Research Progress in the Modification of Quercetin Leading to Anticancer Agents

 ,
,  , ,
, ,  ,
,  and
and
Abstract
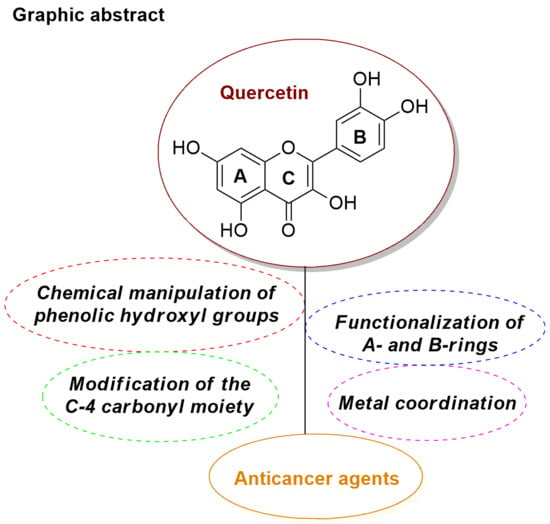
:
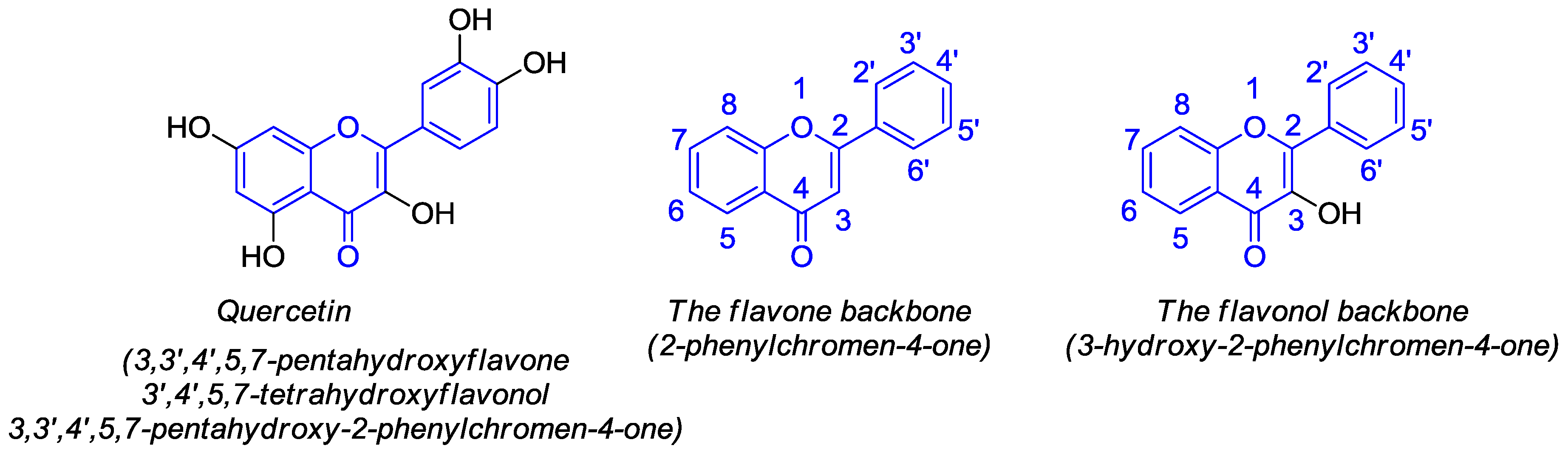
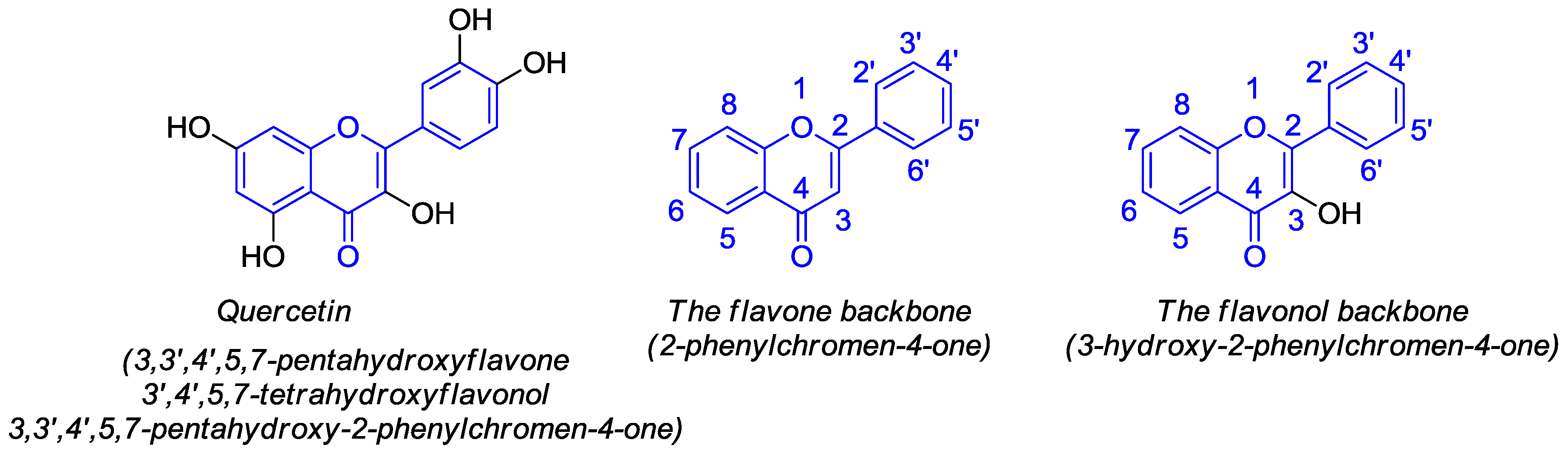
1. Introduction
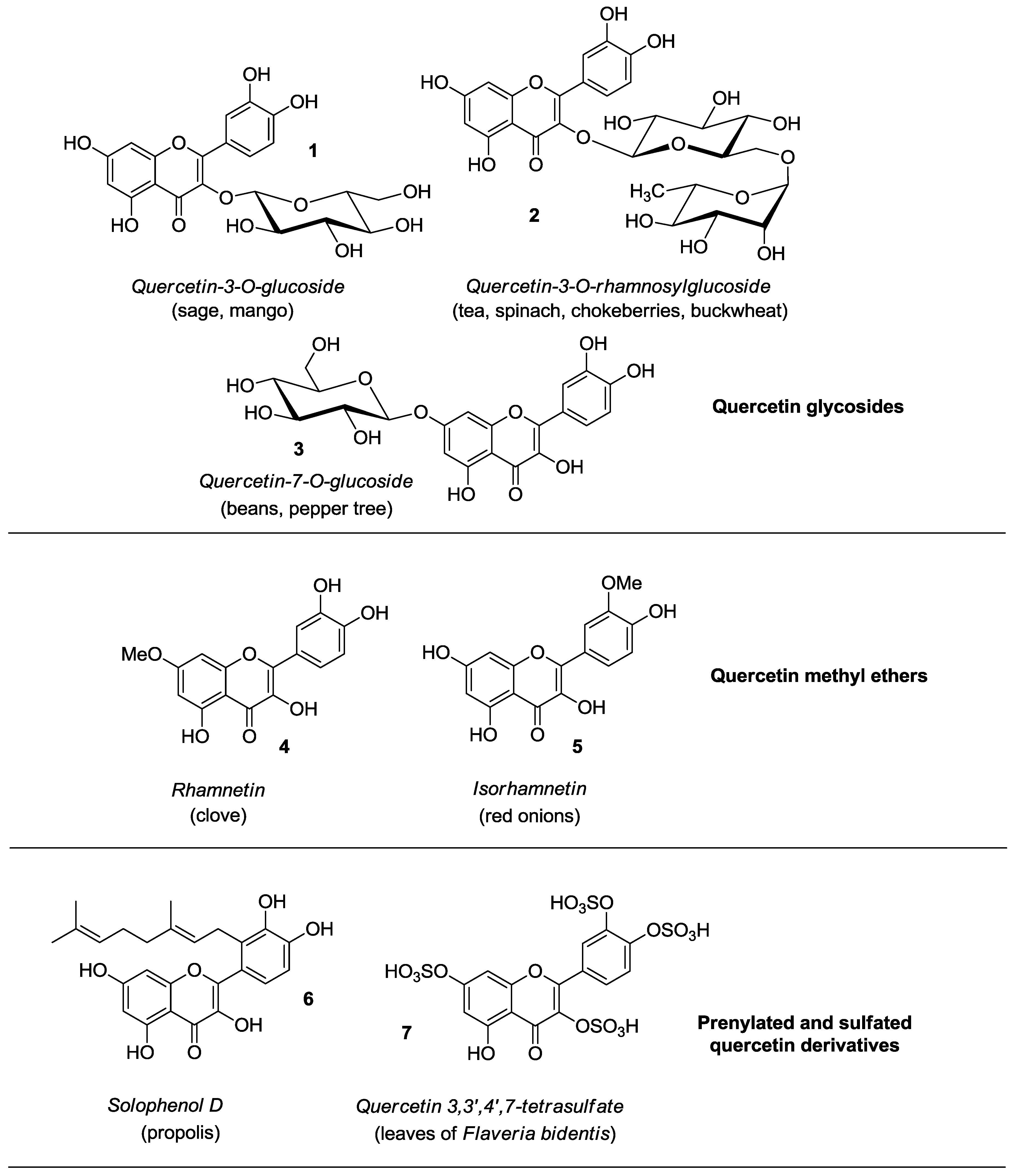
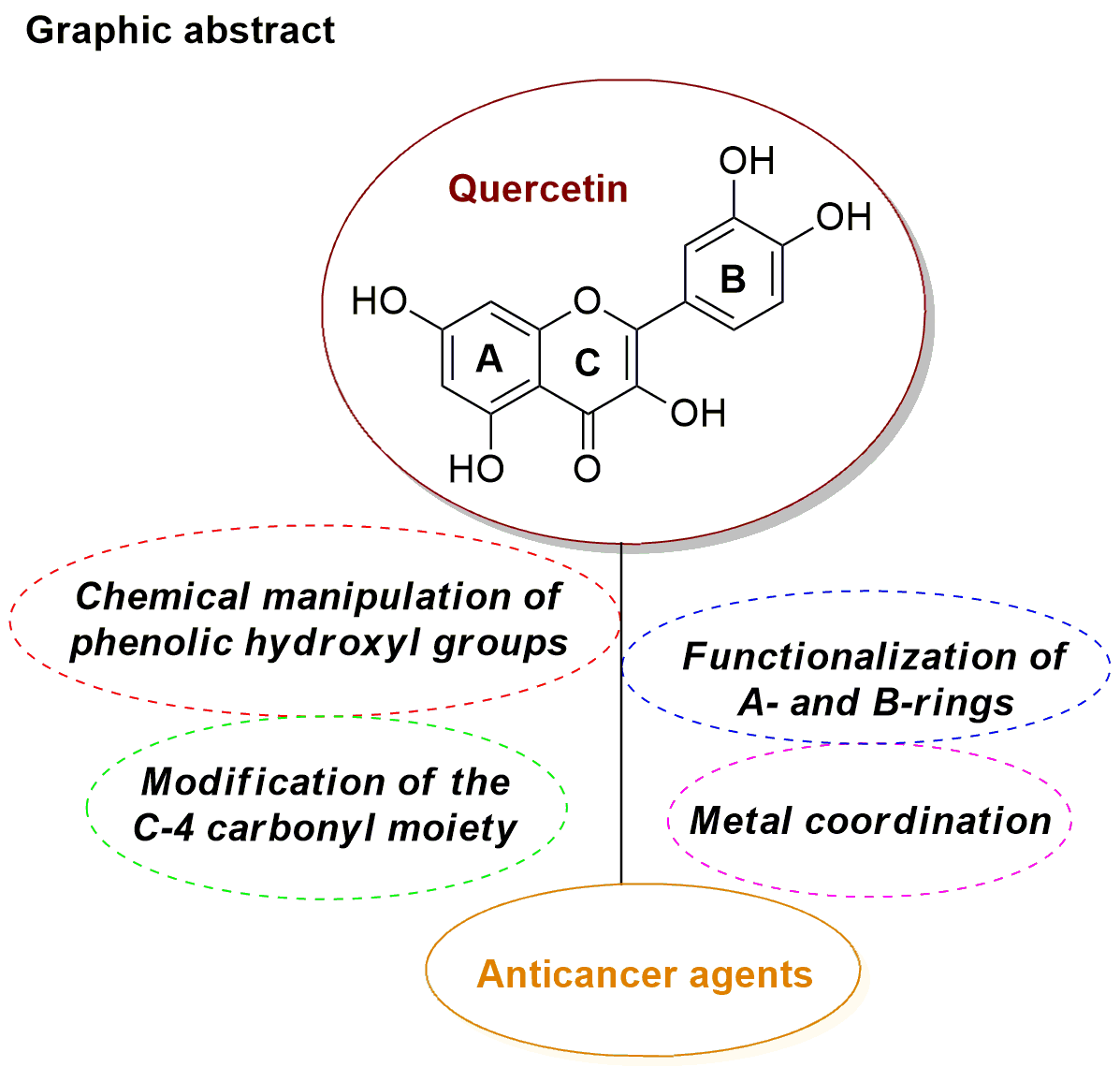
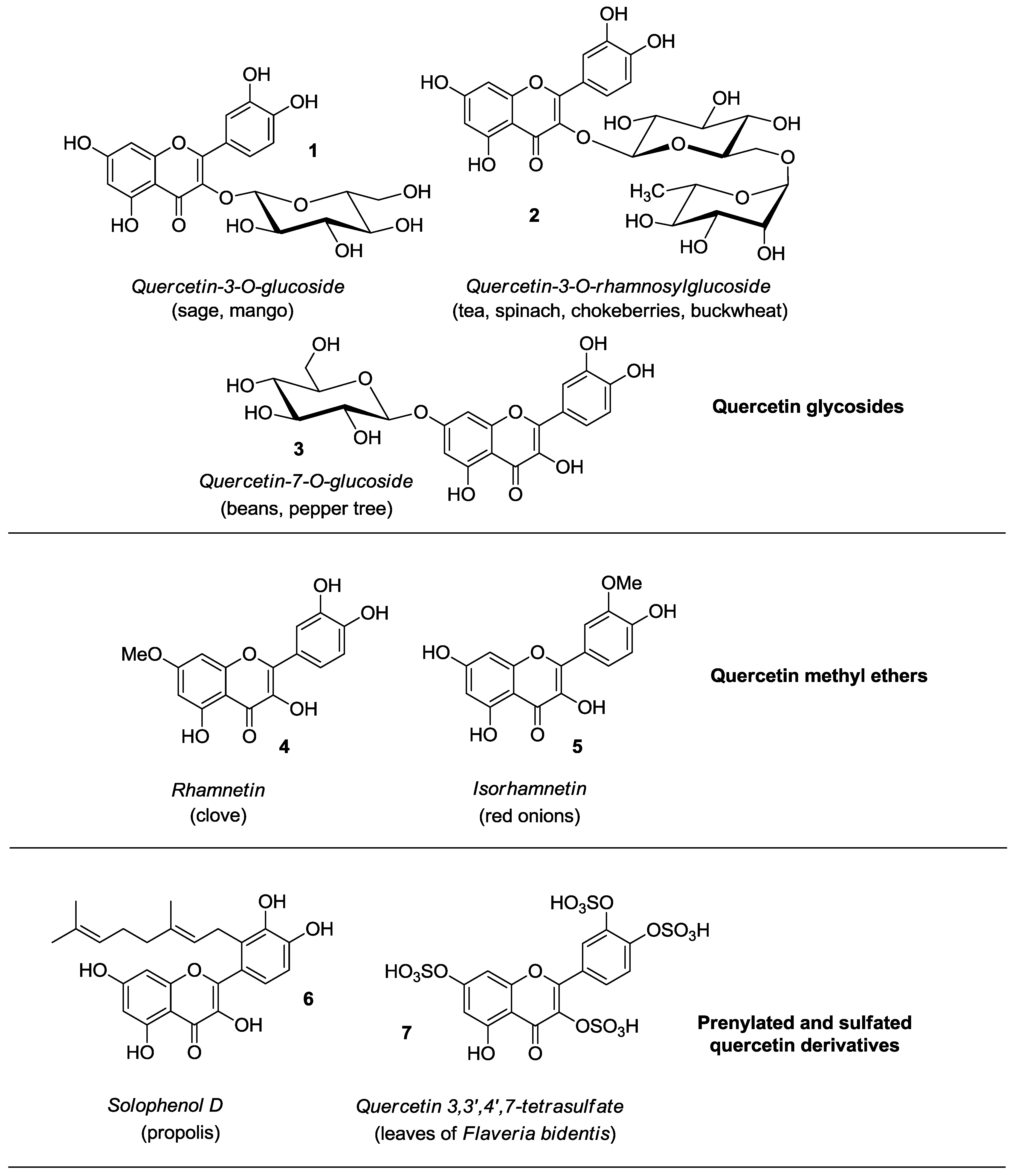
2. Synthesis and Anticancer-Related Activities of Quercetin Derivatives
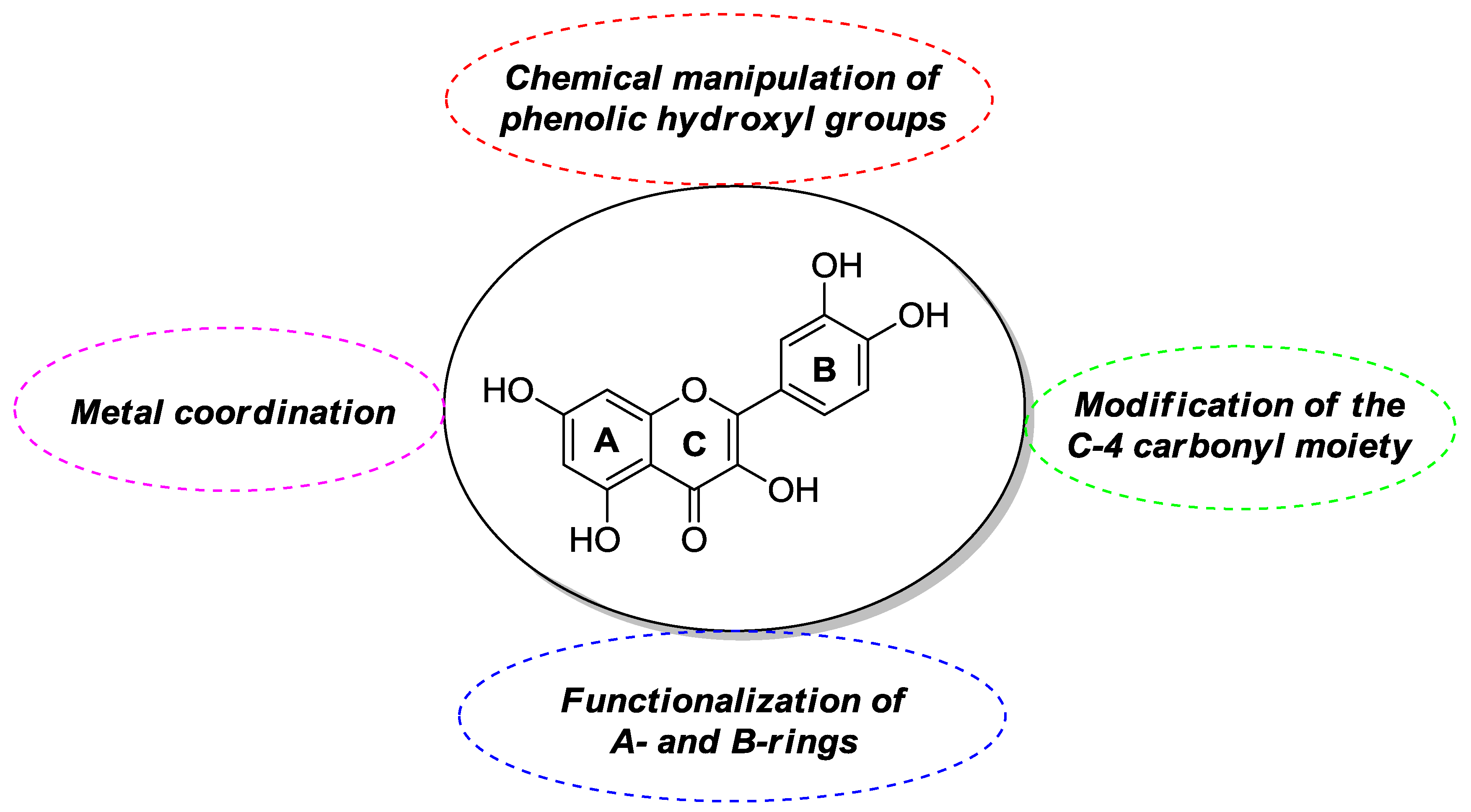
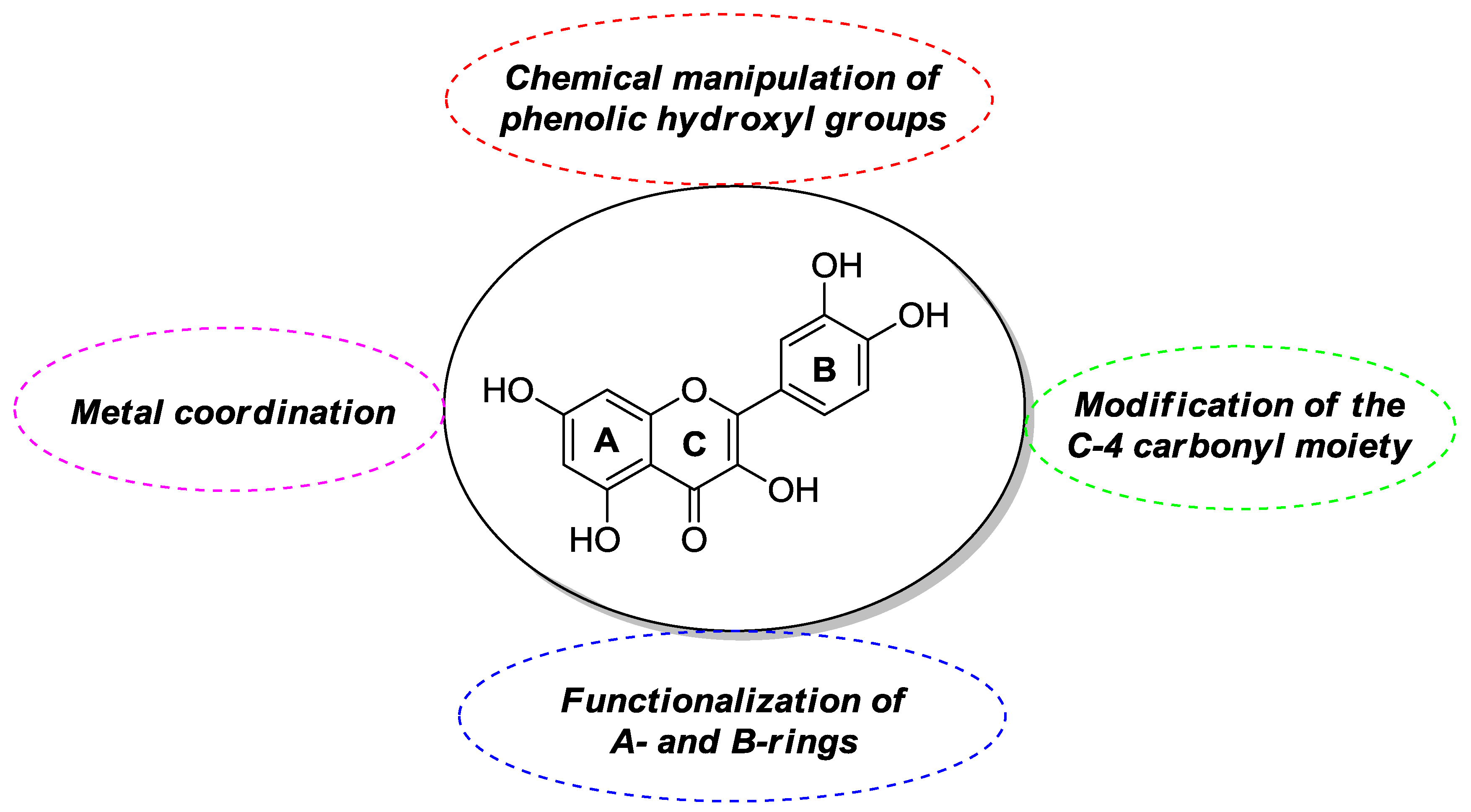
2.1. Chemical Modification of Phenolic Hydroxyl Groups and/or C-4 Carbonyl Moiety
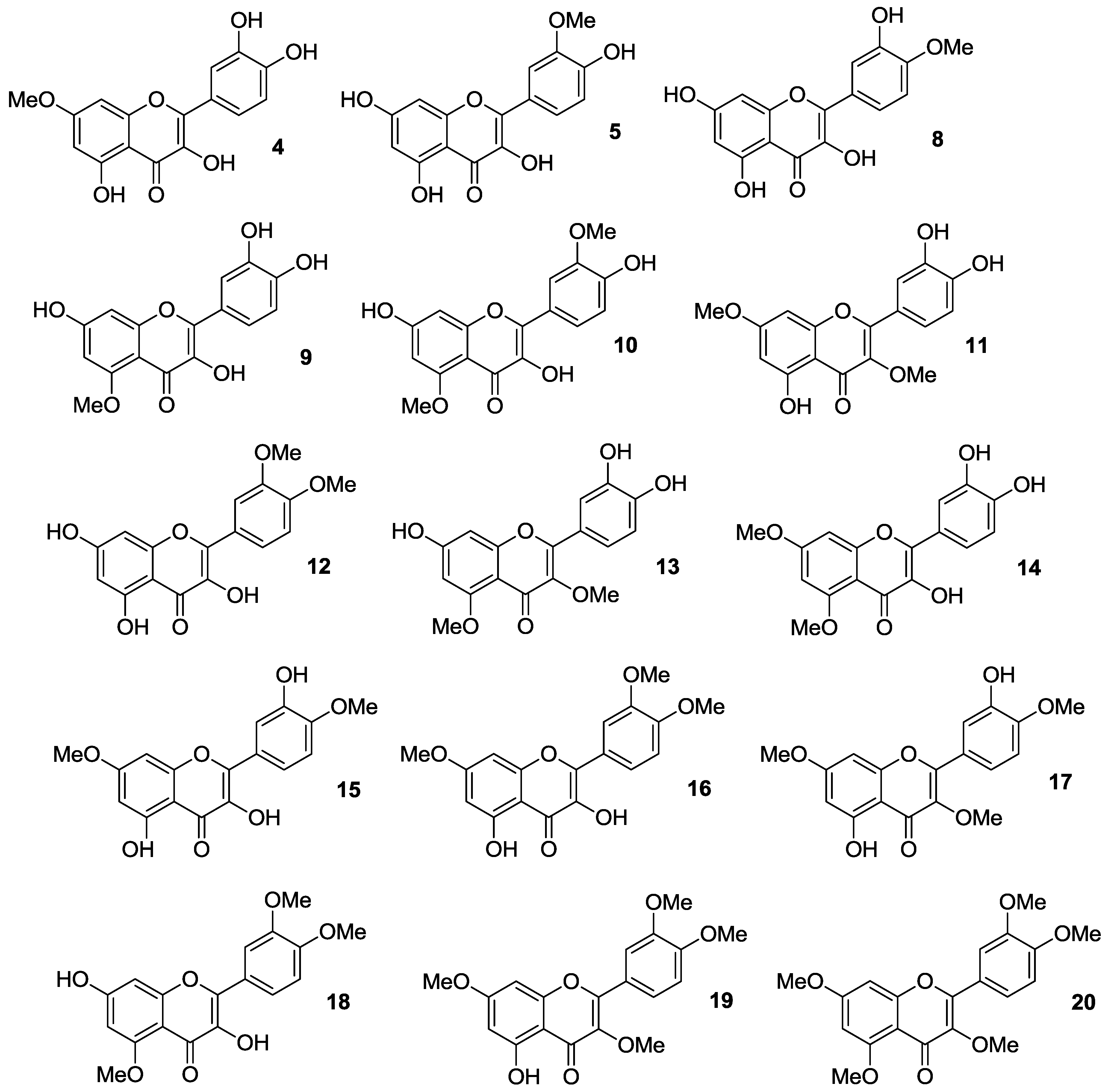
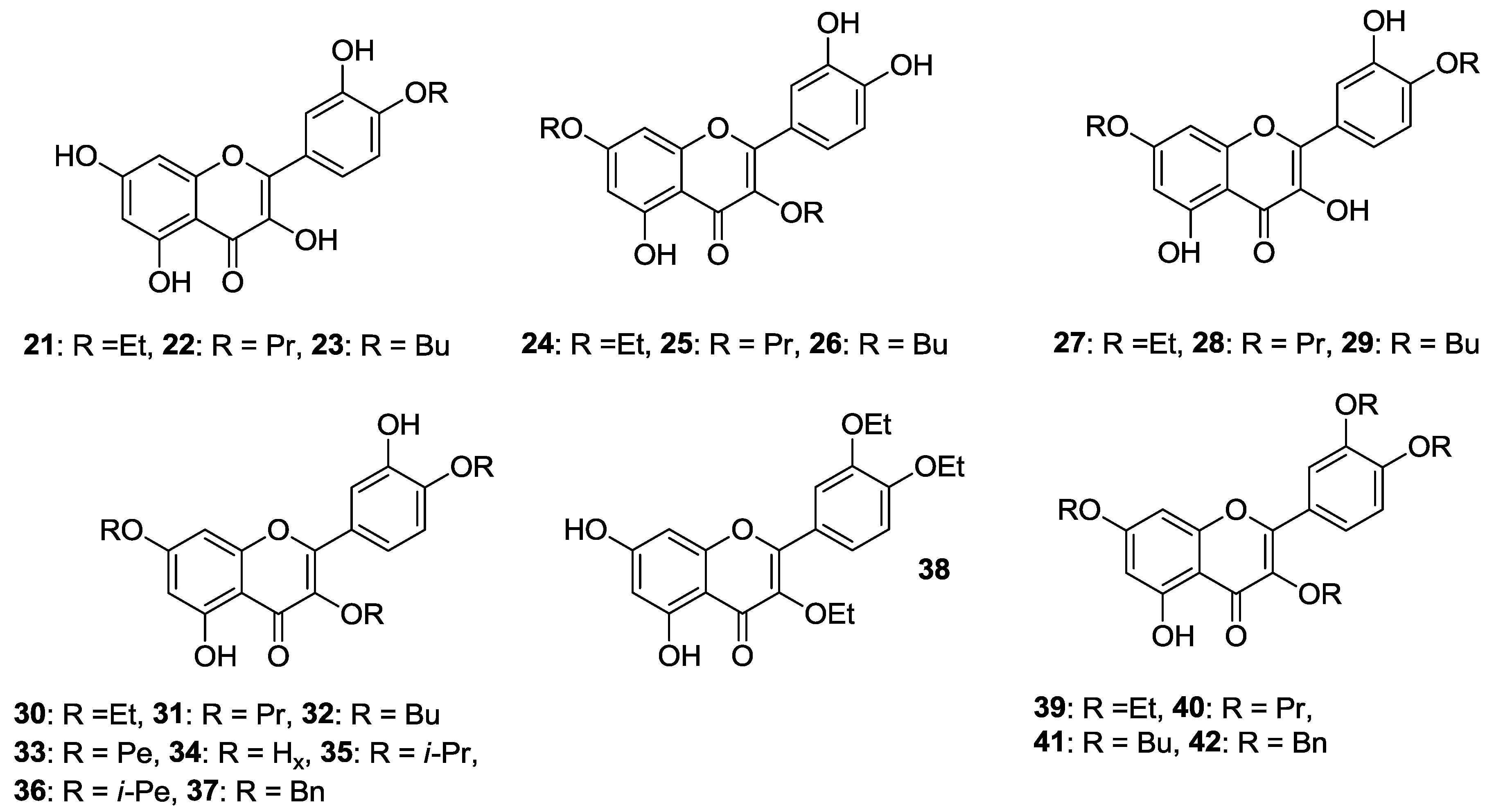
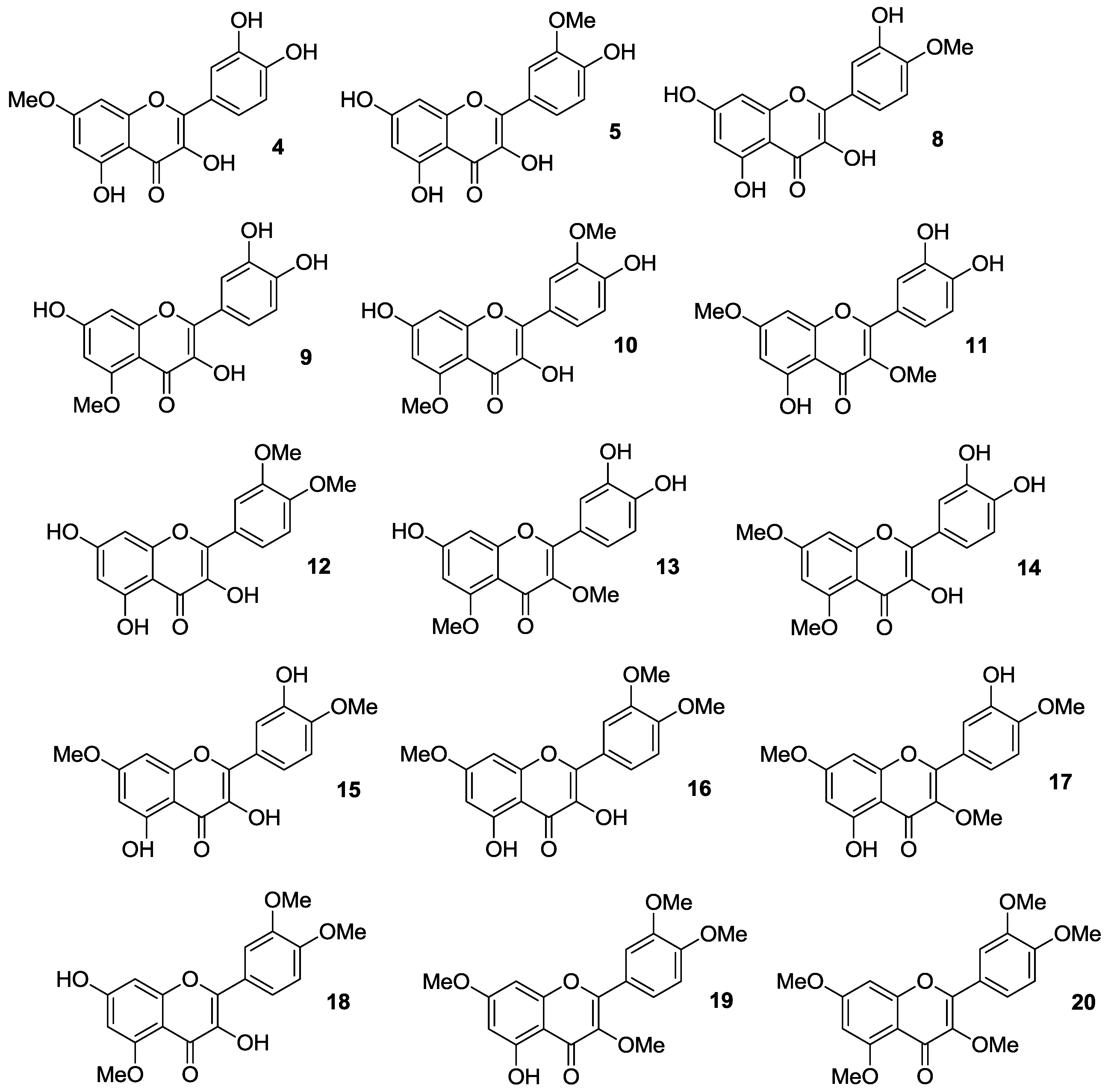
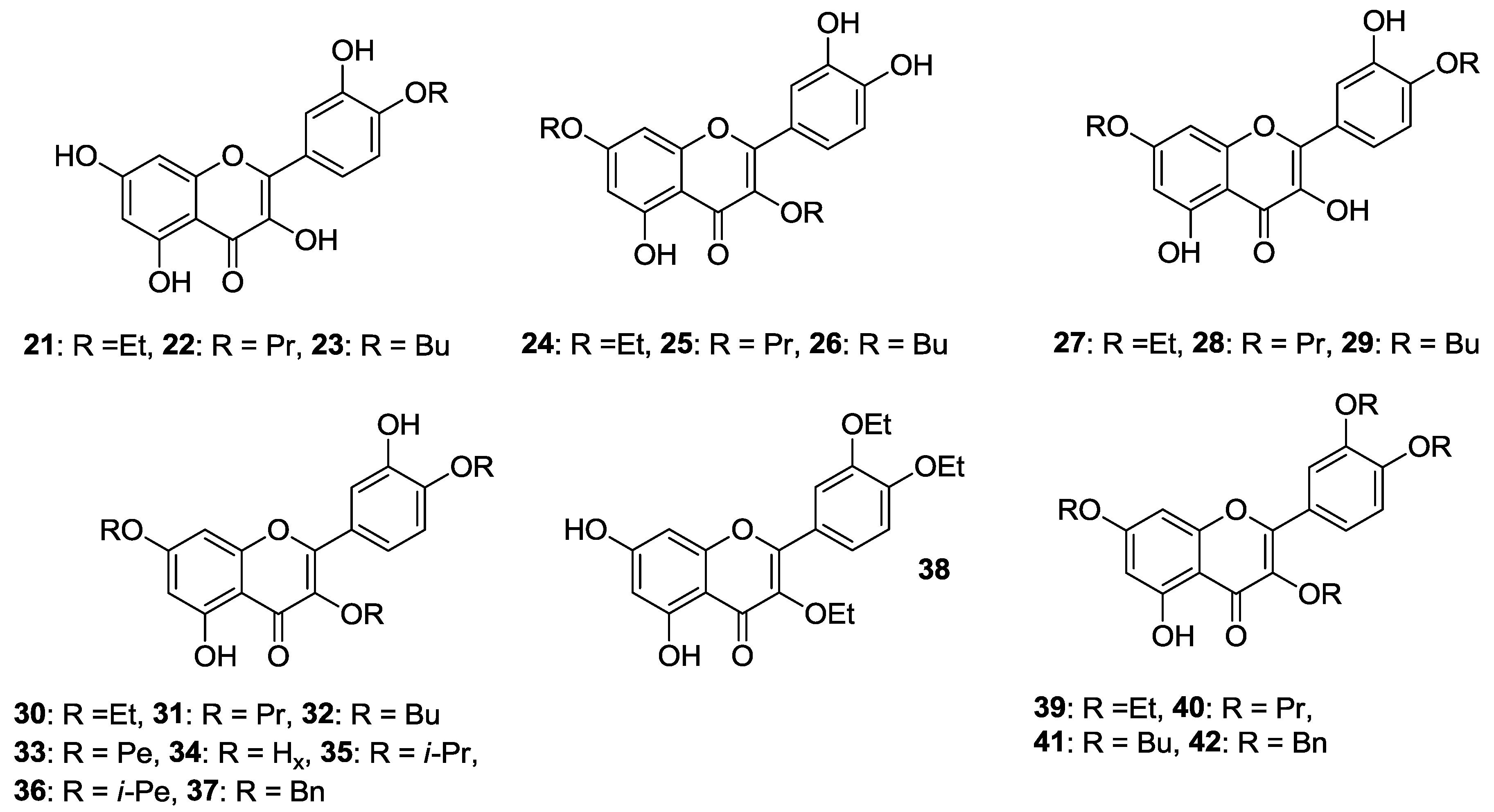
2.1.1. O-Alkylation
2.1.2. O-Alkylation and C-4 Carbonyl Group Modification
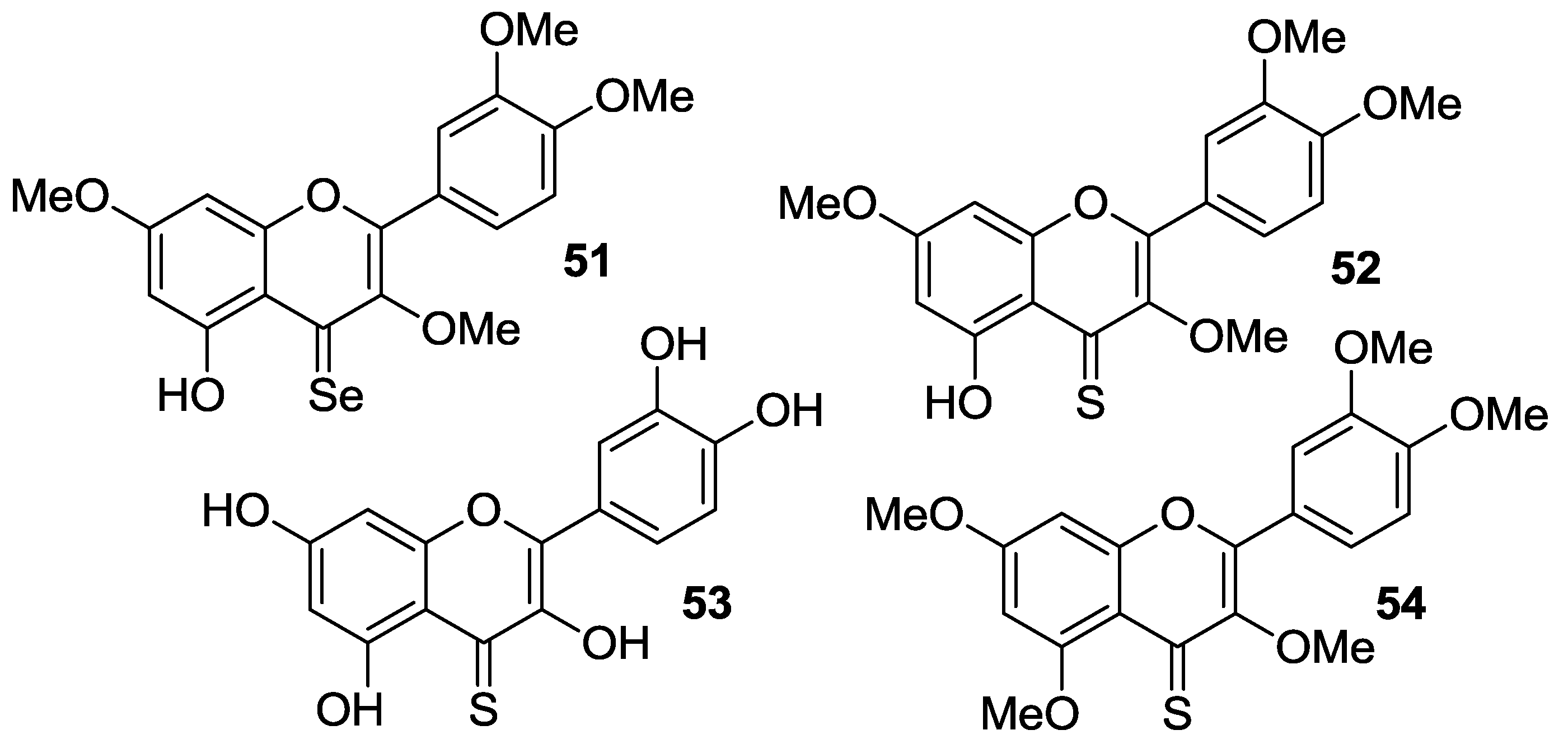
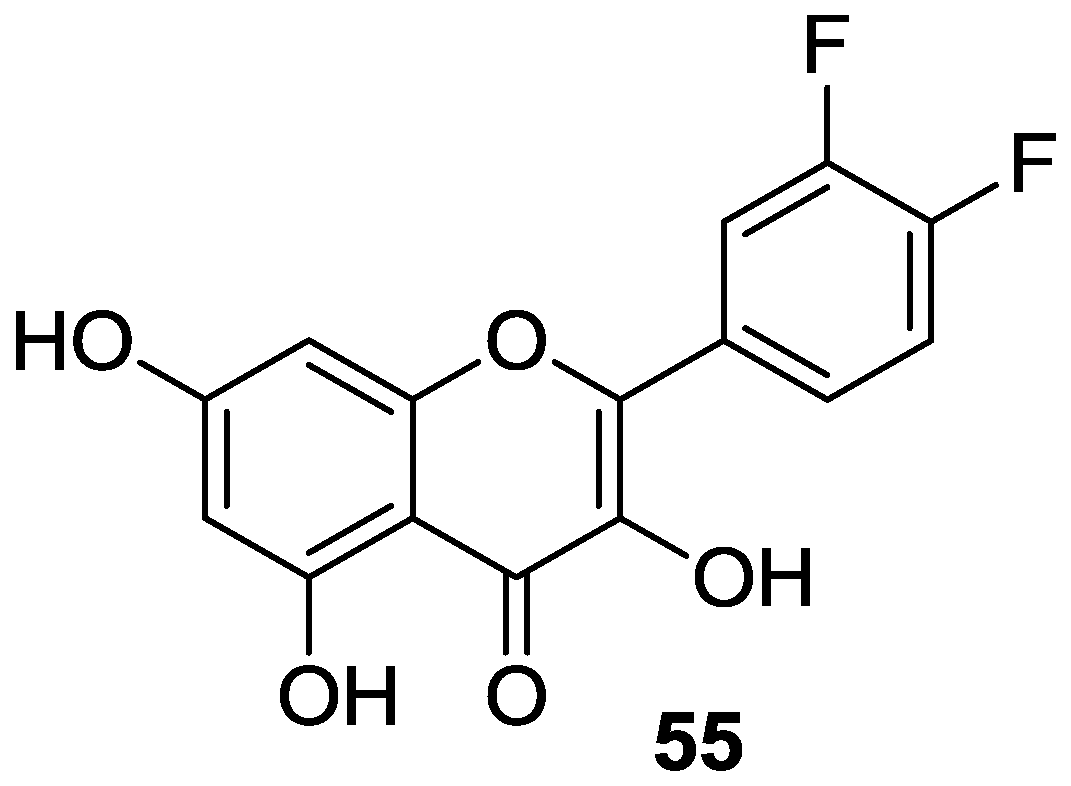
2.1.3. Replacement of Catecholic Hydroxyl Groups
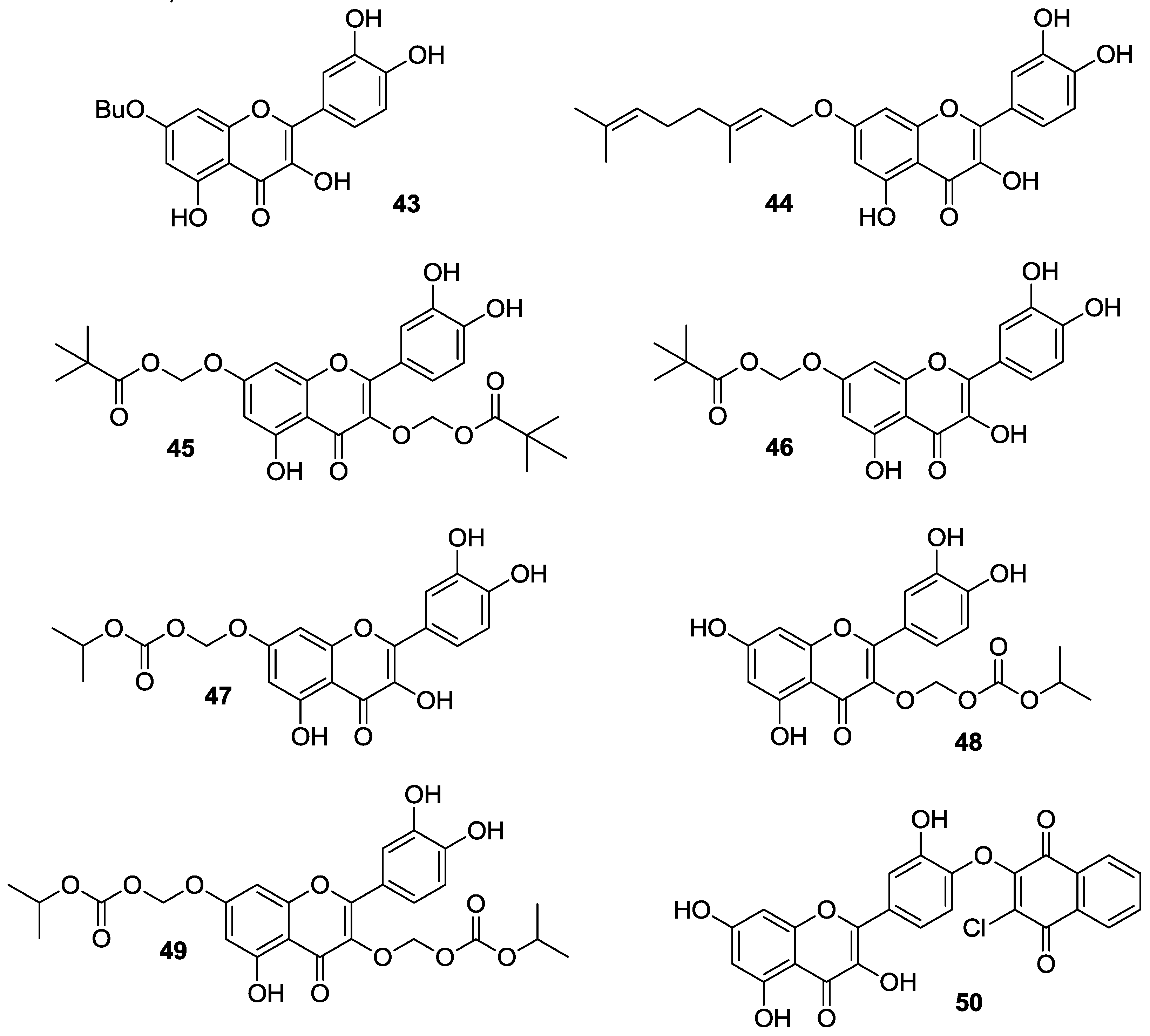
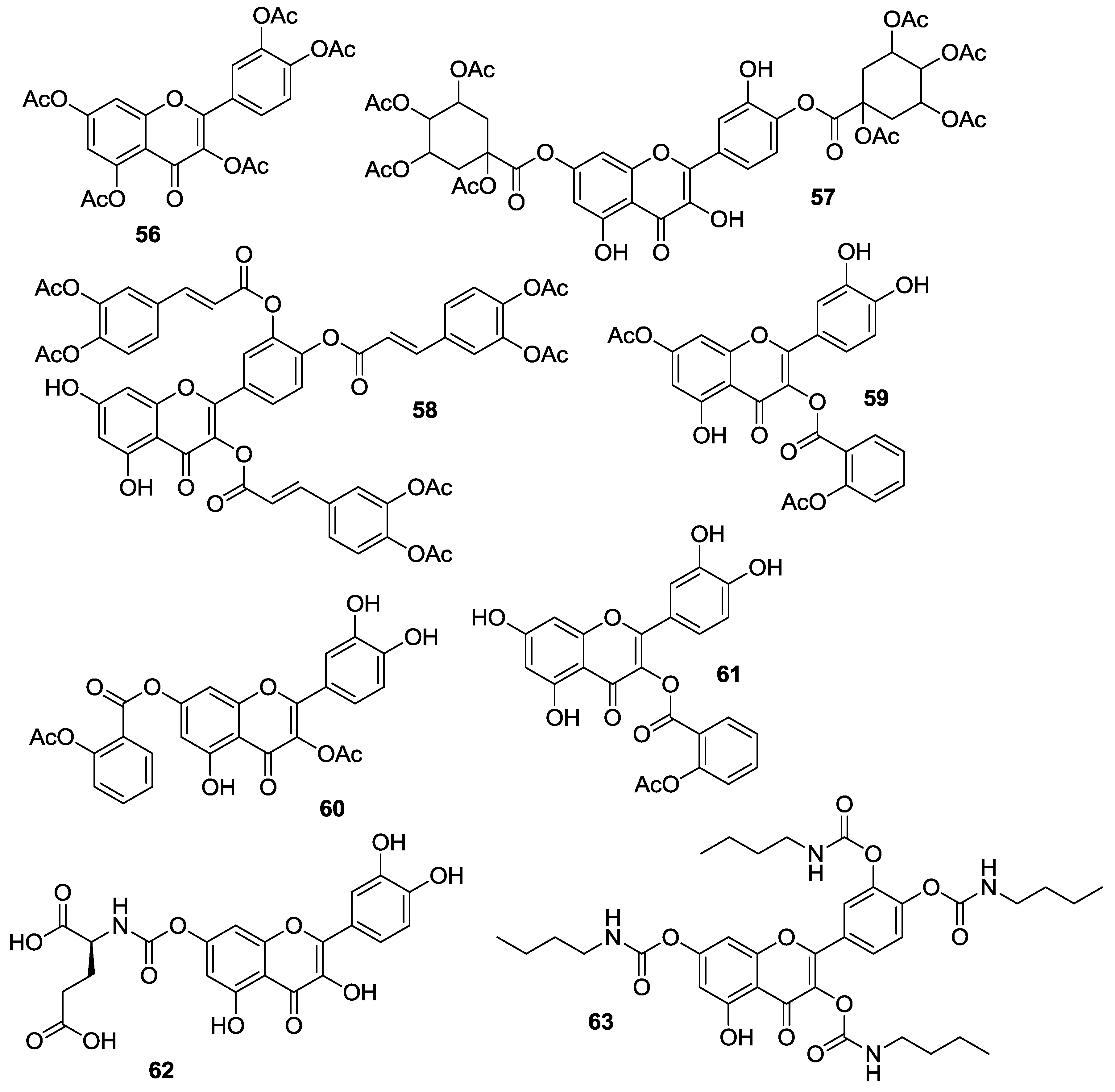
2.1.4. O-Acylation
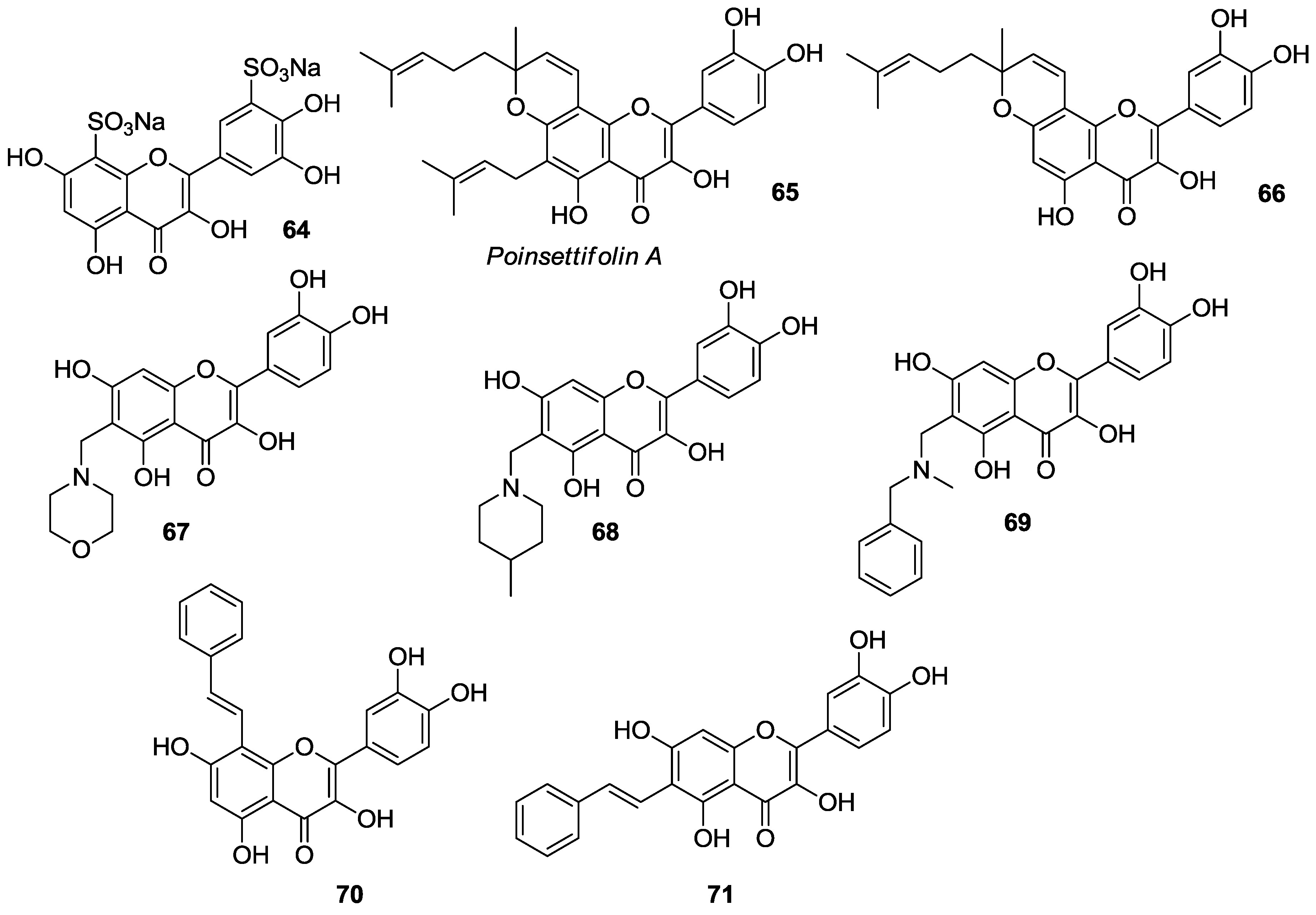
2.2. Functionalization of A- and B-Rings
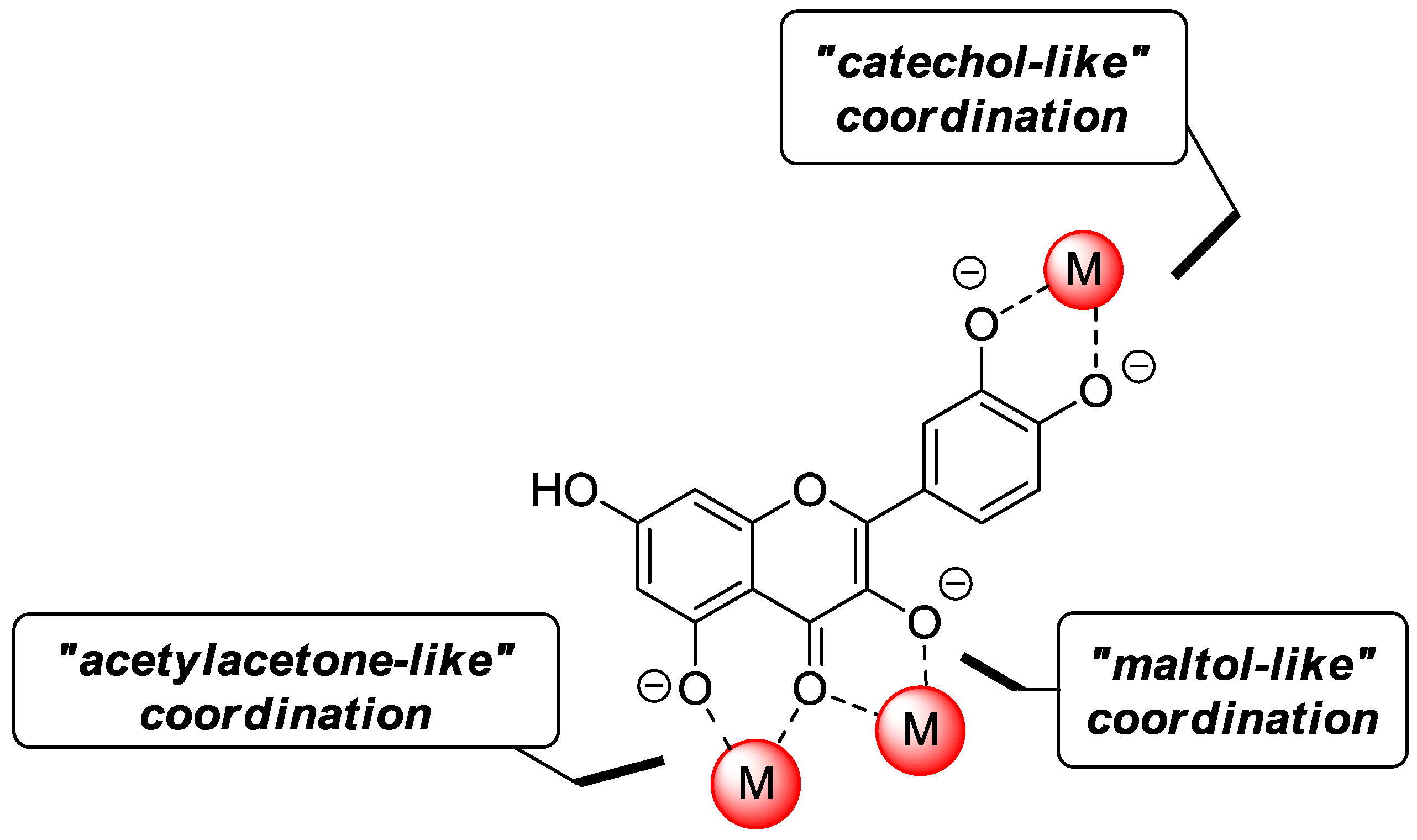
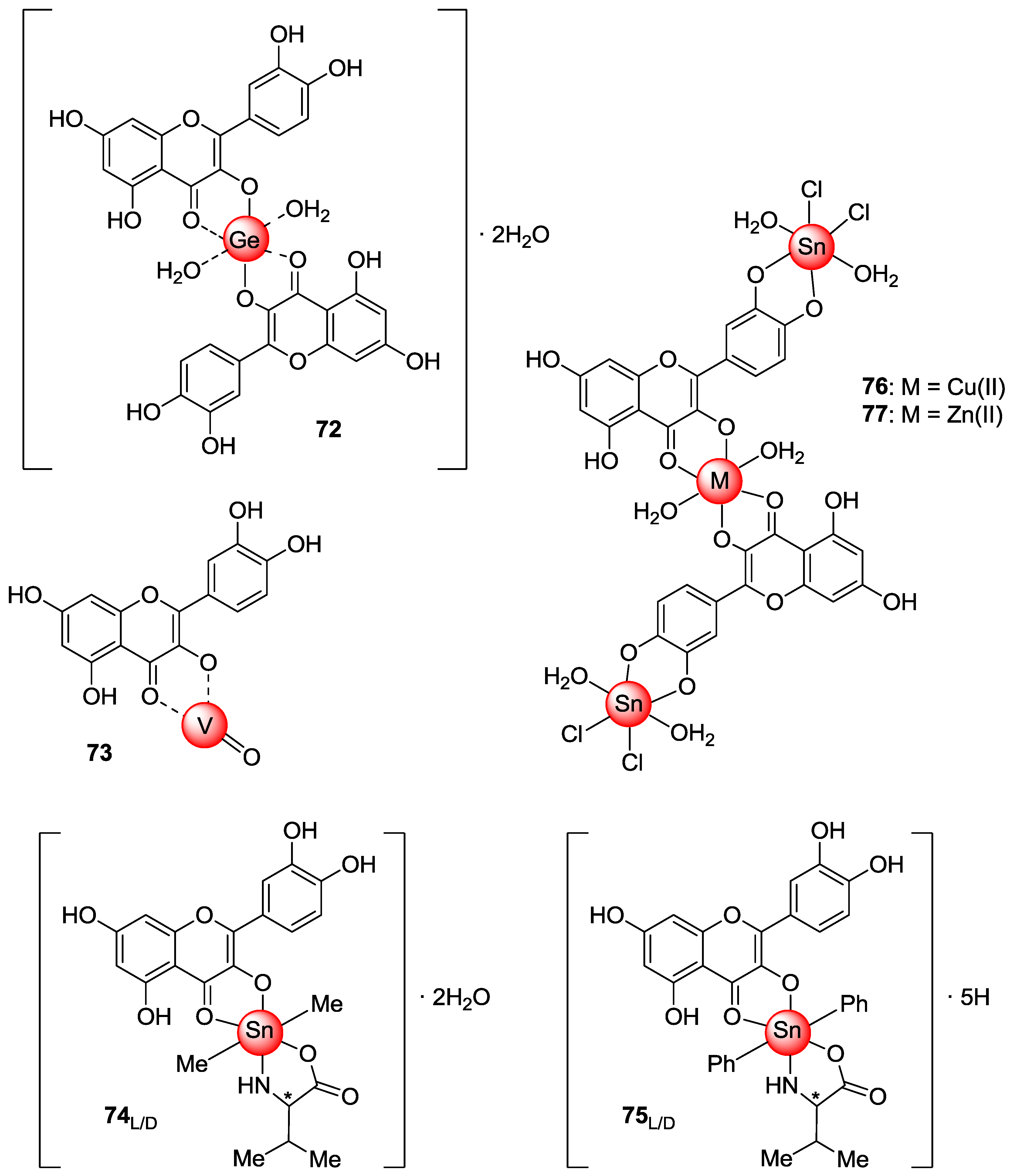
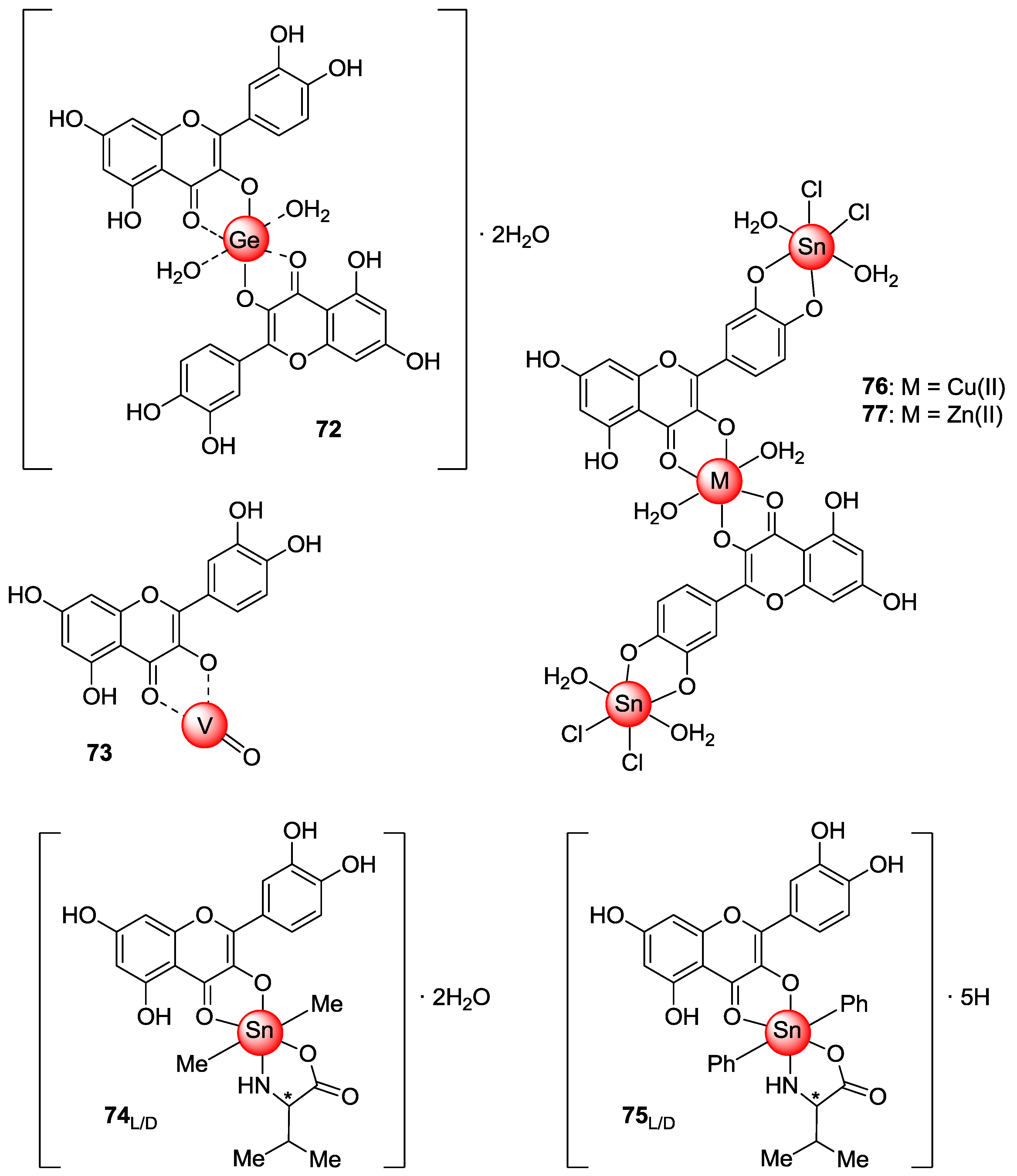
2.3. Metal Coordination
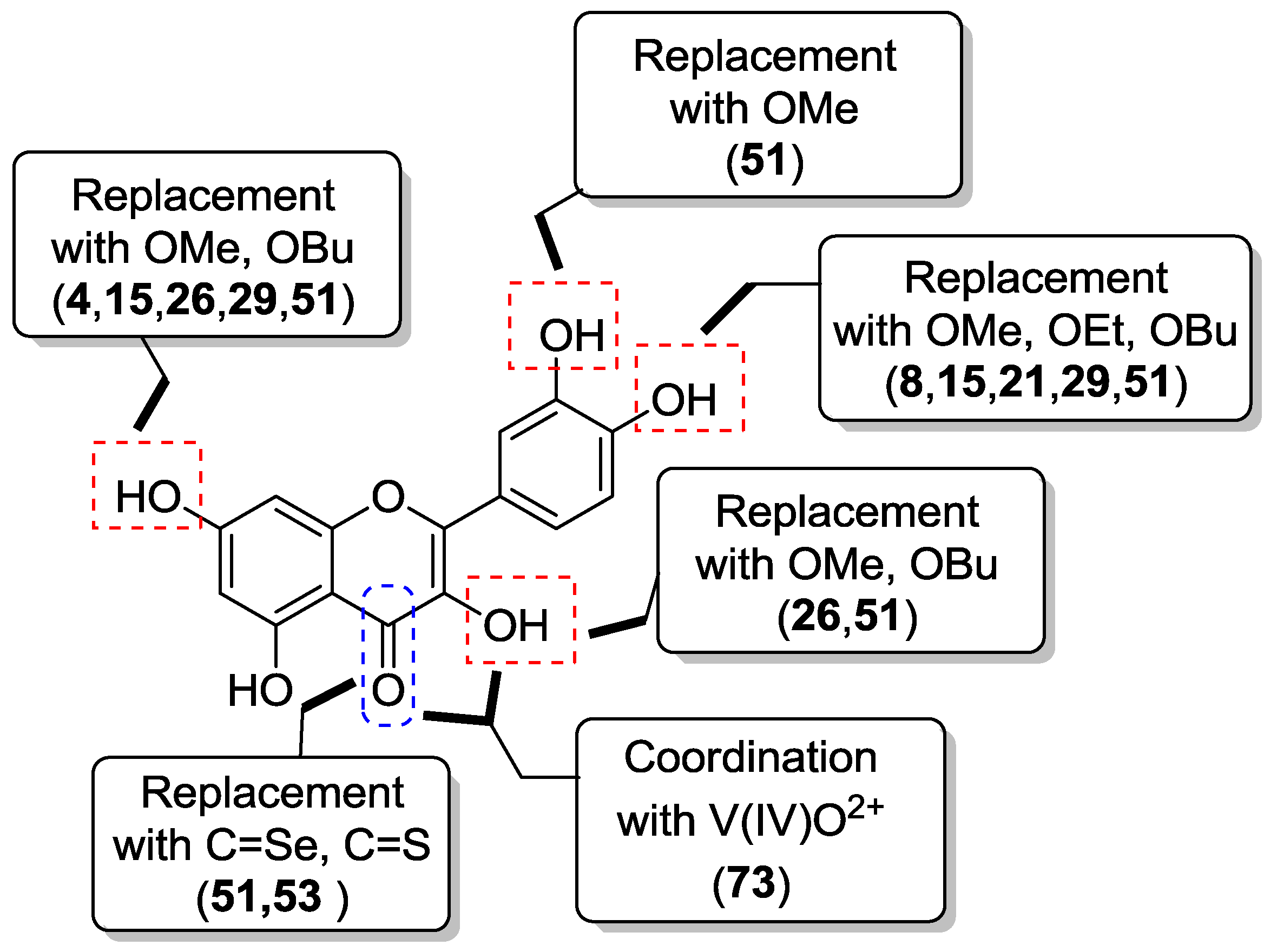
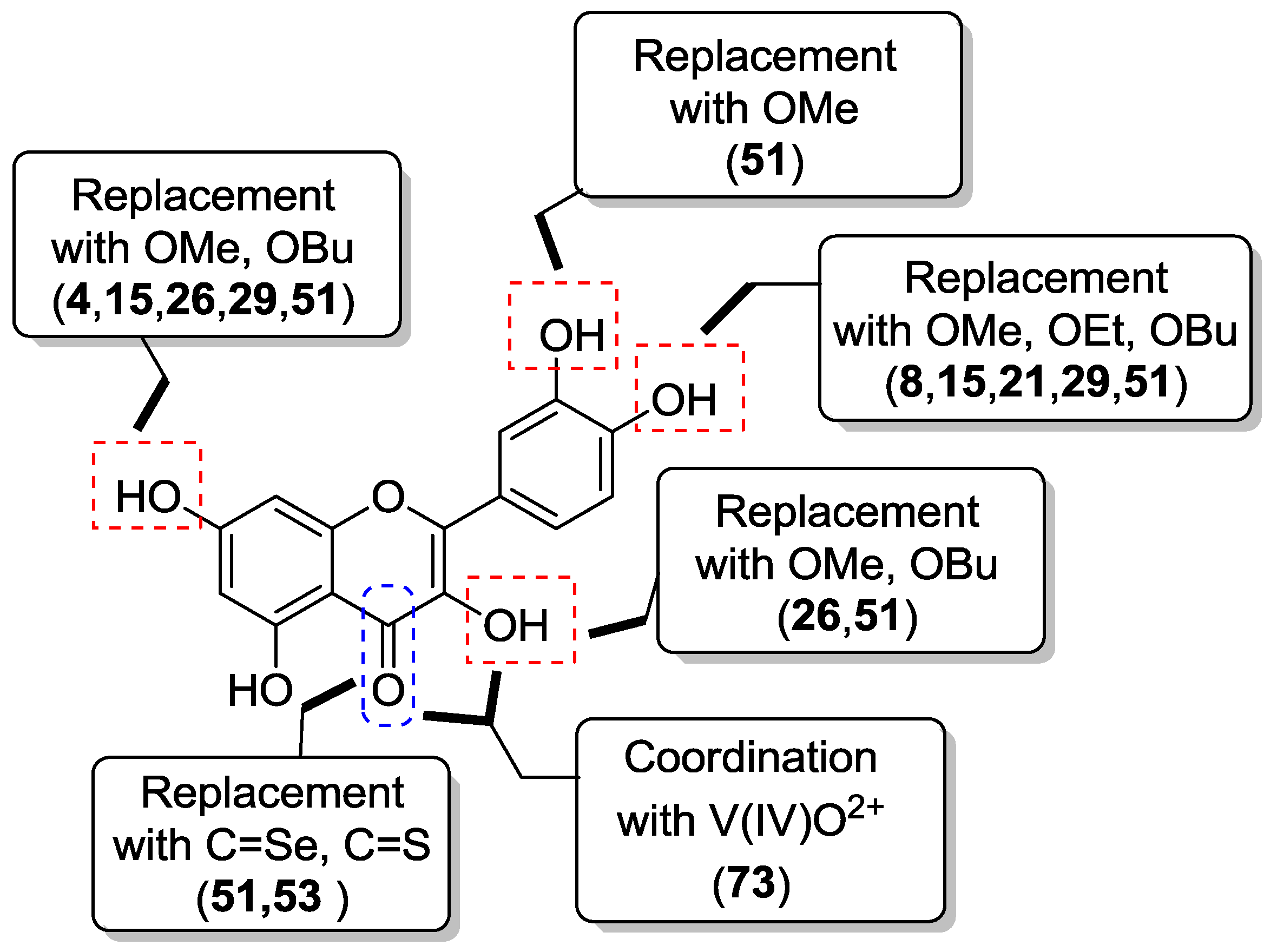
3. Structure-Activity Relationship
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramos, S. Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Mol. Nutr. Food Res. 2008, 52, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Williamson, G. Dietary Intake and Bioavailability of Polyphenols. J. Nutr. 2000, 130, 2073S–2085S. [Google Scholar] [PubMed]
- Sultana, B.; Anwar, F. Flavonols (kaempferol, quercetin, myricetin) contents of selected fruits, vegetables and medicinal plants. Food Chem. 2008, 108, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Materska, M. Quercetin and its Derivatives: Chemical Structure and Bioactivity—A Review. Pol. J. Food Nutr. Sci. 2008, 58, 407–413. [Google Scholar]
- Lu, Y.; Yeap Foo, L. Polyphenolics of Salvia—A review. Phytochemistry 2002, 59, 117–140. [Google Scholar] [CrossRef]
- Berardini, N.; Fezer, R.; Conrad, J.; Beifuss, U.; Carl, R.; Schieber, A. Screening of Mango (Mangifera indica L.) Cultivars for Their Contents of Flavonol O- and Xanthone C-Glycosides, Anthocyanins, and Pectin. J. Agric. Food Chem. 2005, 53, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Kuti, J.O.; Konuru, H.B. Antioxidant Capacity and Phenolic Content in Leaf Extracts of Tree Spinach (Cnidoscolus spp.). J. Agric. Food Chem. 2004, 52, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Materska, M.; Piacente, S.; Stochmal, A.; Pizza, C.; Oleszekc, W.; Perucka, I. Isolation and structure elucidation of flavonoid and phenolic acid glycosides from pericarp of hot pepper fruit Capsicum annuum L. Phytochemistry 2003, 63, 893–898. [Google Scholar] [CrossRef]
- Ryan, D.; Robards, K.; Lavee, S. Determination of phenolic compounds in olives by reversed-phase chromatography and mass spectrometry. J. Chromatogr. A 1999, 832, 87–96. [Google Scholar] [CrossRef]
- Jiang, H.; Engelhardt, U.H.; Thräne, C.; Maiwald, B.; Stark, J. Determination of flavonol glycosides in green tea, oolong tea and black tea by UHPLC compared to HPLC. Food Chem. 2015, 183, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Kulling, S.E.; Rawel, H.M. Chokeberry (Aronia melanocarpa)—A Review on the Characteristic Components and Potential Health Effects. Planta Med. 2008, 74, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Kreft, I.; Fabjan, N.; Yasumoto, K. Rutin content in buckwheat (Fagopyrum esculentum Moench) food materials and products. Food Chem. 2006, 98, 508–512. [Google Scholar] [CrossRef]
