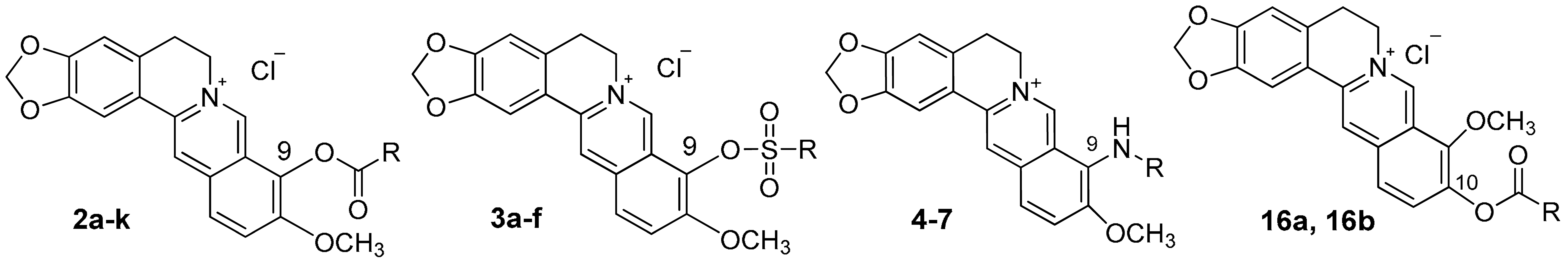
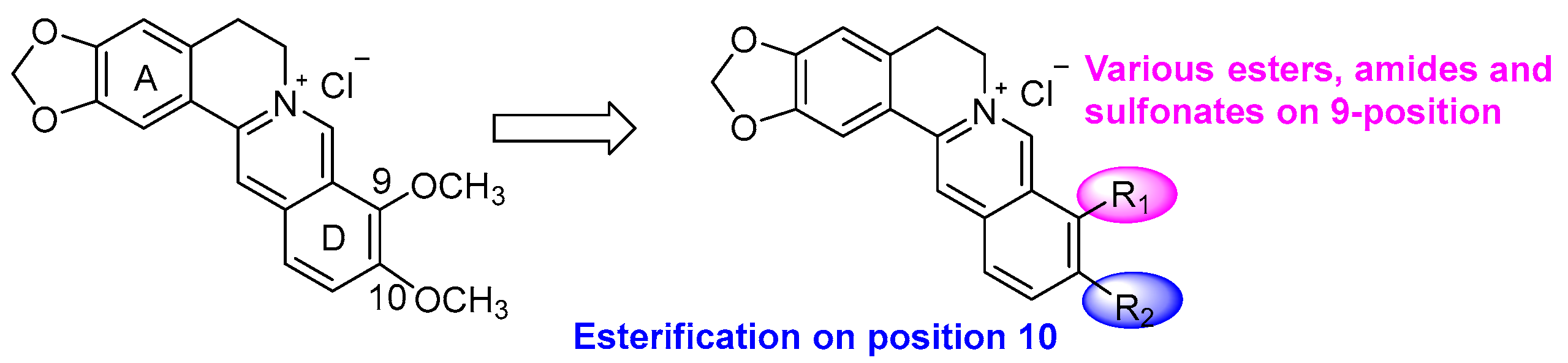
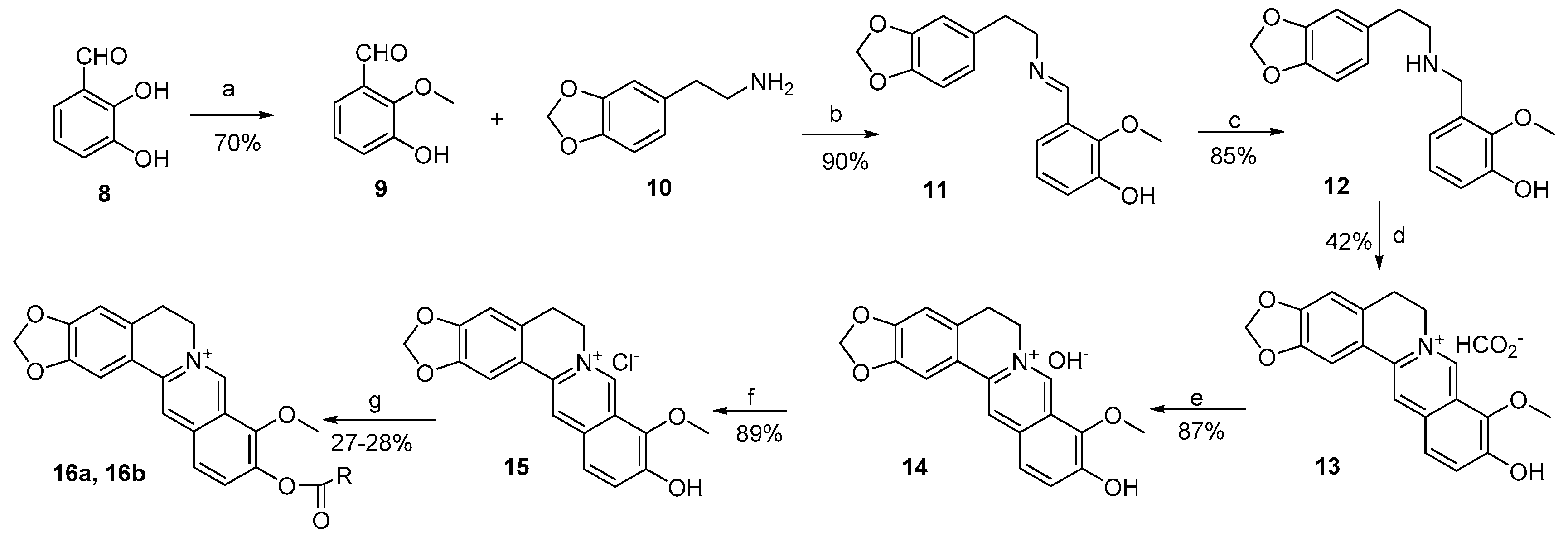
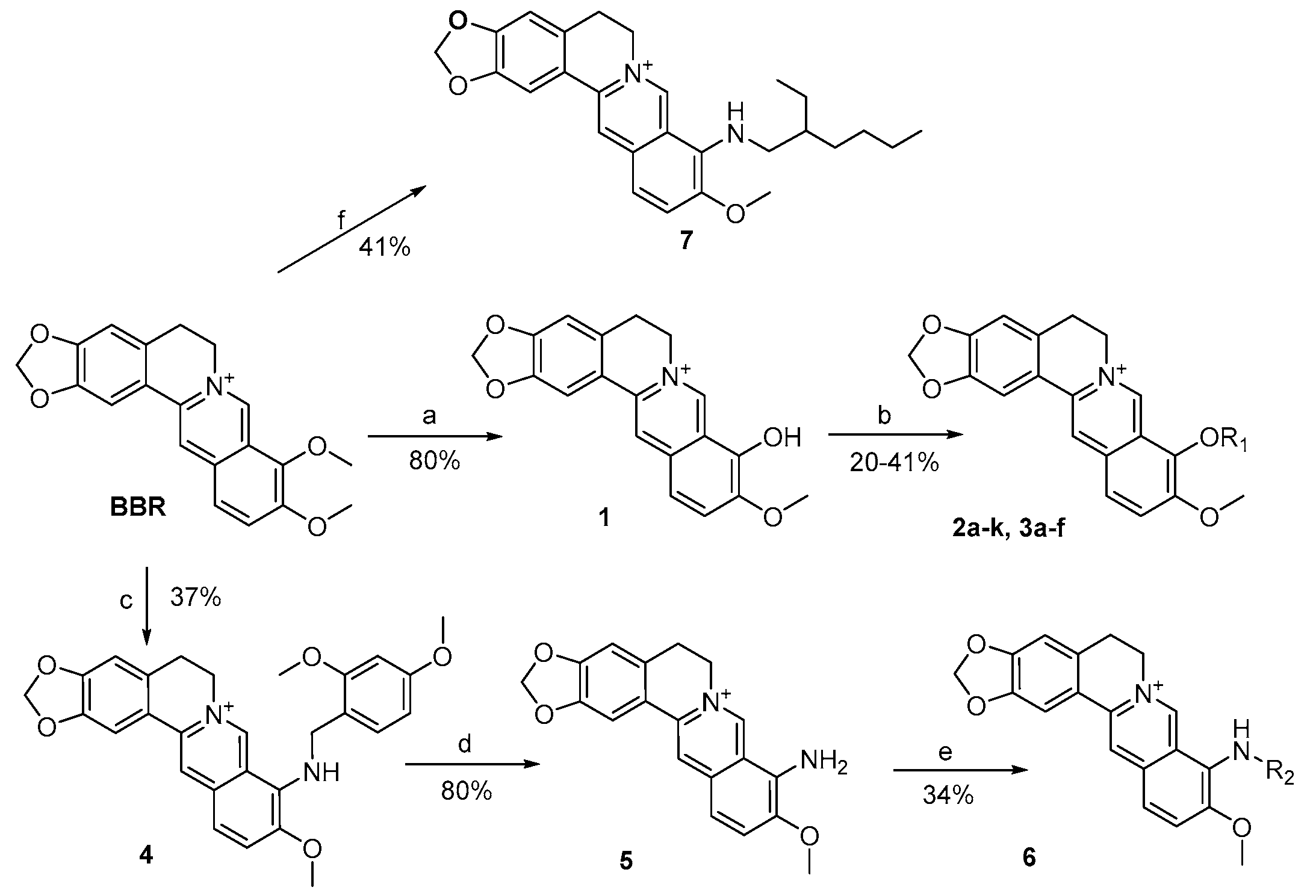
3.2.1. General Procedure for the Synthesis of 2a–k and 3a–f
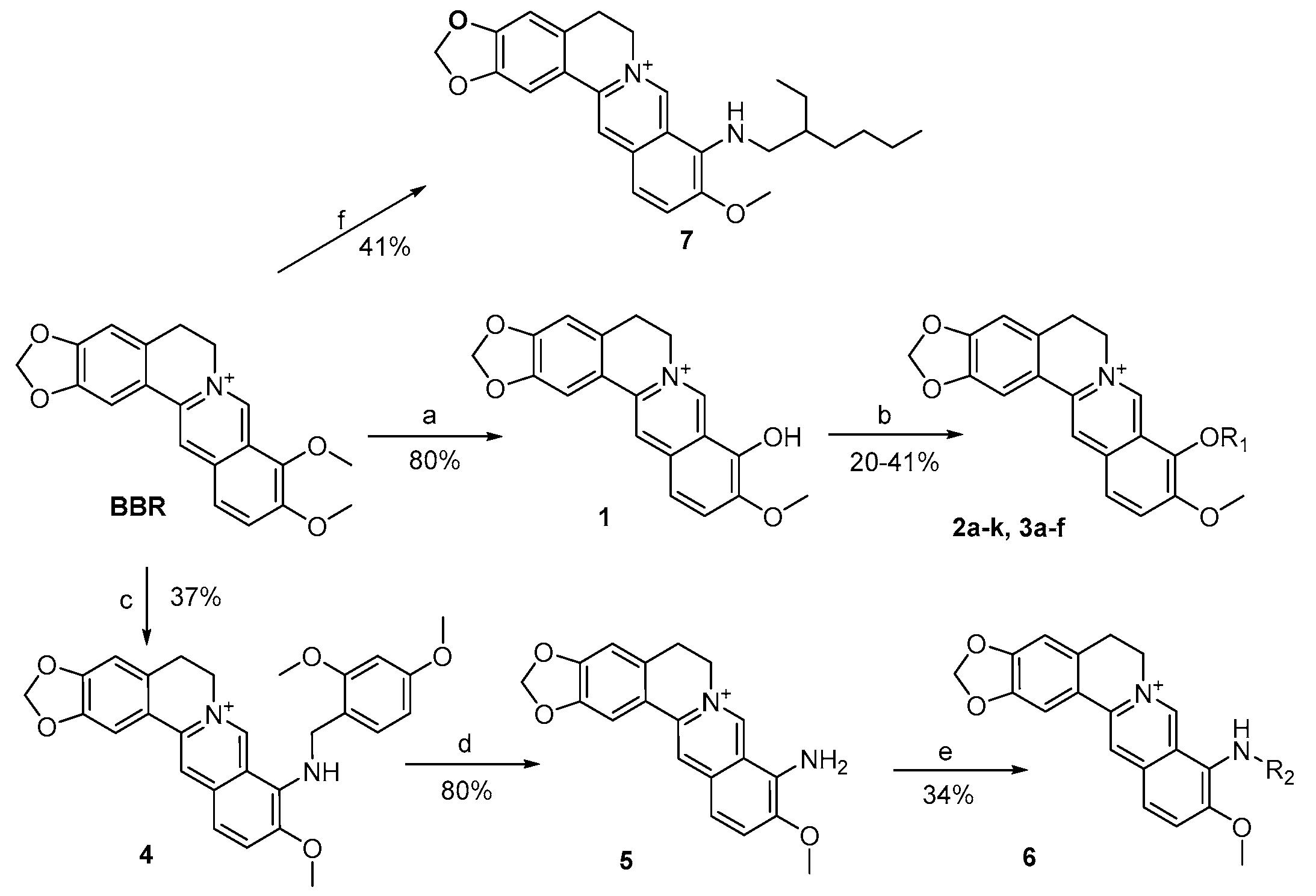
BBR (3.71 g, 10 mmol) was heated at 195–210 °C for 10–15 min under vacuum (30–40 mmHg) to afford the black oil, which was acidified with ethanol/concentrated HCl (95:5). The solvent was removed by evaporation and the residue was collected and then purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient eluent, giving the title compound 1 (2.85 g, 80%) as an orange solid.
To a stirred solution of 1 (100 mg, 0.28 mmol) in anhydrous CH3CN, triethylamine (175 µL, 1.26 mmol) was added and heated to 70 °C. Then the RCOX/RSO2Cl (1.1–1.2 eq) was added and stirred for 5–6 h. The mixture was cooled to precipitate completely, filtrated and washed with CH2Cl2 to afford target compounds 2a–k and 3a–f.
