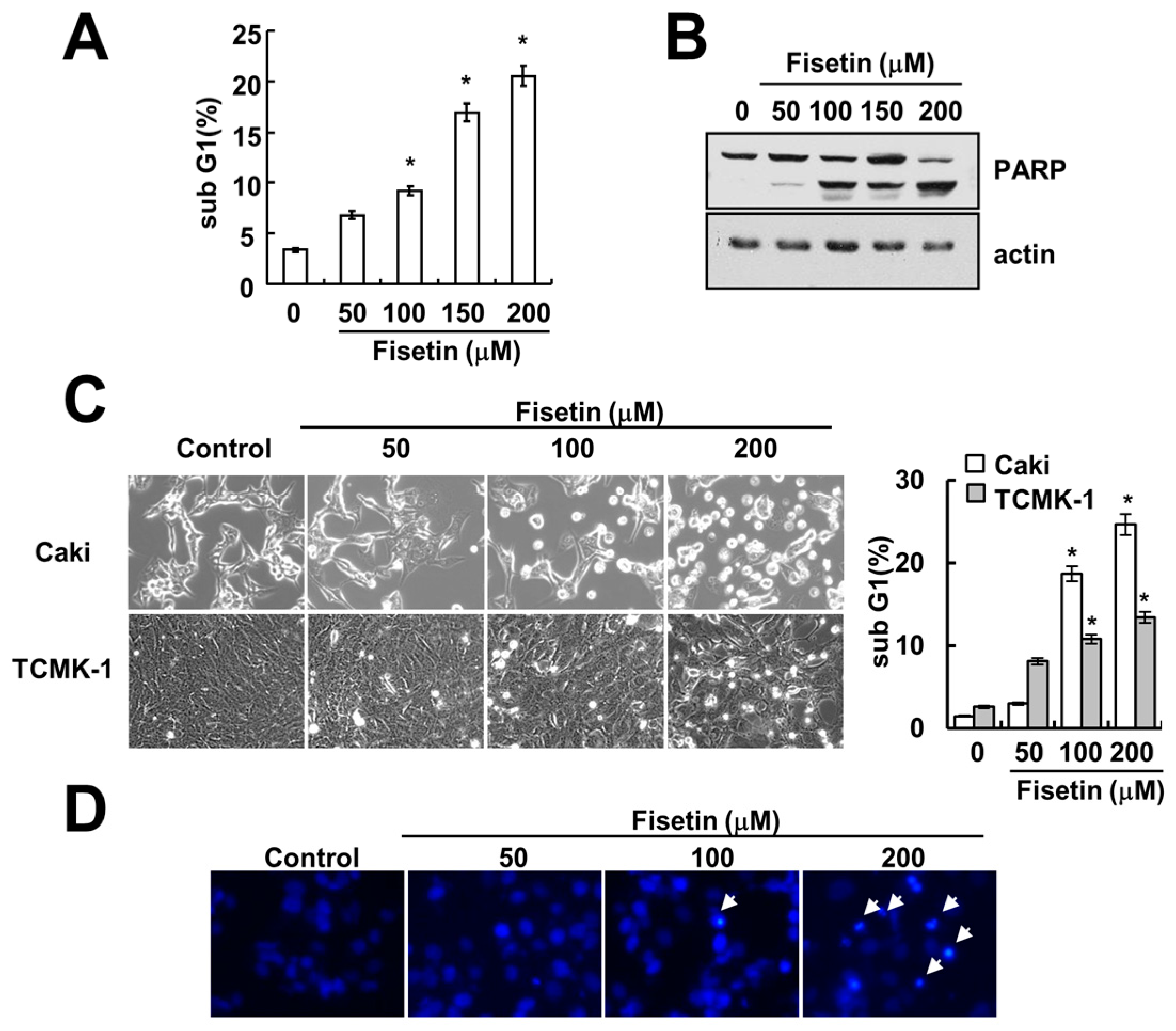
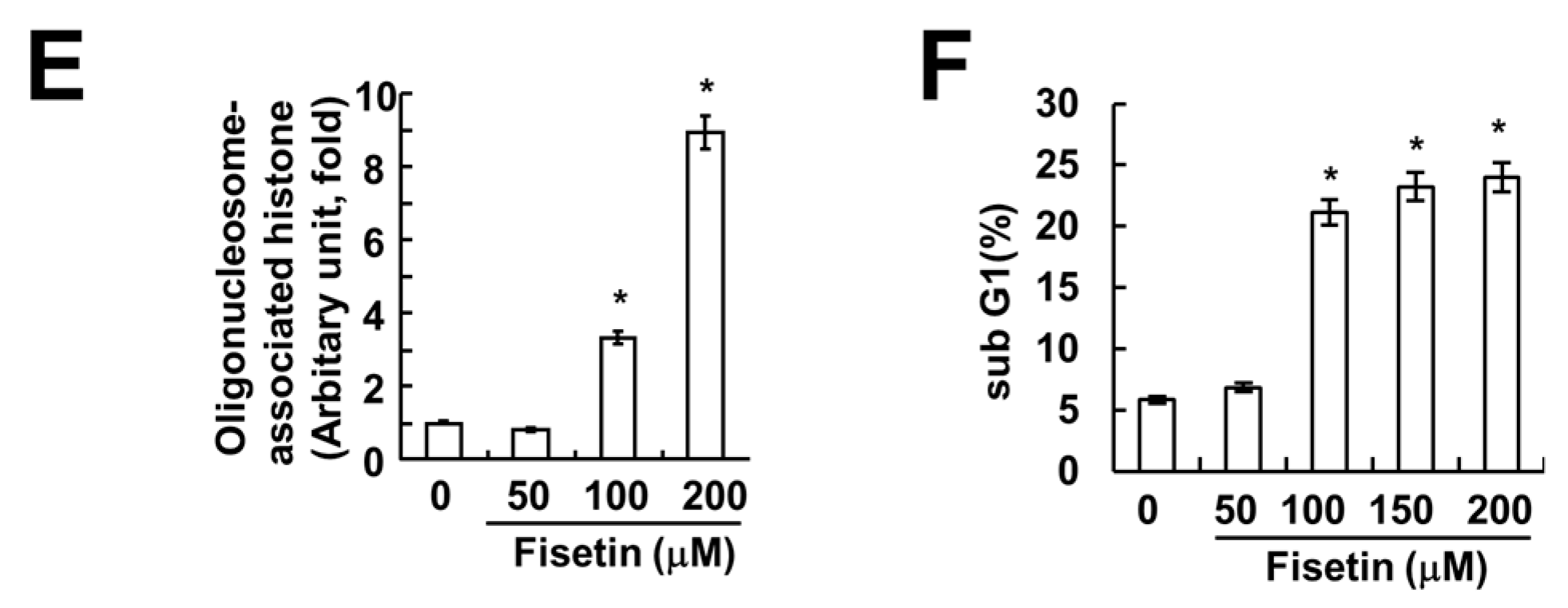
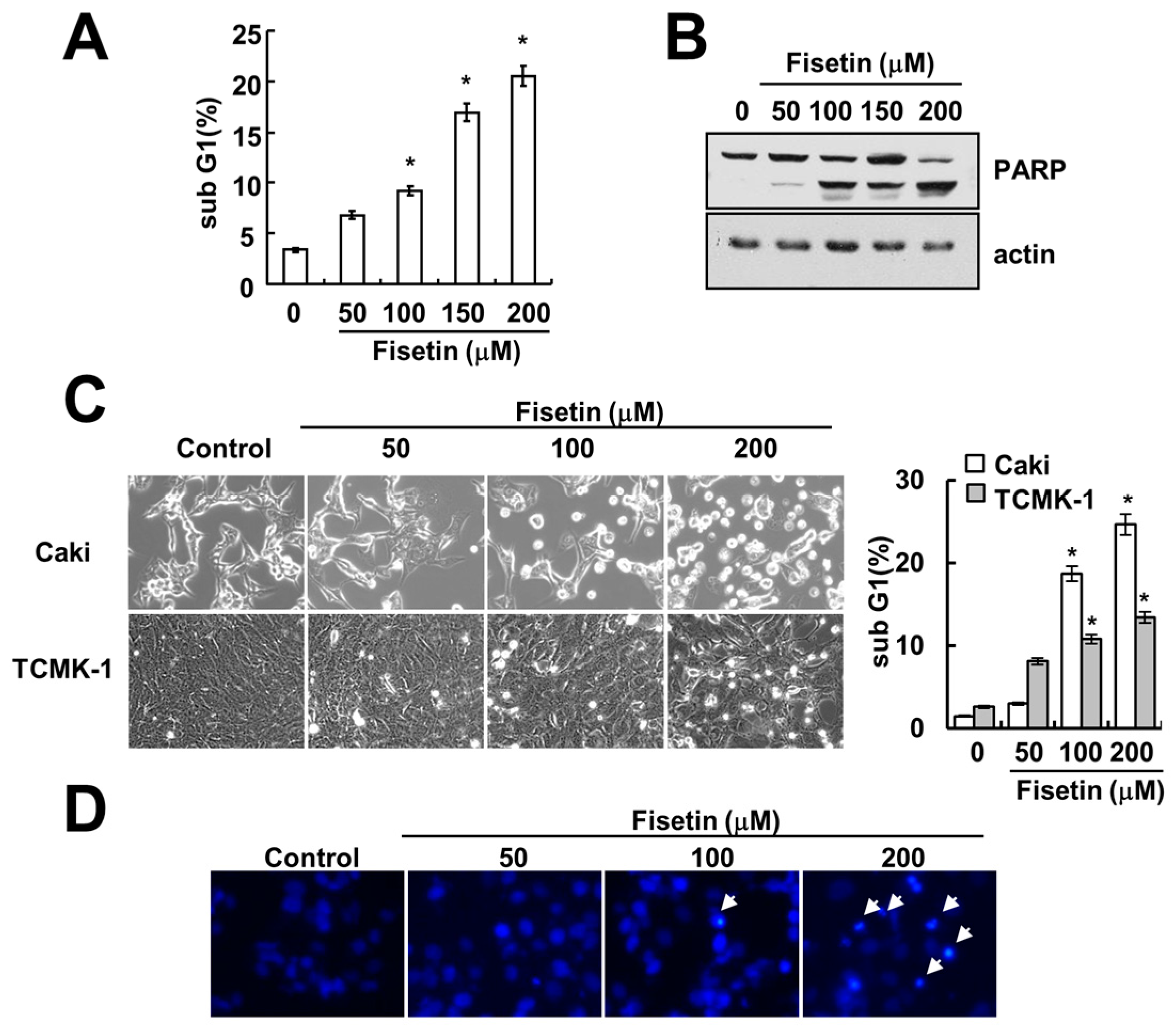
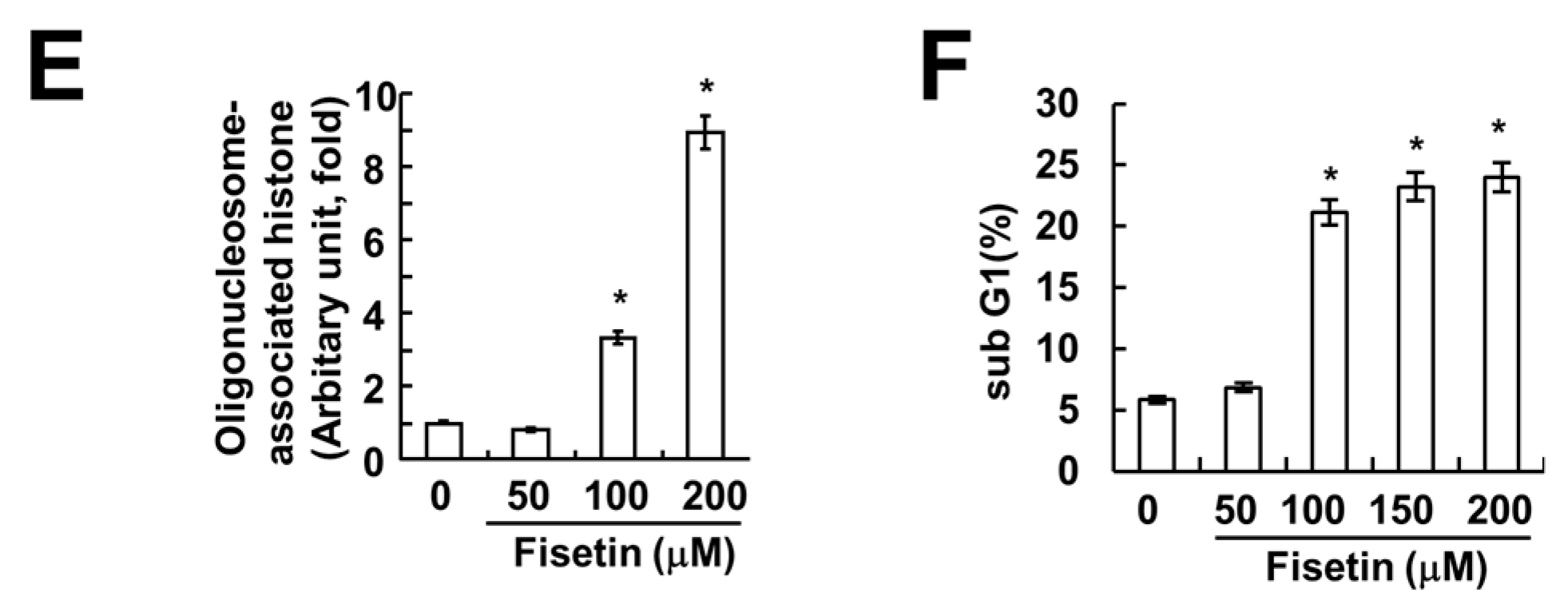
2.1. Fisetin Induces Apoptosis in Human Renal Carcinoma Caki Cells
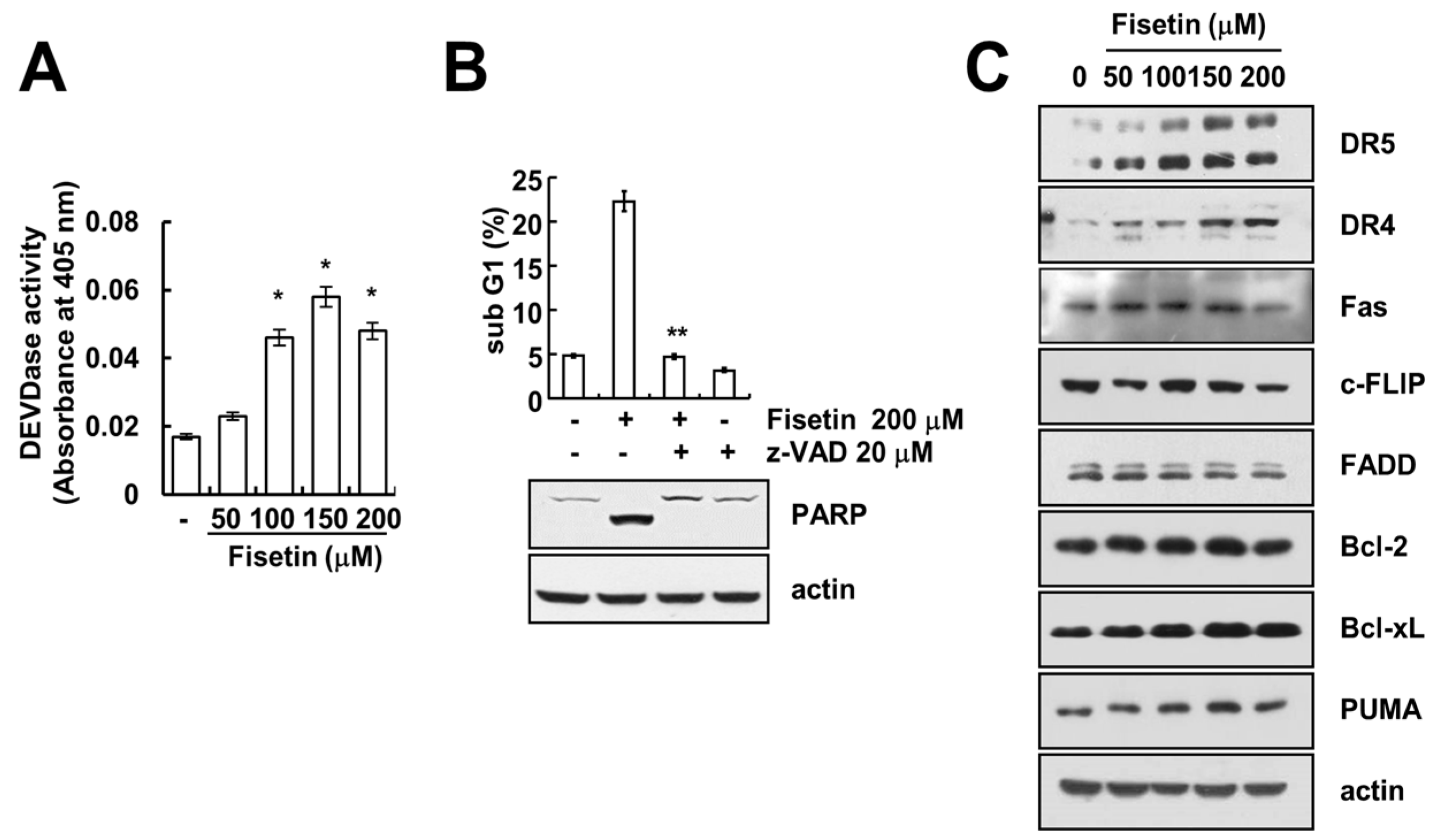
2.2. Caspase Activation Is Involved in Fisetin-Induced Apoptosis
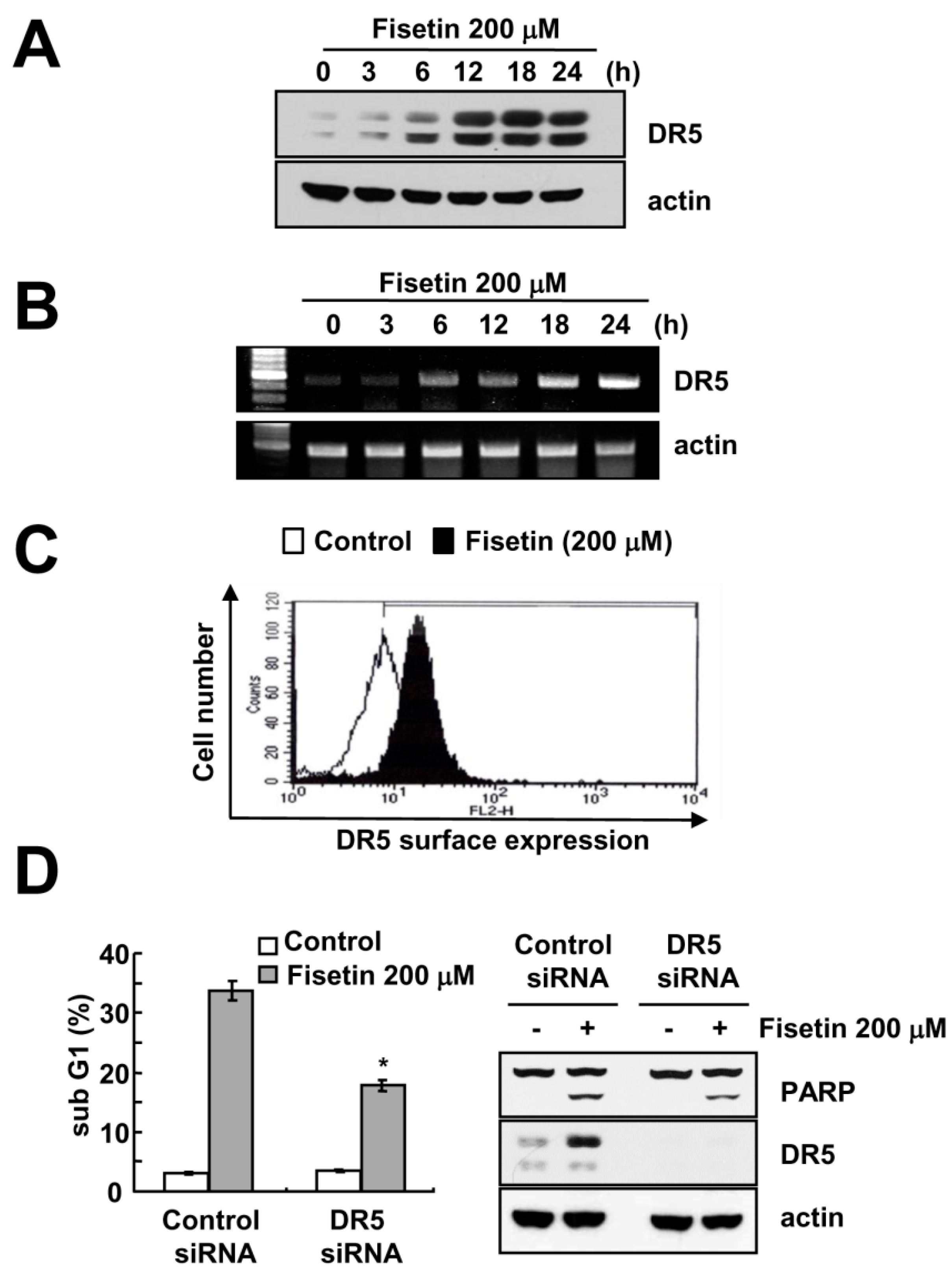
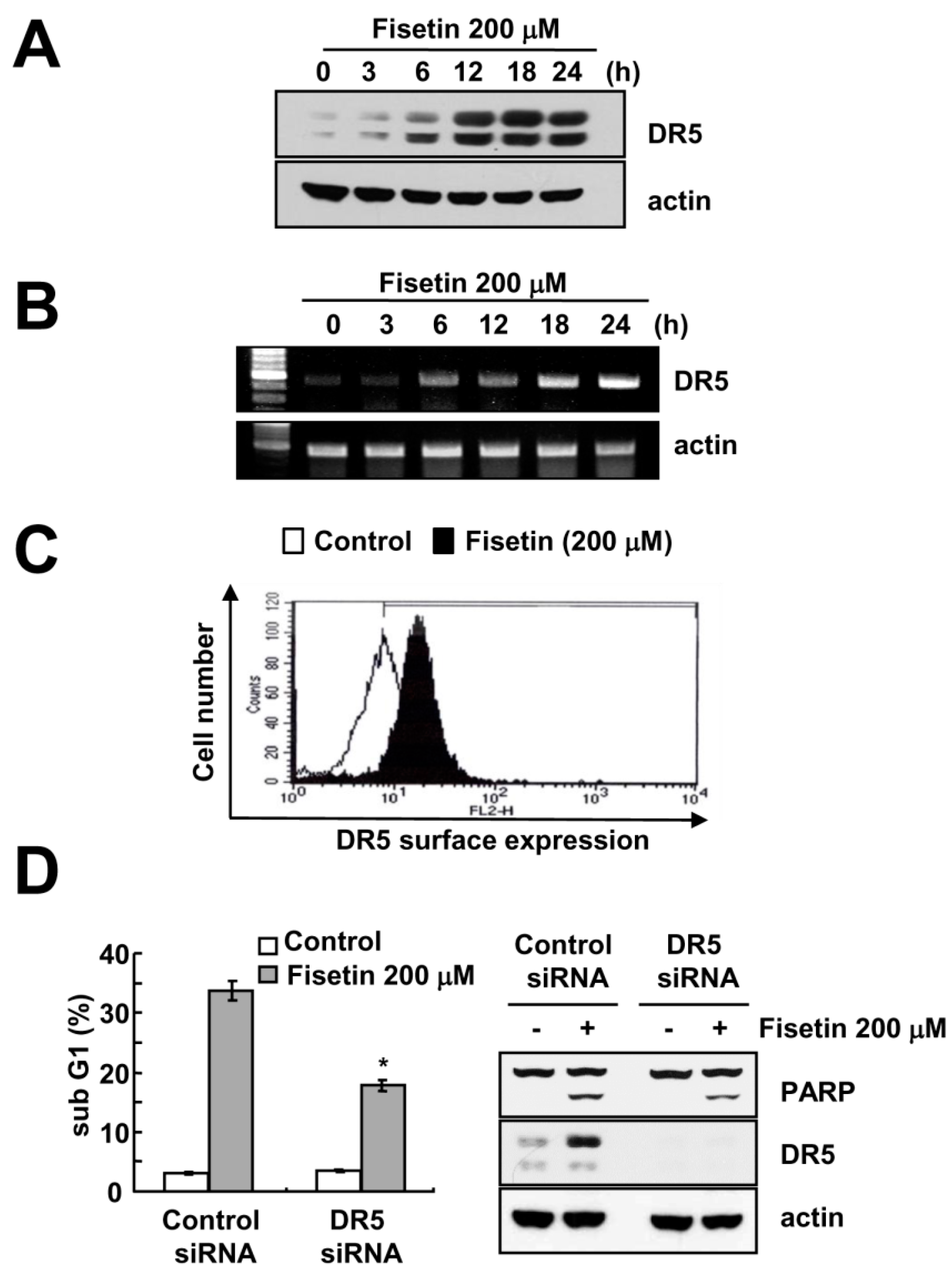
2.3. Fisetin Induced Apoptosis Through Up-Regulation of DR5 Expression
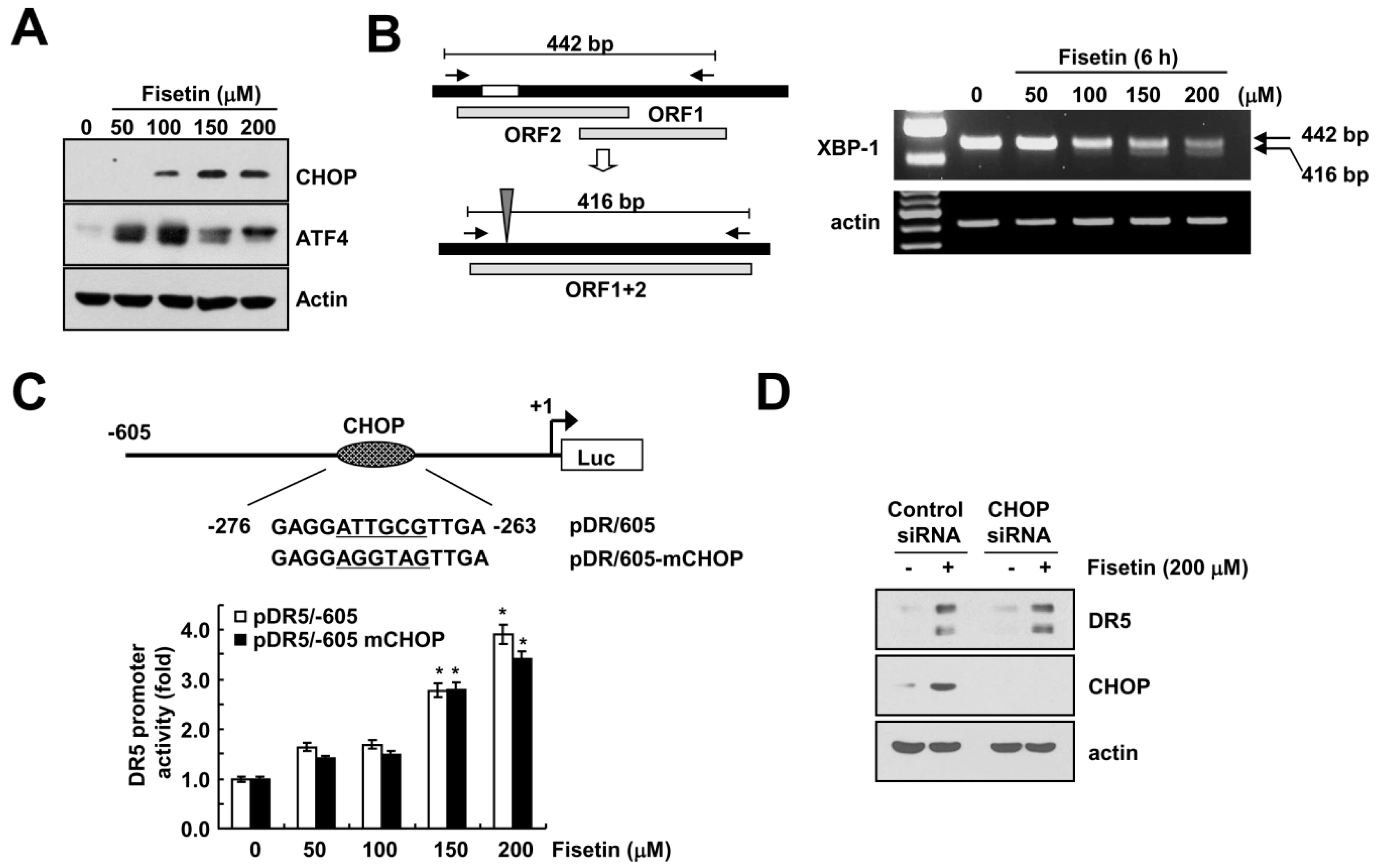
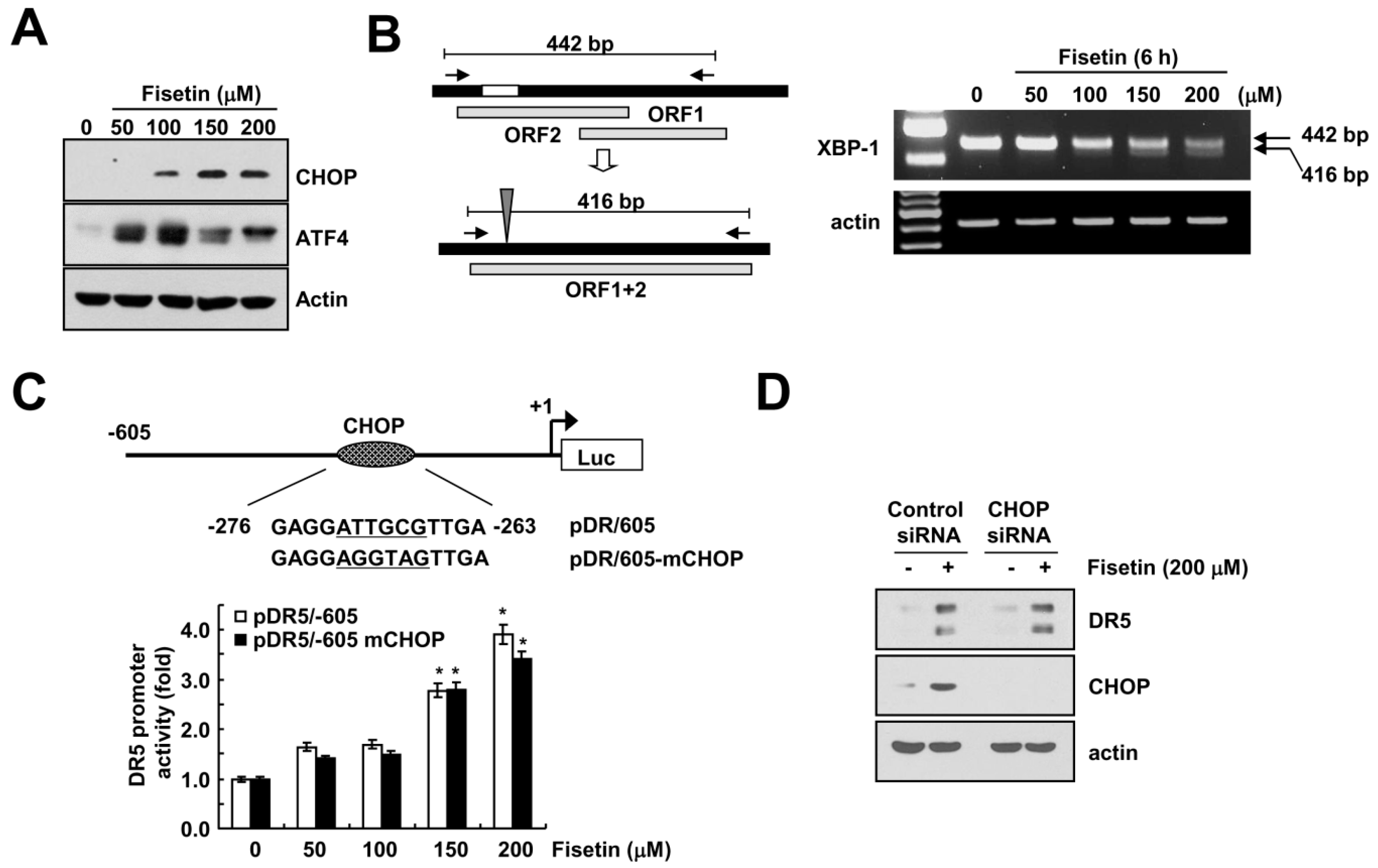
2.4. Endoplasmic Reticulum Stress Has No Effect on Fisetin-Induced DR5 Expression
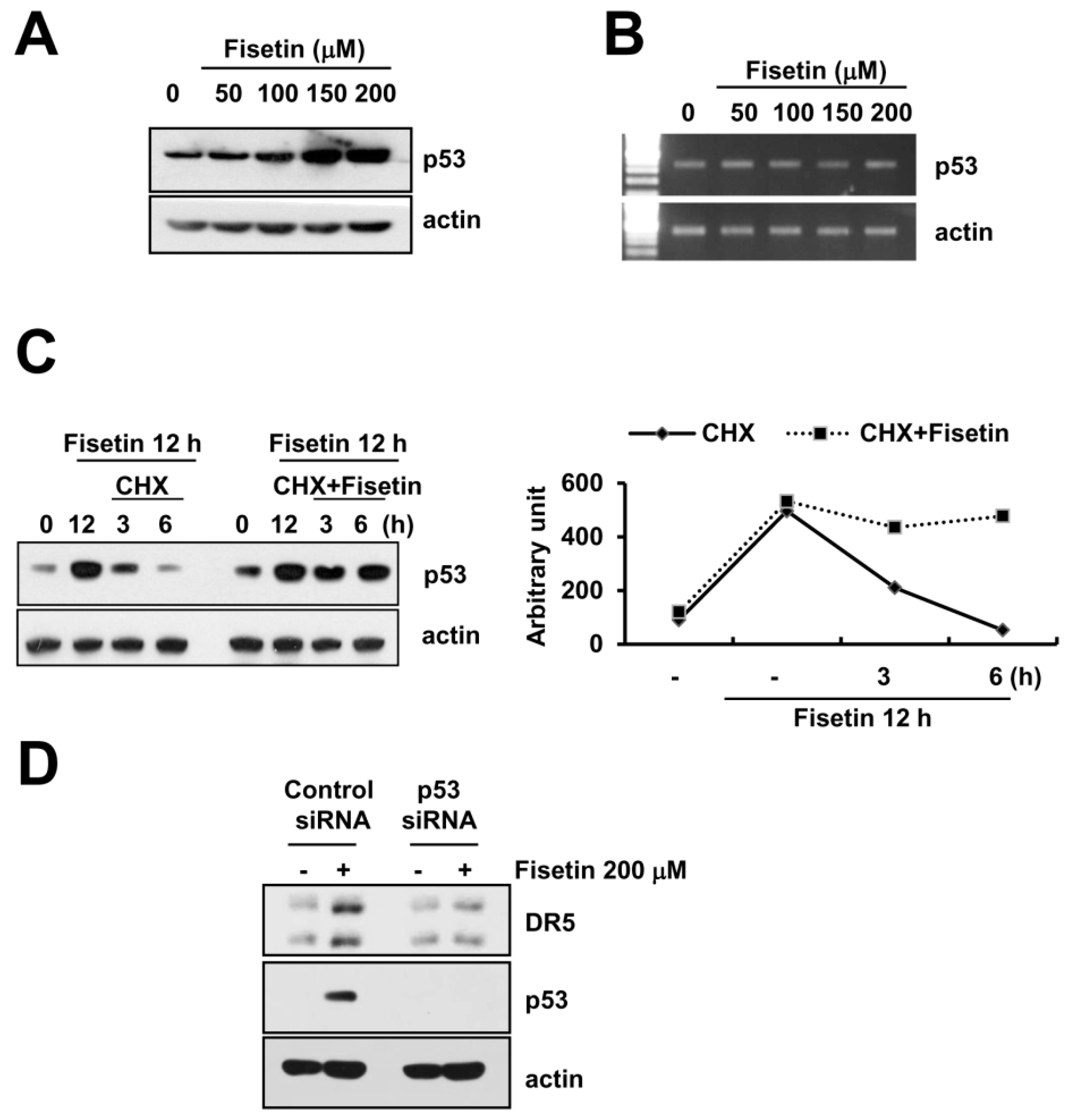
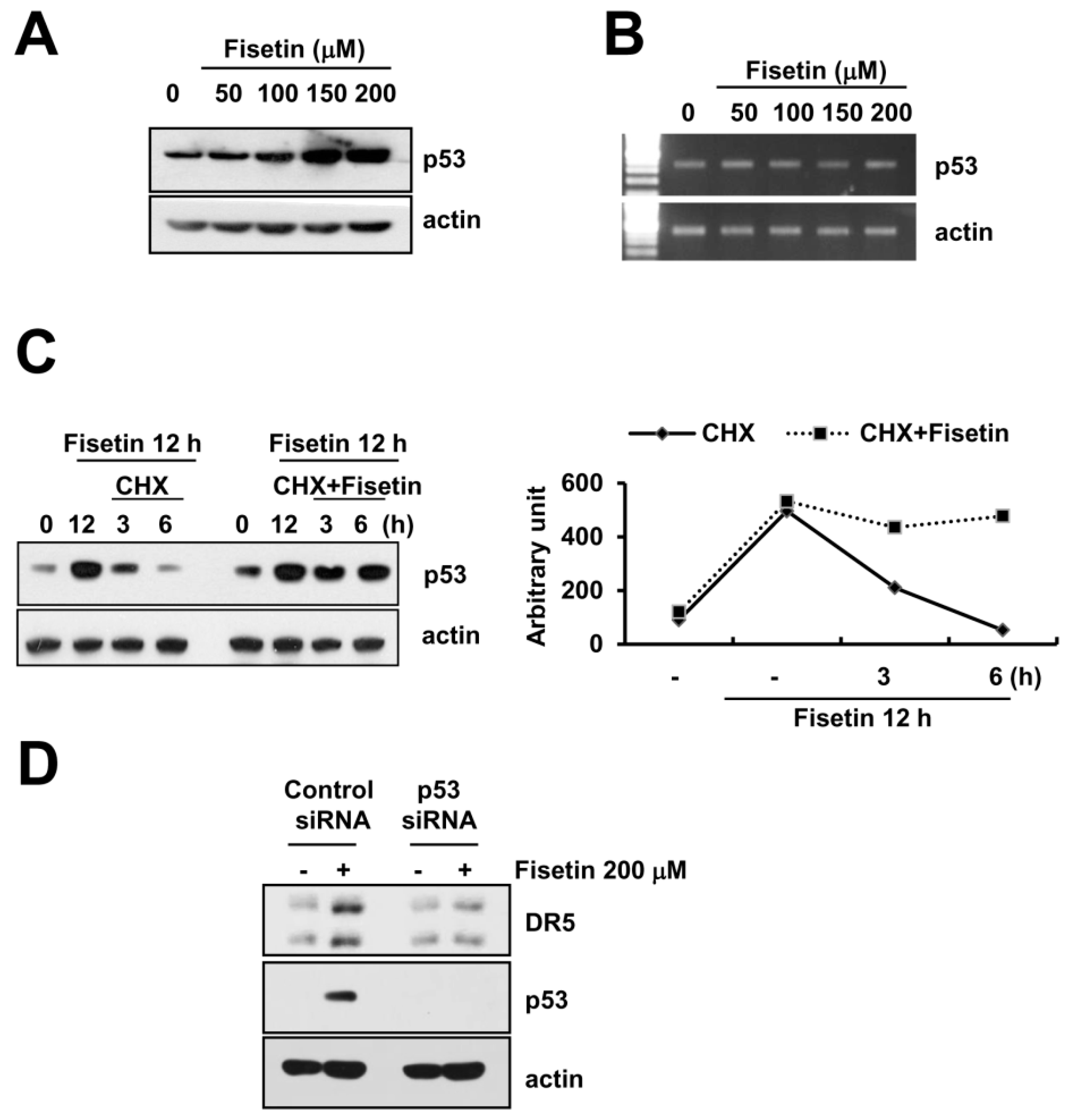
2.5. Fisetin-Induced p53 Expression Is Associated with DR5 Up-Regulation
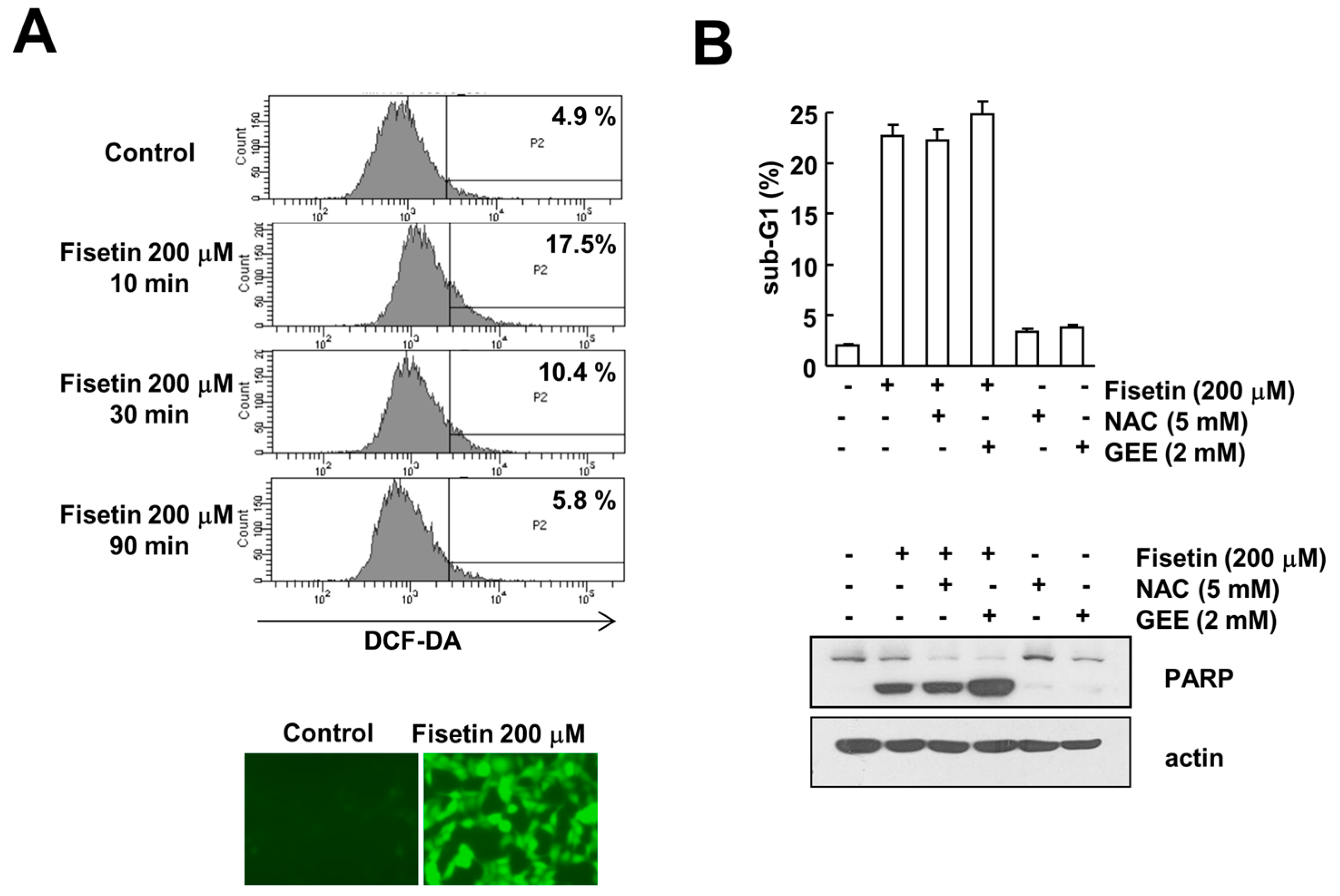
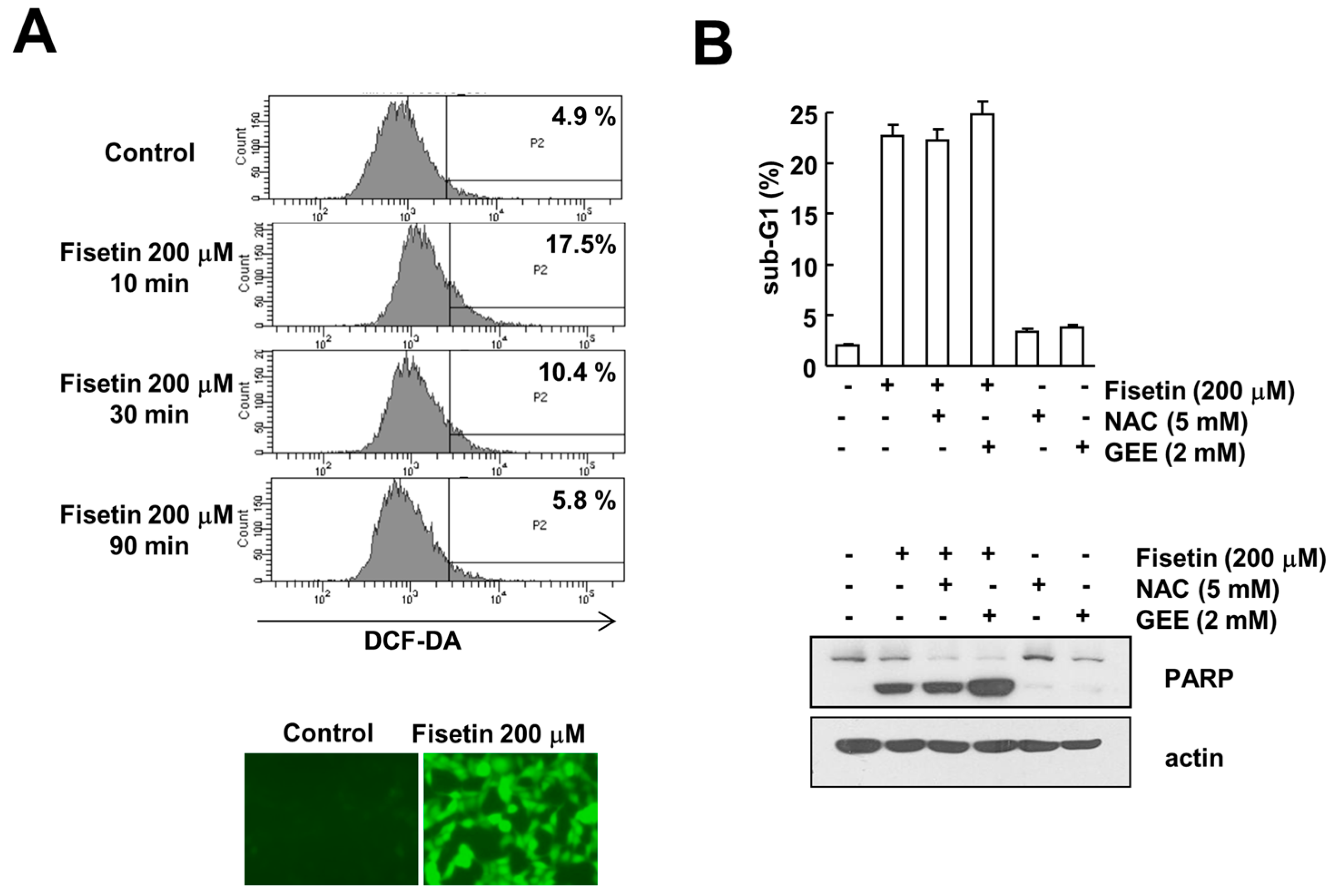
2.6. Fisetin-Induced Reactive Oxygen Species Production Has No Effect on Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Materials
4.2. Flow Cytometry Analysis
4.3. Western Blot Analysis
4.4. 4′,6′-diamidino-2-phenylindole Staining (DAPI) for Nuclei Condensation and Fragmentation