- Chang, Q.; Wong, Y.-S. Identification of Flavonoids in Hakmeitau Beans (Vigna sinensis) by High-Performance Liquid Chromatography-Electrospray Mass Spectrometry (LC-ESI/MS). J. Agric. Food Chem. 2004, 52, 6694–6699. [Google Scholar] [CrossRef] [PubMed]
- Abd-Allah, W.E.; Awad, H.M.; AbdelMohsen, M.M. HPLC Analysis of Quercetin and Antimicrobial Activity of Comparative Methanol Extracts of Shinus molle L. Int. J. Curr. Microbiol. Appl. Sci. 2015, 4, 550–558. [Google Scholar]
- Wang, W.; Sun, C.; Mao, L.; Ma, P.; Liu, F.; Yang, J.; Gao, Y. The biological activities, chemical stability, metabolism and delivery systems of quercetin: A review. Trends Food Sci. Technol. 2016, 56, 21–38. [Google Scholar] [CrossRef]
- Zimmerman, N.P.; Peiffer, D.; Stoner, G.D. Cancer Prevention by Antioxidant Compounds from Berries. In Inflammation, Oxidative Stress, and Cancer: Dietary Approaches for Cancer Prevention; Kong, A.N.T., Ed.; CRC Press: Boca Raton, FL, USA, 2013; pp. 419–432. [Google Scholar]
- D’Andrea, G. Quercetin: A flavonol with multifaceted therapeutic applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Birhman, K.; Raheja, I.; Sharma, S.K.; Kar, H.K. Quercetin: A wonder bioflavonoid with therapeutic potential in disease management. Asian Pac. J. Trop. Dis. 2016, 6, 248–252. [Google Scholar] [CrossRef]
- Maalik, A.; Khan, F.A.; Mumtaz, A.; Mehmood, A.; Azhar, S.; Atif, M.; Karim, S.; Altaf, Y.; Tariq, I. Pharmacological Applications of Quercetin and its Derivatives: A Short Review. Trop. J. Pharm. Res. 2014, 13, 1561–1566. [Google Scholar] [CrossRef]
- Rani, N.; Velan, L.P.T.; Vijaykumar, S.; Arunachalam, A. An insight into the potentially old-wonder molecule-quercetin: The perspectives in foresee. Chin. J. Integr. Med. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Russo, G.L. The flavonoid quercetin in disease prevention and therapy: Facts and fancies. Biochem. Pharmacol. 2012, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sha, P.M.; Vishnu, P.V.; Gayathri, R. Quercetin—A Flavonoid: A Systematic Review. J. Pharm. Sci. Res. 2016, 8, 878–880. [Google Scholar]
- Mukhopadhyay, P.; Prajapati, A.K. Quercetin in anti-diabetic research and strategies for improved quercetin bioavailability using polymer-based carriers—A review. RSC Adv. 2015, 5, 97547–97562. [Google Scholar] [CrossRef]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed. Pharmacother. 2016, 84, 892–908. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.R.; Nabavi, S.M.; Braidy, N.; Setzer, W.N.; Ahmed, T.; Nabavi, S.F. Quercetin and the mitochondria: A mechanistic view. Biotechnol. Adv. 2016, 34, 532–549. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-Y.; Wu, Y.-C.; Chung, J.-G.; Yang, J.-S.; Lu, H.-F.; Tsou, M.-F.; Wood, W.G.; Kuo, S.-J.; Chen, D.-R. Quercetin-induced apoptosis acts through mitochondrial and caspase-3-dependent pathways in human breast cancer MDA-MB-231 cells. Hum. Exp. Toxicol. 2009, 28, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A. Chemistry and biological activities of flavonoids: an overview. Sci. World J. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.S.; Tran, E.; Nguyen, T.T.T.; Ong, C.K.; Lee, S.K.; Lee, J.J.; Ng, C.P.; Leong, C.; Huynh, H. Quercetin-induced growth inhibition and cell death in nasopharyngeal carcinoma cells are associated with increase in Bad and hypophosphorylated retinoblastoma expressions. Oncol. Rep. 2004, 11, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Mittal, S.; Sak, K.; Singhal, P.; Tuli, H.S. Molecular mechanisms of action of quercetin in cancer: Recent advances. Tumor Biol. 2016, 37, 12927–12939. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Niaz, K.; Maqbool, F.; Hassan, F.I.; Abdollahi, M.; Nagulapalli Venkata, K.C.; Nabavi, S.M.; Bishayee, A. Molecular Targets Underlying the Anticancer Effects of Quercetin: An Update. Nutrients 2016, 8, 529. [Google Scholar] [CrossRef] [PubMed]
