Isolation and Characterization of Aphidicolin Derivatives from Tolypocladium inflatum

Abstract
:1. Introduction
2. Results and Discussion
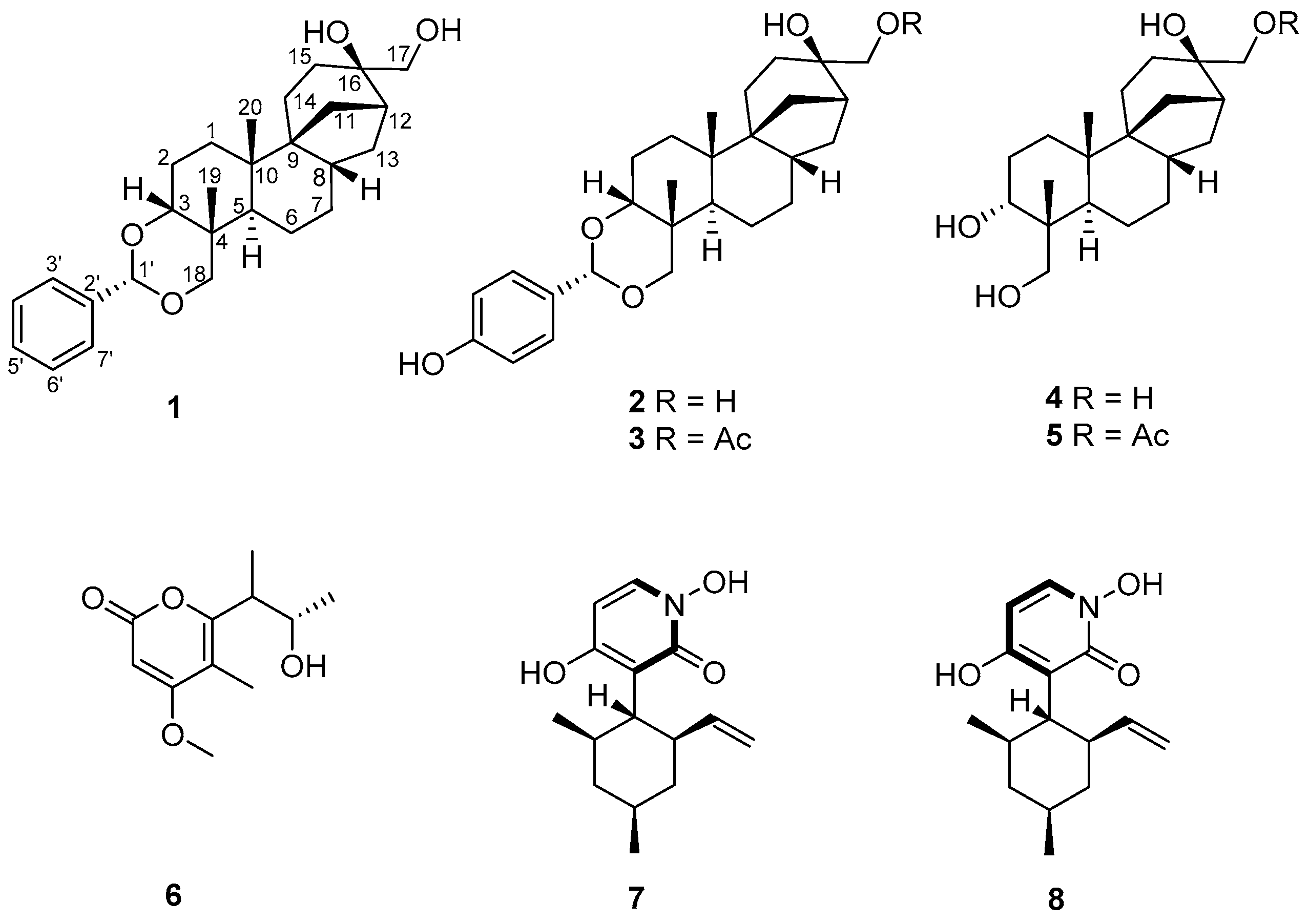
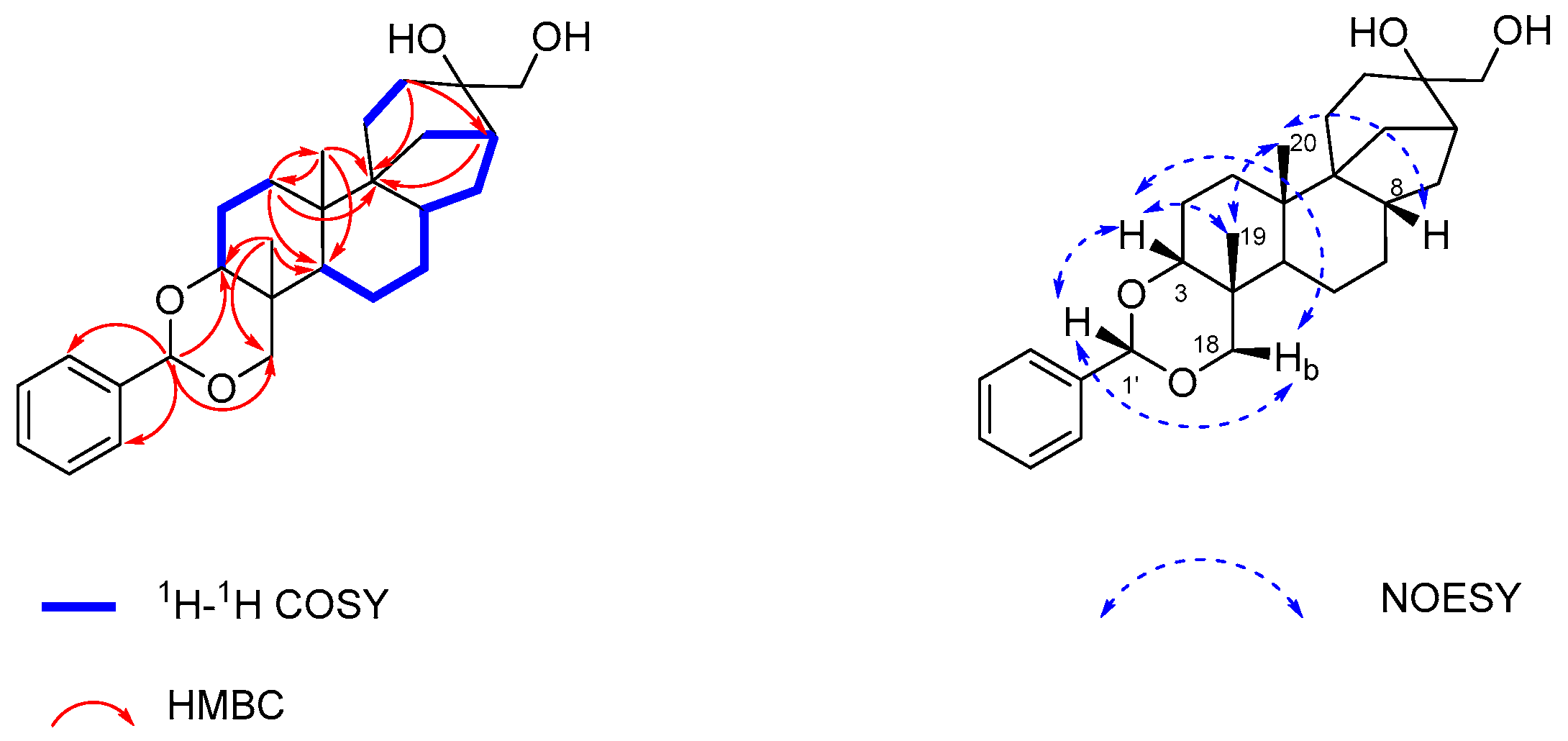
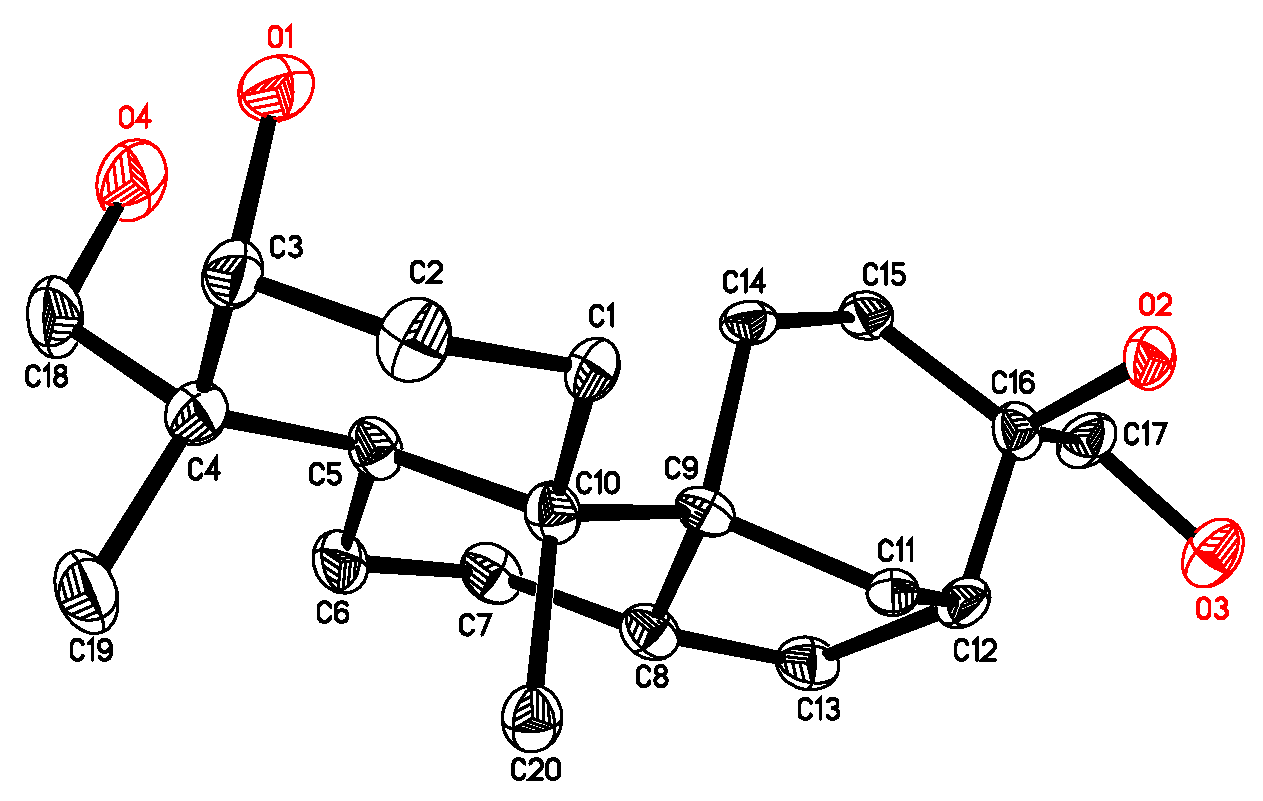
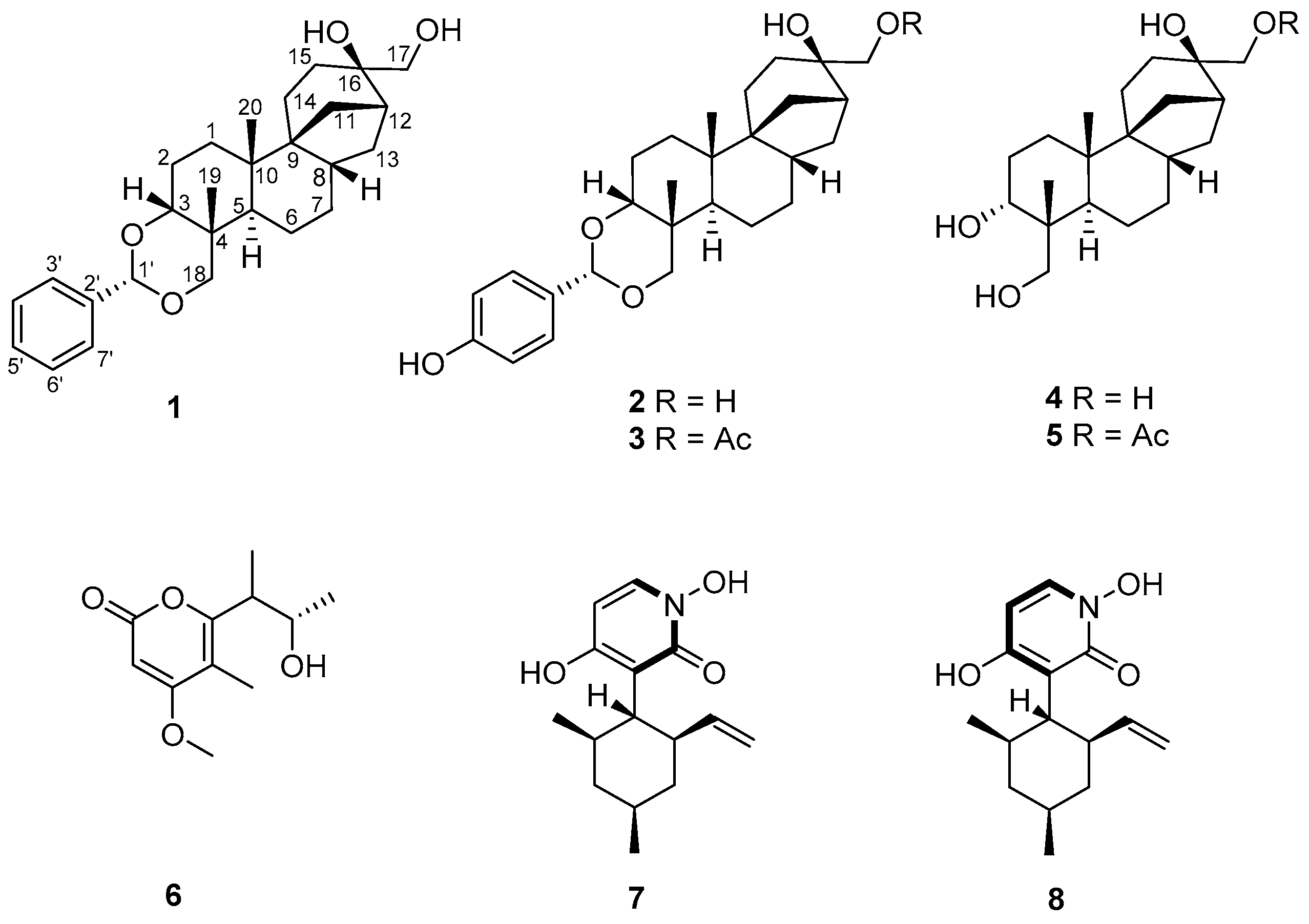
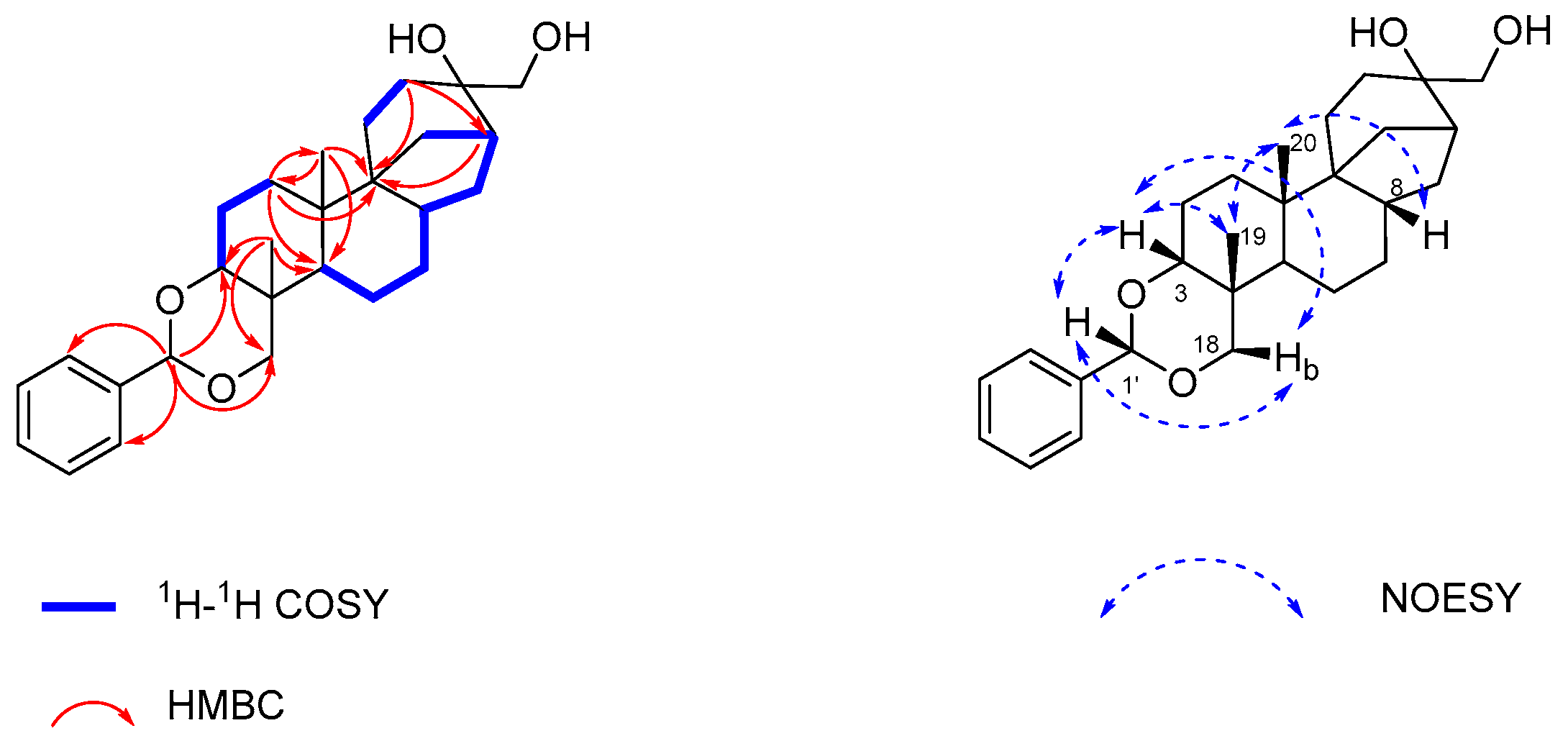
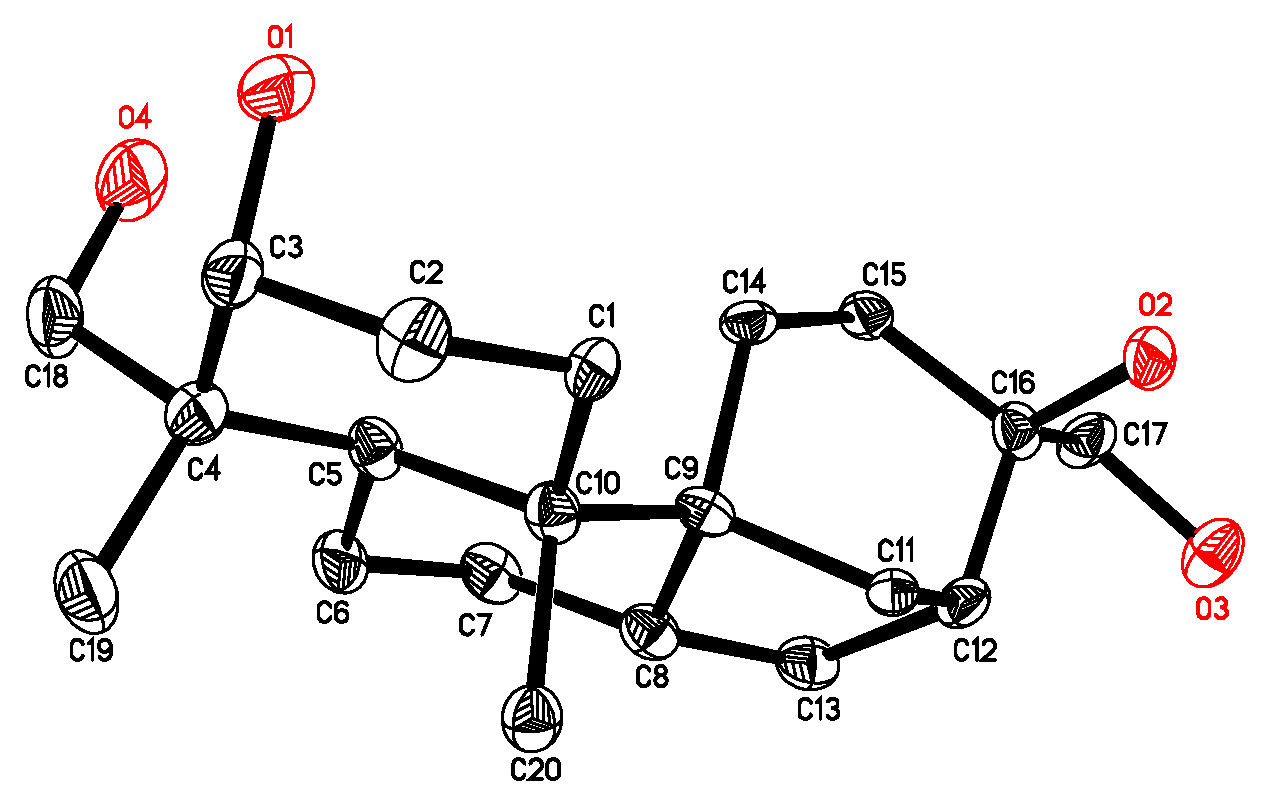
2.1. Isolation and Structure Elucidation of Compound 1
2.2. Bioactivities
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data
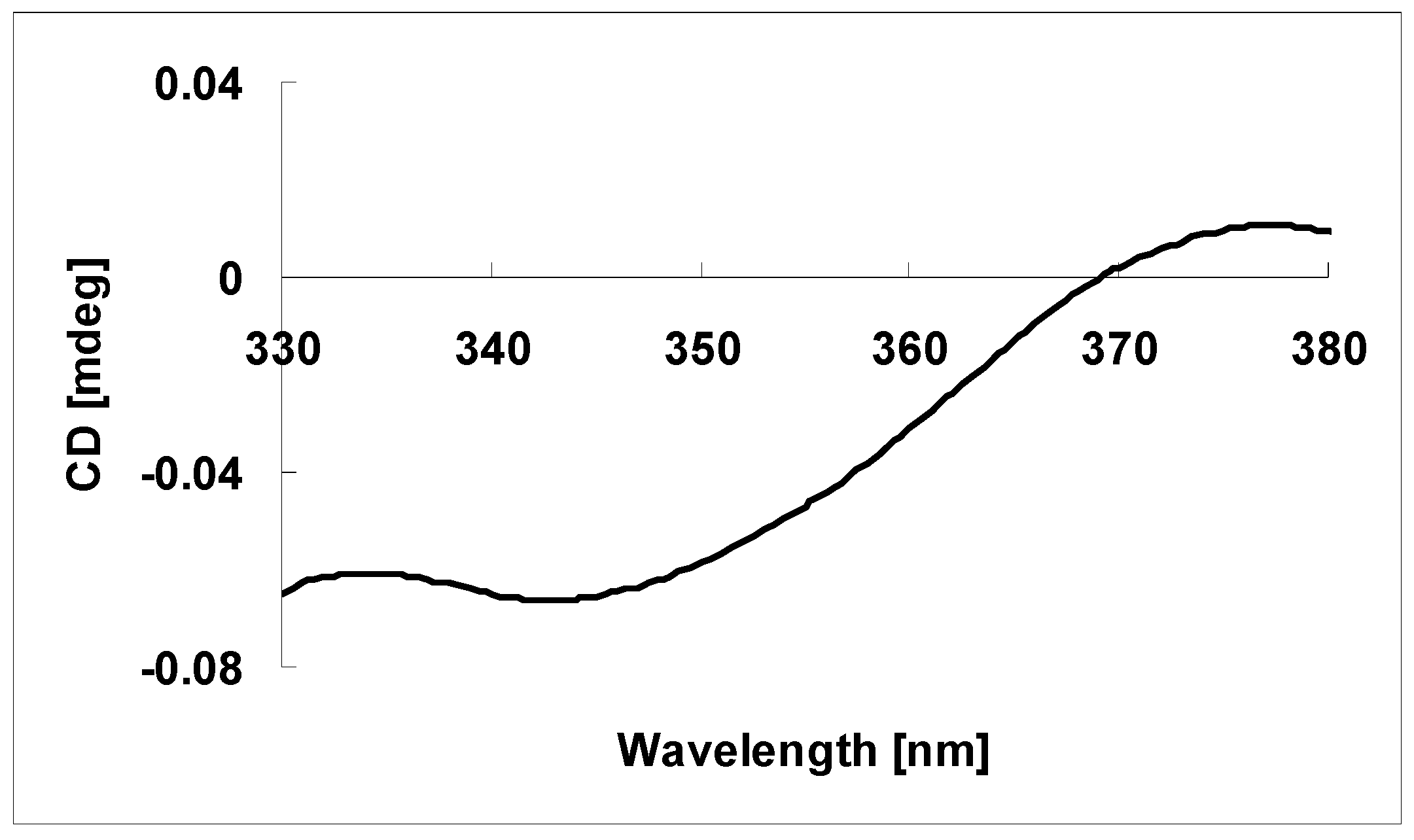
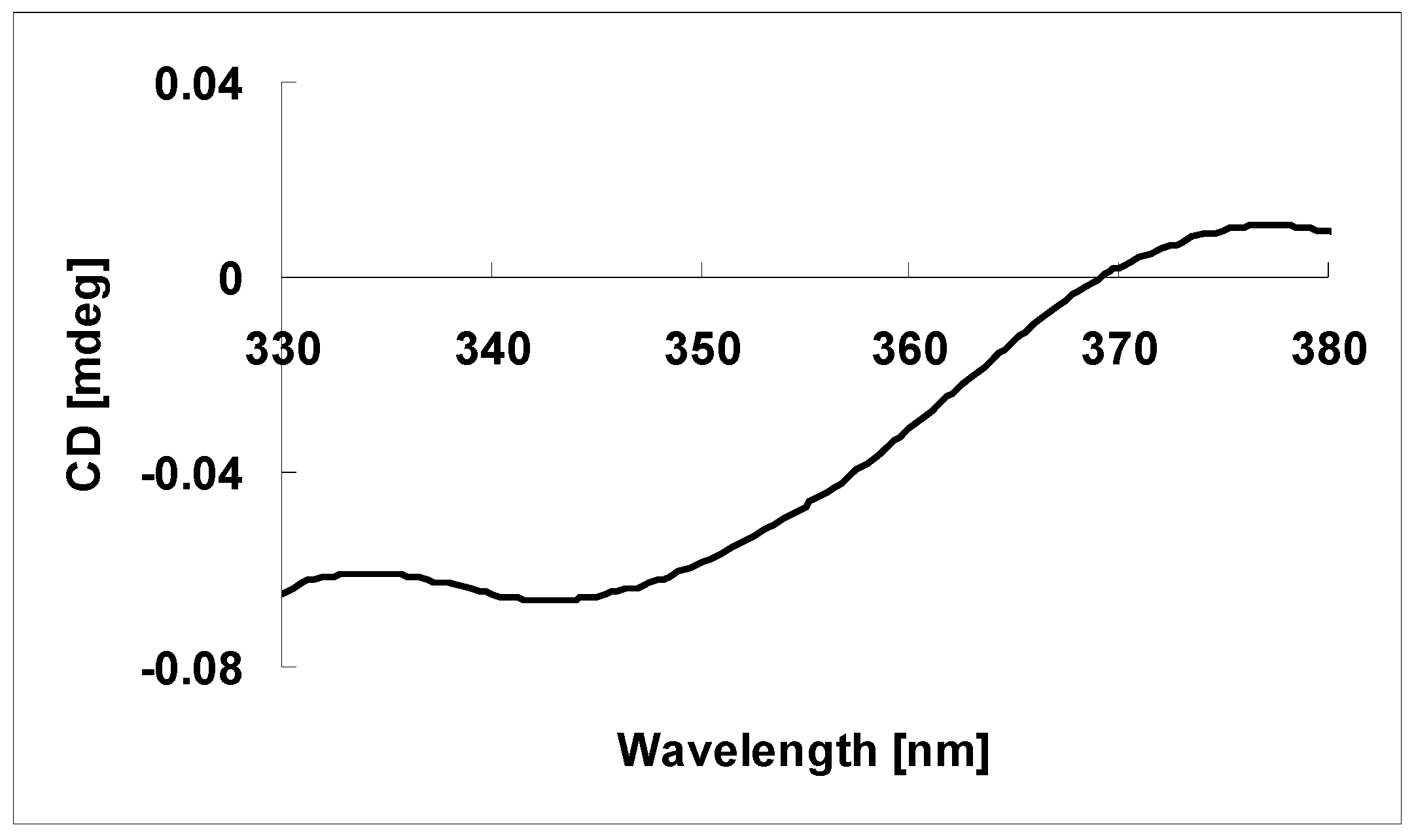