4.5. DNA Fragmentation Assay
4.6. Asp-Glu-Val-Asp-ase (DEVDase) Activity Assay
4.7. Analysis of Cell Surface DR5
4.8. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.9. DNA Transfection and Luciferase Assay
4.10. Small Interfering RNA (siRNA)
4.11. Measurement of Reactive Oxygen Species (ROS)
4.12. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| DR | death receptors |
| CHOP | C/EBP homologous protein |
| ER | endoplasmic reticulum |
| CHX | cycloheximide |
References
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Youns, M.; Hegazy, W.A.H. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef] [PubMed]
- Sabarwal, A.; Agarwal, R.; Singh, R.P. Fisetin inhibits cellular proliferation and induces mitochondria-dependent apoptosis in human gastric cancer cells. Mol. Carcinog. 2017, 56, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.; Ku, S.K.; Bae, J.S. Fisetin inhibits high-glucose-induced vascular inflammation in vitro and in vivo. Inflamm. Res. 2014, 63, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.H.; Jeong, G.S. Fisetin inhibits TNF-alpha-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte HaCaT cells through PI3K/AKT/Nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, S.H.; Son, J.H.; Lee, J.M.; Kang, M.J.; Kim, B.H.; Lee, J.S.; Ryu, J.K.; Kim, Y.T. Fisetin Reduces Cell Viability Through Up-Regulation of Phosphorylation of ERK1/2 in Cholangiocarcinoma Cells. Anticancer Res. 2016, 36, 6109–6116. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hewage, S.R.M.; Ryu, Y.S.; Oh, M.C.; Kwon, T.K.; Chae, S.; Hyun, J.W. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumour Biol. 2016, 37, 9615–9624. [Google Scholar] [CrossRef] [PubMed]
- Su, C.H.; Kuo, C.L.; Lu, K.W.; Yu, F.S.; Ma, Y.S.; Yang, J.L.; Chu, Y.L.; Chueh, F.S.; Liu, K.C.; Chung, J.G. Fisetin-induced apoptosis of human oral cancer SCC-4 cells through reactive oxygen species production, endoplasmic reticulum stress, caspase-, and mitochondria-dependent signaling pathways. Environ. Toxicol. 2017, 32, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Siddiqui, I.A.; Verma, A.K.; Mukhtar, H. Fisetin Enhances Chemotherapeutic Effect of Cabazitaxel against Human Prostate Cancer Cells. Mol. Cancer Ther. 2016, 15, 2863–2874. [Google Scholar] [CrossRef] [PubMed]