- Sak, K. Site-Specific Anticancer Effects of Dietary Flavonoid Quercetin. Nutr. Cancer 2014, 66, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Kaşıkcı, M.B.; Bağdatlıoğlu, N. Bioavailability of Quercetin. Curr. Res. Nutr. Food Sci. 2016, 4, 146–151. [Google Scholar] [CrossRef]
- Matsuo, M.; Sasaki, N.; Saga, K.; Kaneko, T. Cytotoxicity of Flavonoids toward Cultured Normal Human Cells. Biol. Pharm. Bull. 2005, 28, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Harwood, M.; Danielewska-Nikiel, B.; Borzelleca, J.F.; Flamm, G.W.; Williams, G.M.; Lines, T.C. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem. Toxicol. 2007, 45, 2179–2205. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, P.J.; Ferry, D.R.; Anderson, D.; Hussain, S.A.; Young, A.M.; Cook, J.E.; Hodgkin, E.; Seymour, L.W.; Kerr, D.J. Pre-clinical and clinical study of QC12, a water-soluble, pro-drug of quercetin. Ann. Oncol. 2001, 12, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Hirpara, K.V.; Aggarwal, P.; Mukherjee, A.J.; Joshi, N.; Burman, A.C. Quercetin and Its Derivatives: Synthesis, Pharmacological Uses with Special Emphasis on Anti-Tumor Properties and Prodrug with Enhanced Bio-Availability. Anti-Cancer Agents Med. Chem. 2009, 9, 138–161. [Google Scholar] [CrossRef]
- Walle, T. Methylation of Dietary Flavones Increases Their Metabolic Stability and Chemopreventive Effects. Int. J. Mol. Sci. 2009, 10, 5002–5019. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wong, I.L.K.; Jiang, T.; Wang, S.W.; Liu, T.; Wen, B.J.; Chow, L.M.C.; Wan Sheng, B. Synthesis of methylated quercetin derivatives and their reversal activities on P-gp- and BCRP-mediated multidrug resistance tumour cells. Eur. J. Med. Chem. 2012, 54, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Juvale, K.; Stefan, K.; Wiese, M. Synthesis and biological evaluation of flavones and benzoflavones as inhibitors of BCRP/ABCG2. Eur. J. Med. Chem. 2013, 67, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.H.; Li, N.G.; Tang, Y.P.; Shi, Q.P.; Tang, H.; Li, W.; Zhang, X.; Fu, H.-A.; Duan, J.A. Biological Evaluation and SAR Analysis of O-Methylated Analogs of Quercetin as Inhibitors of Cancer Cell Proliferation. Drug Dev. Res. 2014, 75, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.H.; Li, N.G.; Tang, Y.P.; Shi, Q.P.; Zhang, W.; Zhang, P.X.; Dong, Z.X.; Li, W.; Zhang, X.; Fu, H.A.; et al. Synthesis, biological evaluation and SAR analysis of O-alkylated analogs of quercetin for anticancer. Bioorg. Med. Chem. Lett. 2014, 24, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Al-Jabban, S.M.R.; Zhang, X.; Chen, G.; Mekuria, E.A.; Rakotondraibe, L.H.; Chen, Q.-H. Synthesis and Anti-Proliferative Effects of Quercetin Derivatives. Nat. Prod. Commun. 2015, 10, 2113–2118. [Google Scholar] [PubMed]
- Khan, I.; Paul, S.; Jakhar, R.; Bhardwaj, M.; Han, J.; Kang, S.C. Novel quercetin derivative TEF induces ER stress and mitochondria-mediated apoptosis in human colon cancer HCT-116 cells. Biomed. Pharmacother. 2016, 84, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Bao, X.; Zhu, J.; Qu, J.; Sun, Y.; Ma, X.; Wang, E.; Guo, X.; Kang, Q.; Zhen, Y. O-Alkylated derivatives of quercetin induce apoptosis of MCF-7 cells via a caspase-independent mitochondrial pathway. Chem. Biol. Interact. 2015, 242, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.R.; Liao, H.; Qu, J.; Sun, Y.; Guo, X.; Wang, E.X.; Zhen, Y.H. Synthesis, Characterization and Cytotoxicity of Alkylated Quercetin Derivatives. Iran J. Pharm. Res. 2016, 15, 329–335. [Google Scholar] [PubMed]