3.5. Absolute Configuration of the Tertiary Alcohol in 1 [11,12]
3.6. Cytotoxicity Assay [16,17]
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Du, L.; Robles, A.J.; King, J.B.; Powell, D.R.; Miller, A.N.; Mooberry, S.L.; Cichewicz, R.H. Crowdsourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. Int. Ed. 2014, 53, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Gloer, J.B. Antiinsectan natural products from fungal sclerotia. Acc. Chem. Res. 2002, 28, 343–350. [Google Scholar] [CrossRef]
- Meinwald, J.; Eisner, T. Chemical ecology in retrospect and prospect. Proc. Natl. Acad. Sci. USA 2008, 105, 4539–4540. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ren, F.; Che, Y.; Liu, G.; Liu, L. New bergamotane sesquiterpenoids from the plant endophytic fungus Paraconiothyrium brasiliense. Molecules 2014, 20, 14611–14620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Che, Y.; Liu, X. Epicoccins A–D, epipolythiodioxopiperazines from a Cordyceps-colonizing isolate of Epicoccum nigrum. J. Nat. Prod. 2007, 70, 1522–1525. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, X.; Cai, X.; Yu, X.; Liu, X.; Cao, Y.; Che, Y. Isolation and characterization of aphidicolin and chlamydosporol