2,3-Methylenedioxy-9-((cyclopropanecarbonyl)oxy)-10-methoxy protoberberine chloride (2a). Compound 1 (100 mg, 0.28 mmol) was treated with cyclopropanecarbonyl chloride (28 µL, 0.31 mmol) according to the general procedure to give the desired product 2a as a yellow solid, yield: 34%; M.p.: 193–195 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H, CHarom), 9.08 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.98 (t, J = 6.4 Hz, 2H, CH2), 4.04 (s, 3H, OCH3), 3.22 (t, J = 6.4 Hz, 2H, CH2), 2.13 (tt, J = 7.8, 4.7 Hz, 1H, CH), 1.26–1.16 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 172.3, 151.0, 150.6, 148.3, 144.9, 138.7, 134.2, 133.5, 131.5, 127.3, 126.5, 121.8, 121.3, 121.0, 109.1, 106.2, 102.8, 57.9, 56.7, 26.8, 13.4, 10.4(2); HRMS: calcd. for C23H20NO5Cl [M − Cl]+ 390.1336, found 390.1342.
2,3-Methylenedioxy-9-((cyclopentanecarbonyl)oxy)-10-methoxy protoberberine chloride (2b). Compound 1 (100 mg, 0.28 mmol) was treated with cyclopentanecarbonyl chloride (38 µL, 0.31 mmol) according to the general procedure to give the desired product 2b as a yellow solid, yield: 32%; M.p.: 207–209 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.47–3.38 (m, 1H, CH), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.14–2.00 (m, 4H, 2 × CH2), 1.79–1.64 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 174.1, 150.9, 150.6, 148.3, 145.1, 138.7, 134.5, 133.5, 131.5, 127.2, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.9, 56.0, 43.6, 30.1(2), 26.8, 26.1(2); HRMS: calcd. for C25H24NO5Cl [M − Cl]+ 418.1649, found 418.1653.
2,3-Methylenedioxy-9-(2′-cyclopentylacetoxy)-10-methoxy protoberberine chloride (2c). Compound 1 (100 mg, 0.28 mmol) was treated with 2-cyclopentylacetyl chloride (42 µL, 0.31 mmol) according to the general procedure to give the desired product 2c as a yellow solid, yield: 38%; M.p.: 186–188 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.94 (s, 1H, CHarom), 9.06 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.95 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.88 (d, J = 7.3 Hz, 2H, CH2), 2.41–2.30 (m, 1H, CH), 1.96–1.88 (m, 2H, CH2), 1.74–1.63 (m, 2H, CH2), 1.63–1.53 (m, 2H, CH2), 1.40–1.27 (m, 2H, CH2); 13C-NMR (126 MHz, CDCl3) δ 171.5, 151.3, 151.0, 148.7, 147.3, 138.3, 136.3, 133.1, 131.0, 125.9, 125.5, 122.5, 120.2, 119.7, 108.9, 105.3, 102.4, 57.2, 55.3, 40.5, 36.7, 32.6(2), 27.8, 25.4(2); HRMS: calcd. for C26H26NO5Cl [M − Cl]+ 432.1805, found 432.1808.