- Kim, M.K.; Park, K.S.; Lee, C.; Park, H.R.; Choo, H.; Chong, Y. Enhanced Stability and Intracellular Accumulation of Quercetin by Protection of the Chemically or Metabolically Susceptible Hydroxyl Groups with a Pivaloxymethyl (POM) Promoiety. J. Med. Chem. 2010, 53, 8597–8607. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Park, K.S.; Chong, Y. Remarkable Stability and Cytostatic Effect of a Quercetin Conjugate, 3,7-Bis-O-Pivaloxymethyl (POM) Quercetin. ChemMedChem 2012, 7, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Park, K.S.; Choo, H.; Chong, Y. Quercetin-POM (pivaloxymethyl) conjugates: Modulatory activity for P-glycoprotein-based multidrug resistance. Phytomedicine 2015, 22, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, O.; Paprocka, M.; Kozlak, J.; Motohashi, N.; Dus, D.; Michalak, K. Human sarcoma cell lines MES-SA and MES-SA/Dx5 as a model for multidrug resistance modulators screening. Anticancer Res. 2005, 25, 383–389. [Google Scholar] [PubMed]
- Cho, S.Y.; Kim, M.K.; Park, K.S.; Choo, H.; Chong, Y. Quercetin-POC conjugates: Differential stability and bioactivity profiles between breast cancer (MCF-7) and colorectal carcinoma (HCT116) cell lines. Bioorg. Med. Chem. 2013, 21, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Dupont, M.S.; Day, A.J.; Plumb, G.W.; Williamson, G.; Johnson, I.T. Intestinal transport of quercetin glycosides in rats involves both deglycosylation and interaction with the hexose transport pathway. J. Nutr. 2000, 130, 2765–2771. [Google Scholar] [PubMed]
- Enayat, S.; Şeyma Ceyhan, M.; Taşkoparan, B.; Stefek, M.; Banerjee, S. CHNQ, a novel 2-Chloro-1,4-naphthoquinone derivative of quercetin, induces oxidative stress and autophagy both in vitro and in vivo. Arch. Biochem. Biophys. 2016, 596, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Danihelová, M.; Veverka, M.; Šturdík, E.; Jantová, S. Antioxidant action and cytotoxicity on HeLa and NIH-3T3 cells of new quercetin derivatives. Interdiscip. Toxicol. 2013, 6, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.L.; Charneira, C.; Gandin, V.; Ferreira Da Silva, J.L.; Justino, G.C.; Telo, J.P.; Vieia, A.J.; Marzano, C.; Antunes, A.M.M. Selenium-Containing Chrysin and Quercetin Derivatives: Attractive Scaffolds for Cancer Therapy. J. Med. Chem. 2015, 58, 4250–4265. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, D.; Watson, K.A.; Boateng, S.Y.; Green, R.J.; Greco, F.; Osborn, H.M.I. Exploring quercetin and luteolin derivatives as antiangiogenic agents. Eur. J. Med. Chem. 2015, 97, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, D.; Watson, K.; Greco, F.; Osborn, H. Novel synthesised flavone derivatives provide significant insight into the structural features required for enhanced anti-proliferative activity. RSC Adv. 2016, 6, 64544–64556. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, M.K.; Mok, H.; Choo, H.; Chong, Y. Separation of Quercetin’s Biological Activity from Its Oxidative Property through Bioisosteric Replacement of the Catecholic Hydroxyl Groups with Fluorine Atoms. J. Agric. Food Chem. 2012, 60, 6499–6506. [Google Scholar] [CrossRef] [PubMed]
- Dangles, O.; Fargeix, G.; Dufour, C. One-electron oxidation of quercetin and quercetin derivatives in protic and non protic media. J. Chem. Soc. Perkin Trans. 2 1999, 2, 1387–1395. [Google Scholar] [CrossRef]
- Cárdenas, M.; Marder, M.; Blank, V.C.; Roguin, L.P. Antitumor activity of some natural flavonoids and synthetic derivatives on various human and murine cancer cell lines. Bioorg. Med. Chem. 2006, 14, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Salem, J.H.; Chevalot, I.; Harscoat-Schiavo, C.; Paris, C.; Fick, M.; Humeau, C. Biological activities of flavonoids from Nitraria retusa (Forssk.) Asch. and their acylated derivatives. Food Chem. 2011, 124, 486–494. [Google Scholar] [CrossRef]
- Lu, C.; Huang, F.; Li, Z.; Ma, J.; Li, H.; Fang, L. Synthesis and Bioactivity of Quercetin Aspirinates. Bull. Korean Chem. Soc. 2014, 35, 518–520. [Google Scholar] [CrossRef]