- Szliszka, E.; Helewski, K.J.; Mizgala, E.; Krol, W. The dietary flavonol fisetin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells. Int. J. Oncol. 2011, 39, 771–779. [Google Scholar] [PubMed]
- Zou, W.; Liu, X.; Yue, P.; Zhou, Z.; Sporn, M.B.; Lotan, R.; Khuri, F.R.; Sun, S.Y. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res. 2004, 64, 7570–7578. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. el-Deiry, KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, R.; El-Deiry, W.S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000, 19, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Gladden, J.B.; Henson, E.S.; Hu, X.; Villanueva, J.; Haney, N.; Gibson, S.B. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NFkappaB activation in epithelial derived cell lines. Apoptosis 2002, 7, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Nakayama, M.; Nishina, T.; Nakano, H.; Koyanagi, M.; Takeda, K.; Okumura, K.; Yagita, H. Importin beta1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J. Biol. Chem. 2011, 286, 43383–43393. [Google Scholar] [CrossRef] [PubMed]
- Simova, S.; Klima, M.; Cermak, L.; Sourkova, V.; Andera, L. Arf and Rho GAP adapter protein ARAP1 participates in the mobilization of TRAIL-R1/DR4 to the plasma membrane. Apoptosis 2008, 13, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; McDonald, E.R., 3rd; Dicker, D.T.; El-Deiry, W.S. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J. Biol. Chem. 2004, 279, 35829–35839. [Google Scholar] [CrossRef] [PubMed]
- Leithner, K.; Stacher, E.; Wurm, R.; Ploner, F.; Quehenberger, F.; Wohlkoenig, C.; Balint, Z.; Polachova, J.; Olschewski, A.; Samonigg, H.; et al. Nuclear and cytoplasmic death receptor 5 as prognostic factors in patients with non-small cell lung cancer treated with chemotherapy. Lung Cancer 2009, 65, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Zhang, G.; Zhang, Y.; Takeda, K.; Rosado, L.A.R.; Zhang, B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget 2013, 4, 1349–1364. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Limvorasak, S.; Figlin, R.A. Targeted therapies for renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.Y.; Park, J.H. Induction of p53 contributes to apoptosis of HCT-116 human colon cancer cells induced by the dietary compound fisetin. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1060–G1068. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Andera, L.; Alleva, R.; Borghi, B.; Neuzil, J.; Procopio, A. Alpha-tocopheryl succinate induces DR4 and DR5 expression by a p53-dependent route: Implication for sensitisation of resistant cancer cells to TRAIL apoptosis. FEBS Lett. 2006, 580, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.Y.; Jeong, S.J.; Kim, S.H.; Jung, J.H.; Kim, J.H.; Koh, W.; Chen, C.Y.; Kim, S.H. Activation of reactive oxygen species/AMP activated protein kinase signaling mediates fisetin-induced apoptosis in multiple myeloma U266 cells. Cancer Lett. 2012, 319, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.H.; Yang, S.F.; Tsai, S.J.; Hsieh, S.C.; Huang, Y.C.; Bau, D.T.; Hsieh, Y.H. Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase-8-/caspase-3-dependent pathway. Arch. Toxicol. 2012, 86, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Afaq, F.; Johnson, J.J.; Mukhtar, H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis 2009, 30, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheng, Y.; Qu, W.; Sun, Y.; Wang, Z.; Wang, H.; Tian, B. Fisetin, a dietary flavonoid, induces cell cycle arrest and apoptosis through activation of p53 and inhibition of NF-kappa B pathways in bladder cancer cells. Basic Clin. Pharmacol. Toxicol. 2011, 108, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.S.; Lien, G.S.; Shen, S.C.; Yang, L.Y.; Chen, Y.C. N-acetyl-l-cysteine enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells. Mol. Carcinog. 2014, 53 (Suppl. 1), E119–E129. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Fingas, C.D.; Meng, X.W.; Finnberg, N.; El-Deiry, W.S.; Kaufmann, S.H.; Gores, G.J. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 2011, 286, 39336–39348. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Duong, H.Q.; Choi, J.E.; Lee, T.B.; Kang, J.H.; Oh, S.H.; Han, S.I. Lipid raft-dependent death receptor 5 (DR5) expression and activation are critical for ursodeoxycholic acid-induced apoptosis in gastric cancer cells. Carcinogenesis 2011, 32, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Kang, C.H.; Choi, Y.H.; Kim, G.Y. Mangiferin inhibits tumor necrosis factor-alpha-induced