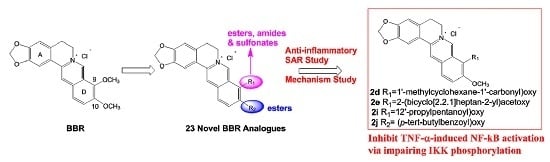
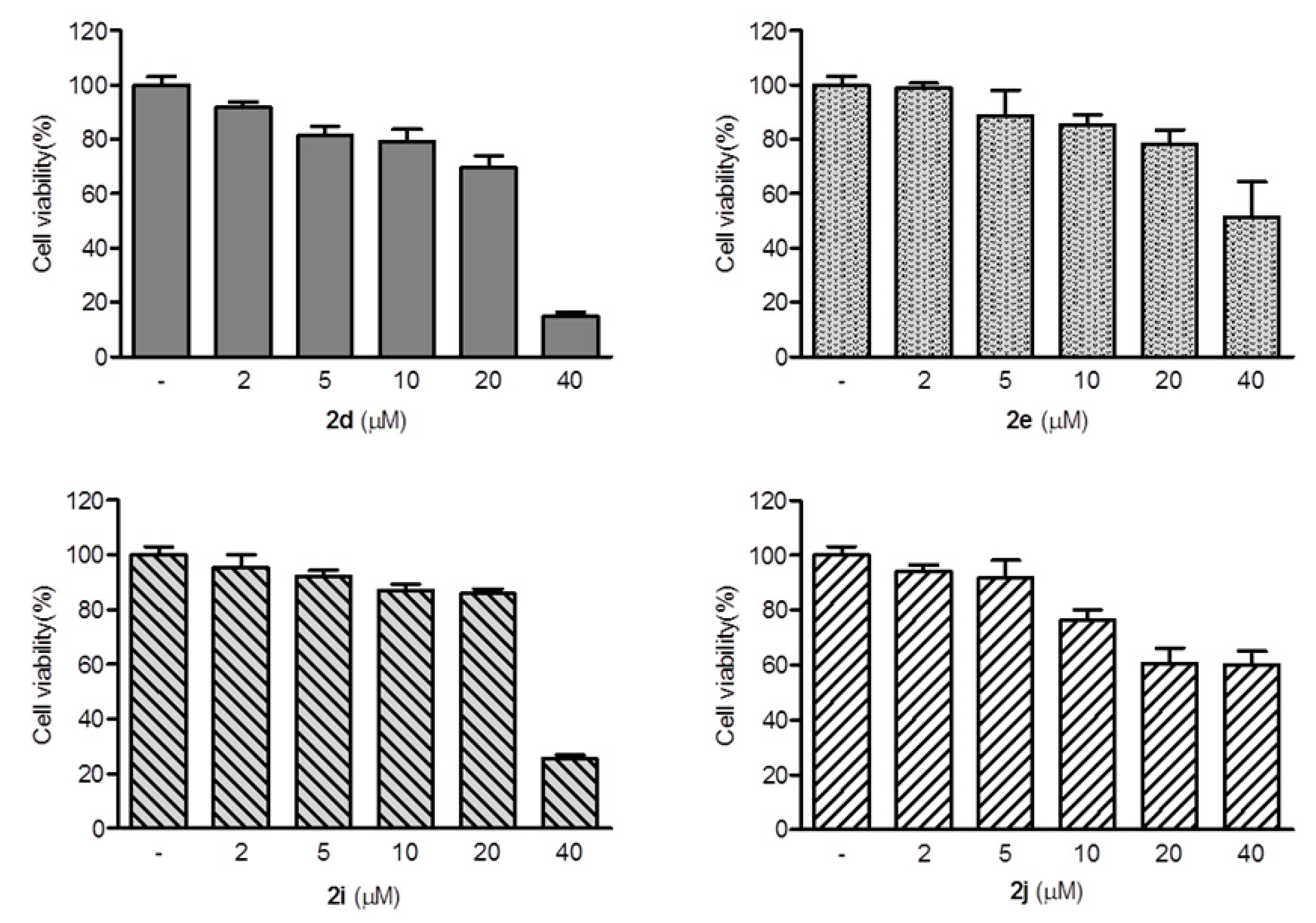
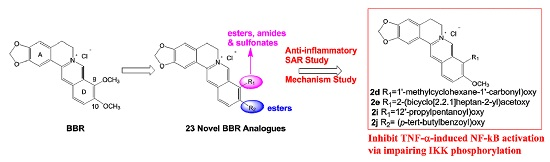
2,3-Methylenedioxy-9-((1′-methylcyclohexane-1′-carbonyl)oxy)-10-methoxy protoberberine chloride (2d). Compound 1 (100 mg, 0.28 mmol) was treated with 1-methyl-1-cyclohexanecarboxylic acid (48 mg, 0.34 mmol) according to the general procedure to give the desired product 2d as a yellow solid, yield: 39%; M.p.: 215–217 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.42 (s, 1H, CHarom), 9.09 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.2 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.21 (t, J = 6.2 Hz, 2H, CH2), 2.24–2.15 (m, 2H, CH2), 1.70–1.34 (m, 11H, 4 × CH2 and CH3); 13C-NMR (126 MHz, DMSO-d6) δ 175.1, 150.8, 150.6, 148.4, 144.4, 138.7, 134.4, 133.7, 131.6, 127.3, 126.6, 121.5, 121.4, 121.0, 109.1, 106.2, 102.8, 57.8, 56.5, 44.0, 35.6(2), 26.8, 26.3, 25.9, 23.1(2); HRMS: calcd. for C27H28NO5Cl [M − Cl]+ 446.1962, found 446.1962.
2,3-Methylenedioxy-9′-(2-(bicyclo[2.2.1]heptan-2-yl)acetoxy)-10-methoxy protoberberine chloride (2e). Compound 1 (100 mg, 0.28 mmol) was treated with 2-norbornaneacetic acid (49 µL, 0.34 mmol) according to the general procedure to give the desired product 2e as a yellow solid, yield: 24%; M.p.: 189–191 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.01–9.94 (m, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.83 (dd, J = 16.0, 7.5 Hz, 1H, CH2), 2.72 (dd, J = 16.0, 7.5 Hz, 1H, CH2), 2.29–2.24 (m, 1H, CH), 2.18–2.14 (m, 1H, CH2), 2.05–1.97 (m, 1H, CH2), 1.62–1.44 (m, 4H, 2 × CH2), 1.29–1.12 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 170.5, 151.0, 150.6, 148.3, 145.1, 138.7, 134.2, 133.5, 131.5, 127.3, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.8, 55.9, 41.2, 40.8, 38.6, 38.0, 36.9, 35.5, 30.1, 28.9, 26.8; HRMS: calcd. for C28H28NO5Cl [M − Cl]+ 458.1962, found 458.1963.