- Kim, M.K.; Park, K.-S.; Yeo, W.-S.; Choo, H.; Chong, Y. In vitro solubility, stability and permeability of novel quercetin-amino acid conjugates. Bioorg. Med. Chem. 2009, 17, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Choo, H.; Chong, Y. Water-Soluble and Cleavable Quercetin-Amino Acid Conjugates as Safe Modulators for P-Glycoprotein-Based Multidrug Resistance. J. Med. Chem. 2014, 57, 7216–7233. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.K.D.; Huynh, T.K.C.; Nguyen, T.D. Synthesis, characterization, anti-inflammatory and anti-proliferative activity against MCF-7 cells of O-alkyl and O-acyl flavonoid derivatives. Bioorg. Chem. 2015, 63, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, M.; Yu, L.; Zhao, Y.; He, N.; Yang, X. Antitumor activities of quercetin and quercetin-5′,8-disulfonate in human colon and breast cancer cell lines. Food Chem. Toxicol. 2012, 50, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Han, Y.; Yang, X.; Sun, Y.; Zhao, Y. Protective Effects of Quercetin and Quercetin-5′,8-Disulfonate against Carbon Tetrachloride-Caused Oxidative Liver Injury in Mice. Molecules 2014, 19, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.P.; Yan, Y.; Xia, J.; Zhang, S.; Wang, M.; Chen, J.; Xu, Y. A phenylacetaldehyde-flavonoid adduct, 8-C-(E-phenylethenyl)-norartocarpetin, exhibits intrinsic apoptosis and MAPK pathways-related anticancer potential on HepG2, SMMC-7721 and QGY-7703. Food Chem. 2016, 197, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
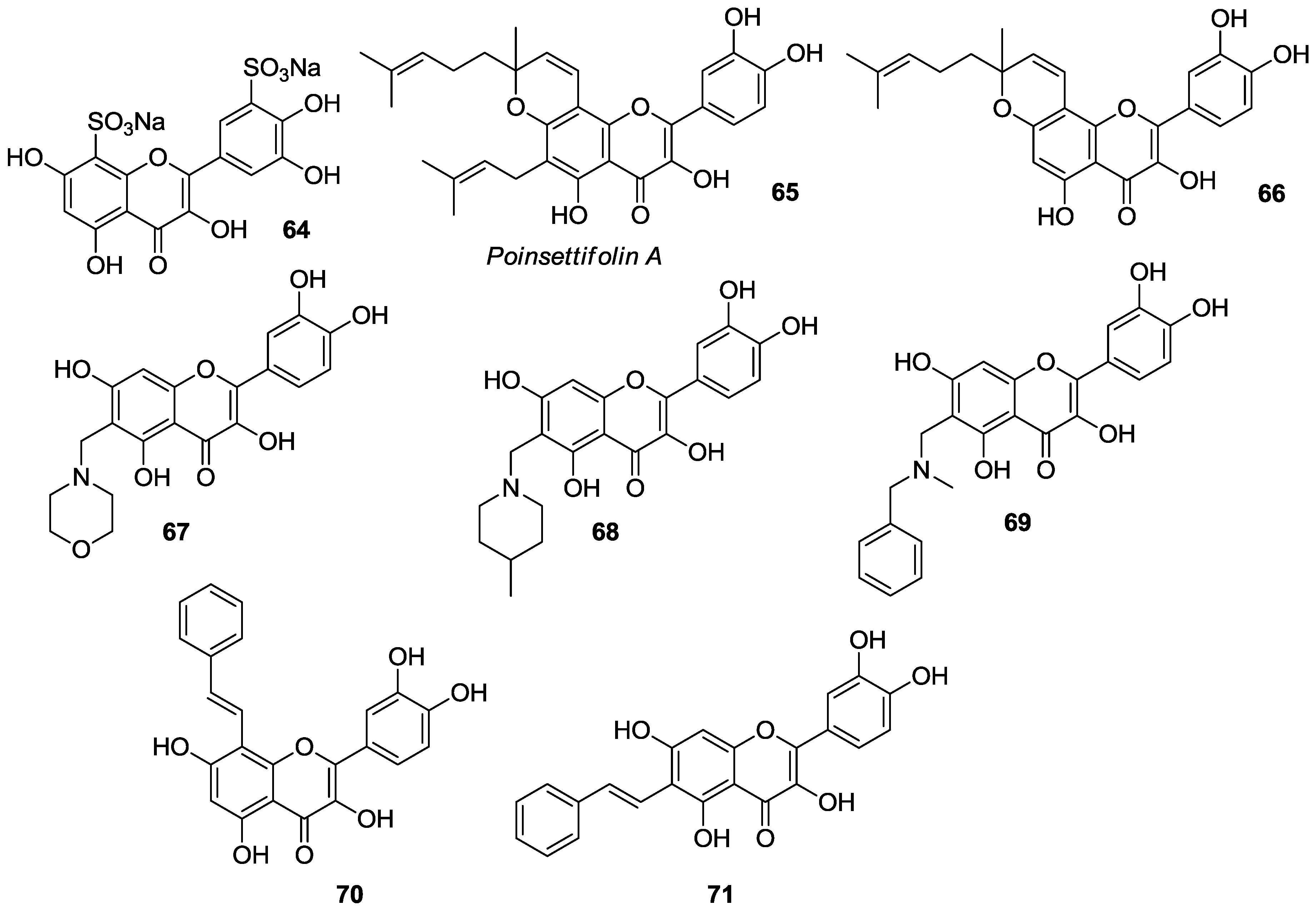
- Tsopmo, A.; Tene, M.; Kamnaing, P.; Ngnokam, D.; Ayafor, J.F.; Sterner, O. Geranylated flavonoids from Dorstenia poinsettifolia. Phytochemistry 1998, 48, 345–348. [Google Scholar] [CrossRef]
- Escobar, Z.; Solano, C.; Larsson, R.; Johansson, M.; Salamanca, E.; Gimenez, A.; Muñoz, E.; Sterner, O. Synthesis of poinsettifolin A. Tetrahedron 2014, 70, 9052–9056. [Google Scholar] [CrossRef]
- Zhan, W.; Lin, S.; Chen, J.; Dong, X.; Chu, J.; Du, W. Design, Synthesis, Biological Evaluation, and Molecular Docking of Novel Benzopyran and Phenylpyrazole Derivatives as Akt Inhibitors. Chem. Biol. Drug Des. 2015, 85, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.C.; Madison, V. AKT crystal structure and AKT-specific inhibitors. Oncogene 2005, 24, 7493–7501. [Google Scholar] [CrossRef] [PubMed]
- Prestayko, A.W.; D’Aoust, J.C.; Issell, B.F.; Crooke, S.T. Cisplatin (cis-diamminedichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–39. [Google Scholar] [CrossRef]
- Giaccone, G. Clinical Perspectives on Platinum Resistance. Drugs 2000, 59 (Suppl. 4), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- McQuitty, R.J. Metal-based drugs. Sci. Prog. 2014, 97, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Thomas, J.A. Kinetically inert transition metal complexes that reversibly bind to DNA. Chem. Soc. Rev. 2003, 32, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, M.; Najam, T.; Shah, S.S.A.; Ahmad, M.M.; Shaheen, S.; Tabassum, R.; Rivera, G. DNA Binding Mode of Transition Metal Complexes, a Relationship to Tumor Cell Toxicity. Curr. Med. Chem. 2014, 21, 3081–3094. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, J.E.N. Molecular aspects on the interaction of quercetin and its metal complexes with DNA. Int. J. Biol. Macromol. 2011, 48, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Zhu, W.; Duan, Y.; Qu, W.; Yan, Z. Synthesis, characterization and antitumor activity of the germanium-quercetin complex. Main Group Met. Chem. 2012, 35, 103–109. [Google Scholar] [CrossRef]
- Tan, J.; Wang, B.C.; Zhu, L. Hydrolytic cleavage of DNA by quercetin manganese(II) complexes. Colloid Surface B 2007, 55, 149–152. [Google Scholar] [CrossRef]