derivatives from Tolypocladium inflatum. J. Nat. Prod. 2011, 74, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.J.; Wood, J.L.; Furst, G.T.; Smith, A.B.I. Aphidicolin synthetic studies, 2.2D NMR analysis of aphidicolin and its degradation products 3α,18-dihydroxy-17-noraphidicolan-16-one and 3α,18-isopropylidenedioxy-17-noraphidicolan-16-one. J. Nat. Prod. 1990, 53, 735–739. [Google Scholar] [CrossRef]
- Hanson, J.R.; Reese, P.B.; Takahashi, J.A.; Wilson, M.R. Biotransformation of some stemodane diterpenoids by Cephalosporium aphidicola. Phytochemistry 1994, 36, 1391–1393. [Google Scholar] [CrossRef]
- Andolfi, A.; Boari, A.; Evidente, M.; Cimmino, A.; Vurro, M.; Ash, G.; Evidente, A. Gulypyrones A and B and Phomentrioloxins B and C produced by Diaporthe gulyae, a potential mycoherbicide for saffron thistle (Carthamus lanatus). J. Nat. Prod. 2015, 78, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Teshima, Y.; Shinya, K.; Shimazu, A.; Furihata, K.; Chul, H.S.; Hayakawa, Y.; Nagai, K.; Seto, H. Isolation and structural elucidation of pyridoxatin, a free radical scavenger of microbial origin. J. Antibiot. 