2,3-Methylenedioxy-9-((3′-chloroadamantane-1′-carbonyl)oxy)-10-methoxy protoberberine chloride (2f). Compound 1 (100 mg, 0.28 mmol) was treated with 3-chloroadamantane-1-carboxylic acid (73 mg, 0.34 mmol) according to the general procedure to give the desired product 2f as a brown solid, yield: 33%; M.p.: 206–208 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.63 (s, 1H, CHarom), 9.08 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.99 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.56 (s, 2H CH2), 2.38–2.31 (m, 2H, CH2), 2.21–2.17 (m, 4H, 2 × CH2), 2.17–2.08 (m, 4H, 2 × CH2), 1.79–1.67 (m, 2H, 2 × CH); 13C-NMR (126 MHz, DMSO-d6) δ 173.2, 150.6, 150.6, 148.4, 144.6, 138.7, 134.3, 133.6, 131.56, 127.4, 126.5, 121.5, 121.4, 121.0, 109.1, 106.2, 102.8, 69.1, 58.0, 56.2, 48.0, 46.7(2), 45.4, 37.2(2), 34.3, 31.2(2), 26.8; HRMS: calcd. for C30H29NO5Cl2 [M − Cl]+ 518.1728, found 518.1733.
2,3-Methylenedioxy-9-pivaloyloxy-10-methoxy protoberberine chloride (2g). Compound 1 (100 mg, 0.28 mmol) was treated with pivaloyl chloride (38 µL, 0.31 mmol) according to the general procedure to give the desired product 2g as a yellow solid, yield: 32%; M.p.: 205–207 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.58 (s, 1H, CHarom), 9.09 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.99 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.22 (t, J = 6.3 Hz, 2H, CH2), 1.47 (s, 9H,(CH3)3); 13C-NMR (151 MHz, CD3OD) δ 175.7, 150.9, 150.9, 148.5, 143.4, 138.8, 134.7, 133.7, 130.6, 126.4, 125.5, 121.7, 120.7, 120.3, 107.9, 105.2, 102.3, 56.3, 56.1, 39.3, 26.6, 26.2(3); HRMS: calcd. for C24H24NO5Cl [M − Cl]+ 406.1649, found 406.1653.
2,3-Methylenedioxy-9-((3′,3′-dimethylbutanoyl)oxy)-10-methoxy protoberberine chloride (2h). Compound 1 (100 mg, 0.28 mmol) was treated with 3,3-dimethylbutyryl chloride (43 µL, 0.31 mmol) according to the general procedure to give the desired product 2h as a yellow solid, yield: 27%; M.p.: 212–214 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.96 (s, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.04 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.75 (s, 2H, CH2), 1.18 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 169.5, 151.0, 150.6, 148.3, 145.1, 138.7, 134.1, 133.6, 131.5, 127.3, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.7, 55.9, 47.6, 31.4, 30.0(3), 26.8; HRMS: calcd. for C25H26NO5Cl [M − Cl]+ 420.1805, found 420.1813.
2,3-Methylenedioxy-9-((2′-propylpentanoyl)oxy)-10-methoxy protoberberine chloride (2i). Compound 1 (100 mg, 0.28 mmol) was treated with 2-propylpentanoyl chloride (58 µL, 0.34 mmol) according to the general procedure to give the desired product 2i as a yellow solid, yield: 24%; M.p.: 199–201 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.68 (s, 1H, CHarom), 9.10 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.23 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.95 (t, J = 6.3 Hz, 2H, CH2), 4.02 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.95 (p, J = 6.6 Hz, 1H, CH), 1.88–1.79 (m, 2H, CH2), 1.72–1.63 (m, 2H, CH2), 1.56–1.42 (m, 4H, 2 × CH2), 0.99 (t, J = 7.3 Hz, 6H, 2 × CH3); 13C-NMR (126 MHz, DMSO-d6) δ 173.4, 150.9, 150.6, 148.4, 144.6, 138.7, 134.1, 133.6, 131.5, 127.3, 126.5, 121.6, 121.3, 121.0, 109.1, 106.2, 102.8, 57.8, 56.2, 44.4, 33.6(2), 26.8, 20.3(2), 14.7(2); HRMS: calcd. for C27H30NO5Cl [M − Cl]+ 448.2118, found 448.2128.