- Naso, L.; Valcarcel, M.; Villacé, P.; Roura-Ferrer, M.; Salado, C.; Ferrer, E.G.; Williams, P.A.M. Specific antitumor activities of natural and oxovanadium(IV) complexed flavonoids in human breast cancer cells. New J. Chem. 2014, 38, 2414–2421. [Google Scholar] [CrossRef]
- Parveen, S.; Tabassum, S.; Arjmand, F. Human Topoisomerase I mediated cytotoxicity profile of l-valine-quercetin diorganotin(IV) antitumor drug entities. J. Organomet. Chem. 2016, 823, 23–33. [Google Scholar] [CrossRef]
- Hong, M.; Yin, H.; Zhang, X.; Li, C.; Yue, C.; Cheng, S. Di- and tri-organotin(IV) complexes with 2-hydroxy-1-naphthaldehyde 5-chloro-2-hydroxybenzoylhydrazone: Synthesis, characterization and in vitro antitumor activities. J. Organomet. Chem. 2013, 724, 23–31. [Google Scholar] [CrossRef]
- Arjmand, F.; Muddassir, M.; Yousuf, I. Design and synthesis of enantiomeric (R)- and (S)-copper (II) and diorganotin (IV)-based antitumor agents: Their in vitro DNA binding profile, cleavage efficiency and cytotoxicity studies. J. Photochem. Photobiol. B 2014, 136, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Yadav, S. Investigation of diorganotin (IV) complexes: Synthesis, characterization, in vitro DNA binding studies and cytotoxicity assessment of di-n-butyltin(IV) complex. Inorg. Chim. Acta 2014, 423, 204–214. [Google Scholar] [CrossRef]
- Saxena, A.K.; Huber, F. Organotin compounds and cancer chemotherapy. Coord. Chem. Rev. 1989, 95, 109–123. [Google Scholar] [CrossRef]
- Tabassum, S.; Zaki, M.; Afzal, M.; Arjmand, F. New modulated design and synthesis of quercetin-CuII/ZnII-Sn2IV scaffold as anticancer agents: In vitro DNA binding profile, DNA cleavage pathway and Topo-I activity. Dalton Trans. 2013, 42, 10029–10041. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anna, M.M.; Censi, V.; Carrozzini, B.; Caliandro, R.; Denora, N.; Franco, M.; Veclani, D.; Melchior, A.; Tolazzi, M.; Mastrorilli, P. Triphenylphosphane Pt(II) complexes containing biologically active natural polyphenols: Synthesis, crystal structure, molecular modeling and cytotoxic studies. J. Inorg. Biochem. 2016, 163, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Seidell, A. Solubilities of Organic Compounds; D. Van Nostrand Company, Inc.: New York, NY, USA, 1940. [Google Scholar]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Diller, D.J.; Hobbs, D.W. Deriving knowledge through data mining high-throughput screening data. J. Med. Chem. 2004, 47, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Suffness, M.; Pezzuto, J.M. Assays related to cancer drug discovery. In Methods in Plant Biochemistry: Assays for Bioactivity; Hostettmann, K., Ed.; Academic Press: London, UK, 1991; pp. 71–133. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 19 | 20 | |
|---|---|---|---|
| Hoechst 33342 | nd 2 | 0.540 ± 0.079 | 0.822 ± 0.169 |
| Pheophorbide A | nd 2 | 0.570 ± 0.093 | 1.880 ± 0.240 |
| Cell Line | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 4 | 8 | 15 | 21 | 26 | 29 | |
| A549 | 6.20 ± 0.51 | 3.08 ± 0.10 | 2.63 ± 0.19 | 3.07 ± 0.02 | 1.24 | 1.13 | 2.06 |
| H157 | 6.00 ± 0.47 | 3.31 ± 0.01 | 3.04 ± 0.02 | 3.45 ± 0.02 | 0.67 | 5.92 | 0.39 |
| H460 | 9.62 ± 0.89 | 3.32 ± 0.02 | 4.45 ± 0.02 | 2.75 ± 0.01 | 1.03 | 3.05 | 3.16 |
| 1944 | 10.18 ± 1.11 | 4.25 ± 0.02 | 3.86 ± 0.02 | 2.86 ± 0.01 | 1.78 | 1.17 | 0.56 |
| H266 | 16.87 ± 1.12 | 13.87 ± 1.16 | 7.57 ± 0.05 | >50 | 2.85 | 8.92 | 15.92 |
| Hop62 | 7.52 ± 0.58 | 6.87 ± 0.04 | 9.93 ± 0.78 | 12.11 ± 1.11 | 4.45 | 4.68 | 4.43 |
| 1299 | 13.60 ± 1.24 | 10.25 ± 0.88 | 21.97 ± 1.78 | 34.82 ± 3.21 | 1.77 | 5.05 | 4.54 |
| 292G | >50 | 27.95 ± 2.17 | 12.54 ± 1.10 | 40.72 ± 3.79 | 4.11 | 8.75 | 17.73 |
| Calu1 | 23.58 ± 1.82 | 23.43 ± 1.76 | 24.95 ± 1.89 | >50 | 1.50 | 5.46 | 6.21 |
| 1792 | 3.85 ± 0.45 | 14.73 ± 1.08 | 4.06 ± 0.03 | 3.36 ± 0.02 | 1.00 | 6.03 | 3.09 |
| M4E | 21.71 ± 1.87 | 10.82 ± 0.98 | 19.91 ± 1.95 | 30.69 ± 2.98 | 6.18 | 11.18 | 6.29 |