matrix metalloproteinase-9 expression and cellular invasion by suppressing nuclear factor-kappaB activity. BMB Rep. 2015, 48, 559–564. [Google Scholar] [CrossRef] [PubMed]
- An, Y.A.; Hwang, J.Y.; Lee, J.S.; Kim, Y.C. Cornus officinalis Methanol Extract Upregulates Melanogenesis in Melan-a Cells. Toxicol. Res. 2015, 31, 165–172. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, K.-j.; Nam, J.-O.; Kwon, T.K. Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells. Molecules 2017, 22, 1285. https://doi.org/10.3390/molecules22081285
Min K-j, Nam J-O, Kwon TK. Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells. Molecules. 2017; 22(8):1285. https://doi.org/10.3390/molecules22081285
Chicago/Turabian StyleMin, Kyoung-jin, Ju-Ock Nam, and Taeg Kyu Kwon. 2017. "Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells" Molecules 22, no. 8: 1285. https://doi.org/10.3390/molecules22081285
APA StyleMin, K.-j., Nam, J.-O., & Kwon, T. K. (2017). Fisetin Induces Apoptosis Through p53-Mediated Up-Regulation of DR5 Expression in Human Renal Carcinoma Caki Cells. Molecules, 22(8), 1285. https://doi.org/10.3390/molecules22081285