2,3-Methylenedioxy-9-((p-tert-butylbenzoyl)oxy)-10-methoxy protoberberine chloride (2j). Compound 1 (100 mg, 0.28 mmol) was treated with 4-tert-butylbenzoyl chloride (61 µL, 0.31 mmol) according to the general procedure to give the desired product 2j as a brown solid, yield: 28%; M.p.: 205–207 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.00 (s, 1H, CHarom), 9.11 (s, 1H, CHarom), 8.35 (d, J = 9.2 Hz, 1H, , CHarom), 8.27 (d, J = 9.2 Hz, 1H, CHarom), 8.20 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.85 (s, 1H, CHarom), 7.72 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.10 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.91 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.20 (t, J = 6.3 Hz, 2H, CH2), 1.38 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 164.0, 158.6, 151.1, 150.6, 148.4, 145.1, 138.8, 134.3, 133.6, 131.5(2), 131.1, 127.5, 126.6(2), 126.5, 125.9, 122.0, 121.3, 121.0, 109.1, 106.2, 102.8, 57.9, 55.8, 35.8, 31.5(3), 26.8; HRMS: calcd. for C30H28NO5Cl [M − Cl]+ 482.1962, found 482.1962.
2,3-Methylenedioxy-9-((3′-tert-butyl-1′-methyl-1H-pyrazole-5′-carbonyl)oxy)-10-methoxy protoberberine chloride (2k). Compound 1 (100 mg, 0.28 mmol) was treated with 5-tert-butyl-2-methylpyrazole-3-carbonyl chloride (59 µL, 0.34 mmol) according to the general procedure to give the desired product 2k as a yellow solid, yield: 42%; M.p.: 207–209 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.03 (s, 1H, CHarom), 9.12 (s, 1H, CHarom), 8.36 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.15 (s, 1H, CHpyrazole), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.92 (t, J = 6.4 Hz, 2H, CH2), 4.13 (s, 3H, NCH3), 4.06 (s, 3H, OCH3), 3.22 (t, J = 6.4 Hz, 2H, CH2), 1.34 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 160.5, 157.2, 151.1, 150.7, 148.4, 145.1, 138.9, 133.6, 133.1, 131.5, 131.2, 127.9, 126.5, 121.8, 121.3, 121.0, 109.6, 109.1, 106.2, 102.8, 58.0, 55.9, 40.0, 32.5, 31.0(3), 26.8; HRMS: calcd. for C28H28N3O5Cl [M − Cl]+ 486.2023, found 486.2027.
2,3-Methylenedioxy-9-((p-nitrophenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3a). Compound 1 (100 mg, 0.28 mmol) was treated with 4-nitrobenzenesulfonyl chloride (75 mg, 0.34 mmol) according to the general procedure to give the desired product 3a as an orange solid, yield: 31%; M.p.: 146–148 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.69 (s, 1H, CHarom), 9.14 (s, 1H, CHarom), 8.52 (d, J = 8.6 Hz, 2H, 2 × CHarom), 8.30 (d, J = 9.3 Hz, 1H, CHarom), 8.27 (d, J = 8.6 Hz, 2H, 2 × CHarom), 8.22 (d, J = 9.3 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.69 (s, 3H, OCH3), 3.22 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.8, 151.8, 150.7, 148.2, 144.2, 140.4, 139.3, 133.9, 131.6, 131.1, 130.8(2), 129.3, 126.5, 125.4(2), 122.1, 121.5, 120.6, 108.9, 106.1, 102.7, 57.3, 56.1, 26.6; HRMS: calcd. for C25H19N2O8SCl [M − Cl]+ 507.0856, found 507.0860.
2,3-Methylenedioxy-9-((p-trifluoromethylphenyl sulfonyl)oxy)-10-methoxy protoberberine chloride (3b). Compound 1 (100 mg, 0.28 mmol) was treated with 4-(trifluoromethyl)benzene-1-sulfonyl chloride (83 mg, 0.34 mmol) according to the general procedure to give the desired product 3b as a yellow solid, yield: 41%; M.p.: 190–191 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H, CHarom), 9.17 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.24–8.20 (m, 4H, 4 × CHarom), 8.15 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.84 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.23 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 152.0, 150.9, 148.4, 144.5, 139.5, 139.2, 135.3, 134.1, 131.7, 131.3, 130.4(2), 129.5, 127.6(2), 126.6, 123.8, 122.3, 121.7, 120.8, 109.1, 106.3, 102.9, 57.4, 56.3, 26.8; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0884.
2,3-Methylenedioxy-9-((m-trifluoromethylphenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3c). Compound 1 (100 mg, 0.28 mmol) was treated with 3-(trifluoromethyl)benzenesulfonyl chloride (55 µL, 0.34 mmol) according to the general procedure to give the desired product 3c as an orange solid, yield: 37%; M.p.: 184–186 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.71 (s, 1H, CHarom), 9.18 (s, 1H, CHarom), 8.31–8.26 (m, 2H, 2 × CHarom), 8.17 (d, J = 9.4 Hz, 1H, CHarom), 8.13 (t, J = 7.7 Hz, 1H, CHarom), 8.07–8.03 (m, 1H, CHarom), 7.99–7.94 (m, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.04 (t, J = 6.4 Hz, 2H, CH2), 3.46 (s, 3H, OCH3), 3.24 (t, J = 6.4 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.5, 150.7, 148.2, 144.2, 139.3, 136.4, 134.0, 133.8, 133.9, 133.0, 131.6, 131.4, 129.7, 129.3, 127.8, 126.3, 122.9, 122.4, 121.6, 120.6, 108.9, 106.1, 102.7, 57.2, 56.2, 26.6; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0883.