| M14 | 12.77 ± 1.08 | 13.32 ± 1.76 | 14.85 ± 1.26 | 4.29 ± 0.02 | 0.94 | 3.47 | 9.35 |
| LOX-IMVI | 4.65 ± 0.28 | >50 | >50 | >50 | 1.14 | 2.55 | 2.36 |
| SKBR | 16.71 ± 1.21 | 6.25 ± 0.03 | 7.97 ± 1.01 | 8.30 ± 0.54 | 3.19 | 4.58 | 4.39 |
| Hela | 3.56 ± 0.28 | 4.26 ± 0.02 | 1.81 ± 0.01 | 1.49 ± 0.01 | 1.20 | 6.09 | 0.87 |
| Cell Line | Compound | ||||
|---|---|---|---|---|---|
| 1 | 11 | 24 | 25 | 26 | |
| PC-3 | >100 | 22.27 ± 6.83 | 13.74 ± 1.62 | 17.68 ± 0.96 | 11.95 ± 1.12 |
| DU145 | >100 | 46.82 ± 3.69 | 12.59 ± 0.96 | 19.25 ± 2.21 | 18.63 ± 6.86 |
| LNCaP | 45.46 ± 1.31 | 13.23 ± 4.75 | 4.20 ± 0.96 | 6.42 ± 2.72 | 6.46 ± 1.10 |
| Cell Line | Compound | ||
|---|---|---|---|
| 1 | 43 | 44 | |
| MCF-7 | 343 | 43.5 ± 2.1 3 | 22.6 ± 2.2 3 |
| 38.6 4 | 20.2 4 | ||
| CaCo-2 | 340 | 66.8 | 43.7 |
| NCI-H446 | 68.9 | 27.6 | |
| A549 | 77.2 | 29.5 | |
| MGC-803 | 80.6 | 25.4 | |
| SGC-7901 | 75.7 | 18.5 | |
| Medium | Compound | |
|---|---|---|
| 1 | 45 | |
| PBS | 10 h | >24 h |
| cDMEM | <0.5 h | 100 h 2 |
| Anticancer Drug | Modulator 2 | |||
|---|---|---|---|---|
| None (IC50) | Verapamil (IC50/FR) | 1 (IC50/FR) | 46 (IC50/FR) | |
| Doxorubicine | 8.18 ± 0.01 | 0.12 ± 0.01/68.3 | 4.26 ± 0.32/1.9 | 0.34 ± 0.09/24.1 |
| Actinomycin D | 13.10 ± 0.34 | 0.23 ± 0.02/57.0 | 4.68 ± 1.00/2.8 | 0.41 ± 0.01/32.0 |
| Vinblastine | 12.25 ± 0.19 | 0.24 ± 0.01/51.0 | 4.90 ± 0.13/2.5 | 0.43 ± 0.06/28.5 |
| Paclitaxel | 10.53 ± 0.21 | 0.22 ± 0.04/47.9 | 4.66 ± 0.11/2.3 | 0.41 ± 0.04/25.7 |
| Medium | Compound | |||
|---|---|---|---|---|
| 1 | 47 | 48 | 49 | |
| PBS | 10 h | >96 h | >96 h | >96 h |
| cDMEM | <0.5 h | 1 h | 54 h | 24 h 2 |
| Cell Line | Compound | |
|---|---|---|
| 1 | 50 | |
| HCT-116 | 117 ± 8.9 | 30.3 ± 1 |
| HT-29 | nc 3 | 23.21 ± 2.4 |
| Cell Line | Compound | |||
|---|---|---|---|---|
| 1 | 51 | 19 | Cisplatin | |
| A375 | 25.13 ± 2.62 | 2.21 ± 1.07 | 69.72 ± 3.32 | 3.12 ± 1.13 |
| HCT-15 | 16.35 ± 2.29 | 2.23 ± 1.01 | 68.11 ± 2.35 | 12.31 ± 1.26 |
| BxPC3 | 24.12 ± 1.85 | 2.42 ± 1.19 | >100 | 11.43 ± 1.29 |
| MCF-7 | 20.90 ± 3.44 | 3.08 ± 1.98 | >100 | 7.6 ± 2.49 |
| MCF-7/ADR | 22.15 ± 2.14 (1.1) | 3.35 ± 1.58 (1.1) | >100 | 8.41 ± 1.22 (16) |
| A431 | 23.04 ± 1.07 | 3.11 ± 1.23 | 57.54 ± 2.28 | 1.62 ± 1.25 |
| A431/Pt | 34.37 ± 3.22 (1.5) | 3.78 ± 1.52 (1.2) | 82.25 ± 2.77 (1.4) | 3.42 ± 1.08 (2.1) |
| 2008 | 21.18 ± 1.84 | 2.09 ± 1.27 | >100 | 2.17 ± 1.37 |
| C13* | 37.62 ± 3.82 (1.8) | 2.29 ± 1.93 (1.1) | >100 | 22.26 ± 1.86 (10.3) |
| Cell Line | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 56 | 57 | 58 | 59 | 60 | 61 | |
| HeLa | 35.5 ± 1.1 2 | 29.6 ± 1.9 2 | 19.5 ± 0.8 2 | 16.5 ± 1.5 2 | |||
| NIH-3T3 | 20.9 ± 0.9 2 | 15.5 ± 0.7 2 | 16.1 ± 0.4 2 | 10.6 ± 0.1 2 | |||
| HL-60 | >100 | 68.71 3 | 69.29 3 | >100 3 | |||
| HepG2 | >100 | 54.22 3 | 38.49 3 | 55.80 3 | |||
| Anticancer Drug | Modulator 2 | |||
|---|---|---|---|---|
| None | Verapamil | 1 | 62 | |
| Doxorubicine | 8.20 | 0.12 | 4.26 | 0.14 |
| Actinomycin D | 0.13 | 0.23 | 4.68 | 0.34 |
| Vinblastine | 0.11 | 0.24 | 4.90 | 0.33 |
| Paclitaxel | 0.10 | 0.22 | 4.66 | 0.32 |
| Medium | Compound | |
|---|---|---|
| 1 | 62 | |
| PBS | 10.3 h | >72 h |
| cRPMI | 0.4 h | 9.3 h |
| Cell Line | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 64 2 | 67 3,4 | 68 3,4 | 69 3,4 | 70 5 | 71 5 | |
| LoVo | 40.2 2 | 28.0 | |||||
| MCF-7 | 30.8 2 | 19.9 | |||||
| HL-60 | 16.65 ± 1.5 (11.45 ± 0.9) | 14.22 ± 1.1 (11.45 ± 0.9) | 7.56 ± 0.4 (11.45 ± 0.9) | ||||
| OVCAR-8 | >50 (7.74 ± 0.5) | >50 (7.74 ± 0.5) | 16.69 ± 2.0 (7.74 ± 0.5) | ||||
| PC-3 | 42.74 ± 3.8 (9.49 ± 0.8) | 32.28 ± 2.2 (9.49 ± 0.8) | 15.45 ± 1.8 (9.49 ± 0.8) | ||||
| HepG2 | 22.16 ± 0.63 5,6 | 12.50 ± 0.9 (10.17 ± 0.6) | 18.57 ± 1.1 (10.17 ± 0.6) | 7.20 ± 0.5 (10.17 ± 0.6) | >40 | 25.76 ± 1.12 | |
| SMMC-7721 | >40 5 | >40 | >40 | ||||