2,3-Methylenedioxy-9-((o-trifluoromethylphenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3d). Compound 1 (100 mg, 0.28 mmol) was treated with 2-(trifluoromethyl)benzenesulfonyl chloride (52 µL, 0.34 mmol) according to the general procedure to give the desired product 3d as an orange solid, yield: 38%; M.p.: 183–185 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H, CHarom), 9.17 (s, 1H, CHarom), 8.36–8.29 (m, 3H, 3 × CHarom), 8.24 (d, J = 1.8 Hz, 1H, CHarom), 8.23 (d, J = 9.4 Hz, 1H, CHarom), 8.01 (t, J = 7.9 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.66 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.8, 150.7, 148.2, 144.4, 139.2, 136.3, 134.0, 133.2, 132.8, 132.0, 131.5, 131.1, 130.7, 129.3, 126.3, 125.6, 123.6, 122.2, 121.5, 120.6, 108.9, 106.1, 102.7, 57.3, 56.1, 26.6; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0880.
2,3-Methylenedioxy-9-((D-(+)-10′-camphorsulfonyl)oxy)-10-methoxy protoberberine chloride (3e). Compound 1 (100 mg, 0.28 mmol) was treated with D(+)-10-camphorsulfonyl chloride (85 mg, 0.34mmol) according to the general procedure to give the desired product 3e as a yellow solid, yield: 20%; M.p.: 193–195 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.77 (s, 1H, CHarom), 9.14 (s, 1H, CHarom), 8.37 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.01 (t, J = 6.4 Hz, 2H, CH2), 4.20 (d, J = 15.0 Hz, 1H ,COCH2), 4.13 (s, 3H, OCH2), 3.98 (d, J = 15.0 Hz, 1H, COCH2), 3.23 (t, J = 6.4 Hz, 2H, CH2), 2.48–2.40 (m, 1H, CH2), 2.39–2.30 (m, 1H, CH2), 2.14 (t, J = 4.5 Hz, 1H, CH), 2.07–1.98 (m, 2H, CH2), 1.73–1.64 (m, 1H, CH2), 1.53–1.44 (m, 1H, CH2), 1.10 (s, 3H, CH3), 0.91 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6) δ 214.2, 152.3, 150.8, 148.4, 144.8, 139.1, 134.0, 132.1, 131.7, 128.6, 127.1, 122.5, 121.5, 120.8, 109.1, 106.2, 102.8, 58.5, 58.1, 56.4, 50.6, 48.8, 42.9, 42.6, 26.9, 26.8, 25.9, 20.0, 19.9; HRMS: calcd. for C29H30NO7SCl [M − Cl]+ 536.1737, found 536.1745.
2,3-Methylenedioxy-9-((L-(−)-10′-camphorsulfonyl)oxy)-10-methoxy protoberberine chloride (3f). Compound 1 (100 mg, 0.28 mmol) was treated with L(−)-10-camphorsulfonyl chloride (85 mg, 0.34 mmol) according to the general procedure to give the desired product 3f as a yellow solid, yield: 21%; M.p.: 201–203 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.76 (s, 1H, CHarom), 9.13 (s, 1H, CHarom), 8.37 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.01 (t, J = 6.4 Hz, 2H, CH2), 4.20 (d, J = 15.0 Hz, 1H, COCH2), 3.97 (d, J = 15.0 Hz, 1H, COCH2), 3.23 (t, J = 6.4 Hz, 2H, CH2), 2.47–2.40 (m, 1H, CH2), 2.38–2.31 (m, 1H, CH2), 2.14 (t, J = 4.5 Hz, 1H, CH), 2.06–1.98 (m, 2H, CH2), 1.73–1.64 (m, 1H, CH2), 1.53–1.46 (m, 1H, CH2), 1.10 (s, 3H), 0.91 (s, 3H); 13C-NMR (126 MHz, DMSO-d6) δ 214.2, 152.3, 150.8, 148.4, 144.8, 139.3, 134.0, 132.1, 131.7, 128.6, 127.9, 122.5, 121.5, 120.8, 109.1, 106.2, 102.8, 58.5, 58.1, 56.4, 50.6, 48.8, 42.9, 42.6, 26.9, 26.8, 25.9, 20.0, 19.9; HRMS: calcd. for C29H30NO7SCl [M − Cl]+ 536.1737, found 536.1742.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}