| QGY-7703 | 18.90 ± 0.48 5 | 13.66 ± 0.90 | 17.70 ± 0.49 | ||||
| Cell Line | Concentration | |
|---|---|---|
| 10 μM | 100 μM | |
| EC9706 | 8.67 ± 5.16 | 35.12 ± 3.80 2 |
| PC-3 | 7.28 ± 4.18 | 29.72 ± 2.84 3 |
| HeLa | 3.86 ± 3.85 | 33.72 ± 6.07 2 |
| SPC-A-1 | 5.04 ± 0.76 | 47.24 ± 2.09 2 |
| Cell Line | Compound | ||
|---|---|---|---|
| 1 | 73 | V(IV)O2+ | |
| MDA-MB231 | 49.6 ± 6.0 | 10.2 ± 8.0 | 49.0 ± 2.5 |
| SKBR3 | 25.8 ± 4.5 | 22.8 ± 7.6 | 95.7 ± 4.8 |
| MDA-MB468 | 23.8 ± 5.2 | 7.4 ± 5.4 | 19.4 ± 2.0 |
| T47D | 81.5 ± 4.8 | 4.8 ± 7.6 | >100 |
| Cell Line | Compound | ||
|---|---|---|---|
| ADR | 74L | 75L | |
| HeLa | <0.0184 | <0.0166 | <0.0144 |
| MCF-7 | <0.0184 | <0.0166 | <0.0144 |
| MIA-Pa-Ca-2 | <0.0184 | <0.0166 | <0.0144 |
| HepG2 | <0.0184 | 0.0757 | <0.0144 |
| Cell Line | Compound | ||
|---|---|---|---|
| ADR | 76 | 77 | |
| U373MG | <17.2 | <8.7 | 42.5 |
| PC-3 | <17.2 | <8.7 | 40.0 |
| Hop62 | <17.2 | <8.7 | 36.5 |
| HL-60 | <17.2 | <8.7 | 31.9 |
| HCT-15 | <17.2 | <8.7 | 42.8 |
| A2780 | 56.0 | >69.6 | 41.1 |
| HeLa | <17.2 | <8.7 | 7.7 |
| Compound | Solubility (Solvent) | Stability (Solvent) |
|---|---|---|
| 1 | 198 μM (water) [89] | 10 h (PBS); <0.5 h (cDMEM) [47,50] 10.3 h (PBS); 0.4 h (cRPMI) [63] |
| 43/44 | 180 μM (DMEM) [45] | |
| 45 | >24 h (PBS); 100 h (cDMEM) 1 [47] | |
| 47 | >96 h (PBS); 1 h (cDMEM) [50] | |
| 48 | >96 h (PBS); 54 h (cDMEM) [50] | |
| 49 | >96 h (PBS); 24 h (cDMEM) 2 [50] | |
| 62 | up to 400 μM (aqueous) [63] | >72 h (PBS); 9.3 h (cRPMI) [63] |
| Cell Line | Compound | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 8 | 15 | 21 | 26 | 29 | 51 2 | 73 | |
| A549 | 6.20 ± 0.51 | 3.08 ± 0.10 | 2.63 ± 0.19 | 3.07 ± 0.02 | 1.24 | 1.13 | 2.06 | ||
| H157 | 6.00 ± 0.47 | 3.31 ± 0.01 | 3.04 ± 0.02 | 3.45 ± 0.02 | 0.67 | 0.39 | |||
| H460 | 9.62 ± 0.89 | 3.32 ± 0.02 | 4.45 ± 0.02 | 2.75 ± 0.01 | 1.03 | 3.05 | 3.16 | ||
| 1944 | 10.18 ± 1.11 | 4.25 ± 0.02 | 3.86 ± 0.02 | 2.86 ± 0.01 | 1.78 | 1.17 | 0.56 | ||
| H266 | 16.87 ± 1.12 | 2.85 | |||||||
| Hop62 | 7.52 ± 0.58 | 4.45 | 4.68 | 4.43 | |||||
| 1299 | 13.60 ± 1.24 | 1.77 | 4.54 | ||||||
| 292G | >50 | 4.11 | |||||||
| Calu1 | 23.58 ± 1.82 | 1.50 | |||||||
| 1792 | 3.85 ± 0.45 | 4.06 ± 0.03 | 3.36 ± 0.02 | 1.00 | 3.09 | ||||
| M4E | 21.71 ± 1.87 | ||||||||
| M14 | 12.77 ± 1.08 | 4.29 ± 0.02 | 0.94 | 3.47 | |||||
| LOX-IMVI | 4.65 ± 0.28 | 1.14 | 2.55 | 2.36 | |||||
| SKBR | 16.71 ± 1.21 | 3.19 | 4.58 | 4.39 | |||||
| Hela | 3.56 ± 0.28 | 4.26 ± 0.02 | 1.81 ± 0.01 | 1.49 ± 0.01 | 1.20 | 0.87 | |||
| A375 | 25.13 ± 2.62 | 2.21 ± 1.07 | |||||||
| HCT-15 | 2.23 ± 1.01 | ||||||||
| BxPC3 | 2.42 ± 1.19 | ||||||||
| MCF-7 | 3.08 ± 1.98 | ||||||||
| A431 | 3.11 ± 1.23 | ||||||||
| 2008 | 2.09 ± 1.27 | ||||||||
| T47D | 4.8 ± 7.6 | ||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massi, A.; Bortolini, O.; Ragno, D.; Bernardi, T.; Sacchetti, G.; Tacchini, M.; De Risi, C. Research Progress in the Modification of Quercetin Leading to Anticancer Agents. Molecules 2017, 22, 1270. https://doi.org/10.3390/molecules22081270
Massi A, Bortolini O, Ragno D, Bernardi T, Sacchetti G, Tacchini M, De Risi C. Research Progress in the Modification of Quercetin Leading to Anticancer Agents. Molecules. 2017; 22(8):1270. https://doi.org/10.3390/molecules22081270
Chicago/Turabian StyleMassi, Alessandro, Olga Bortolini, Daniele Ragno, Tatiana Bernardi, Gianni Sacchetti, Massimo Tacchini, and Carmela De Risi. 2017. "Research Progress in the Modification of Quercetin Leading to Anticancer Agents" Molecules 22, no. 8: 1270. https://doi.org/10.3390/molecules22081270
APA StyleMassi, A., Bortolini, O., Ragno, D., Bernardi, T., Sacchetti, G., Tacchini, M., & De Risi, C. (2017). Research Progress in the Modification of Quercetin Leading to Anticancer Agents. Molecules, 22(8), 1270. https://doi.org/10.3390/molecules22081270