1991, 44, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Frelek, J.; Szczepek, W.J. [Rh2(OCOCF3)4] as an auxiliary chromophore in chiroptical studies on steroidal alcohols. Tetrahedron Asymmetry 1999, 10, 1507–1520. [Google Scholar] [CrossRef]
- Gerards, M.; Snatzke, G. Circular dichroism, XCIII determination of the absolute configuration of alcohols, olefins, epoxides, and ethers from the CD of their “in situ” complexes with [Rh2(O2CCF3)4]. Tetrahedron Asymmetry 1990, 1, 221–236. [Google Scholar] [CrossRef]
- Crystallographic data for 4 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 816540). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
- Sheldrick, G.M. SHELXL-97, Program for X-ray Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1999. [Google Scholar]
- Zhang, N.; Chen, Y.; Jiang, R.; Li, E.; Chen, X.; Xi, Z.; Guo, Y.; Liu, X.; Zhou, Y.; Che, Y.; et al. PARP and RIP 1 are required for autophagy induced by 11′-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011, 7, 1–15. [Google Scholar] [CrossRef]
- Jeong, C.H.; Bode, A.M.; Pugliese, A.; Cho, Y.Y.; Kim, H.G.; Shim, J.H.; Jeon, Y.J.; Li, H.; Jiang, H.; Dong, Z.; et al. [6]-Gingerol suppresses colon cancer growth by targeting leukotriene A4 hydrolase. Cancer Res. 2009, 69, 5584–5591. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos. | δC a, Mult. | δH b (J in Hz) | HMBC b |
|---|---|---|---|
| 1a | 27.9, CH2 | 2.20, td (11.0, 3.0) | 9, 20 |
| 1b | 0.96, dt (11.0, 2.5) | 3, 5 | |
| 2a | 27.8, CH2 | 1.67–1.72 c | 4, 10 |
| 2b | 1.30–1.36 c | ||
| 3 | 81.8, CH | 3.66, t (3.0) | 1, 5, 18, 19, 1′ |
| 4 | 35.7, qC | ||
| 5 | 34.6, CH | 2.82, dd (10.5, 2.5) | 1, 7, 9, 19, 20 |
| 6a | 23.2, CH2 | 1.63, m | 8 |
| 6b | 1.28–1.36 c | ||
| 7a | 25.2, CH2 | 1.92–2.02 c | 9 |
| 7b | 1.68–1.70 c | 5, 9 | |
| 8 | 41.1, CH | 1.96–2.08 c | 6, 10, 12 |
| 9 | 49.9, qC | ||
| 10 | 40.6, qC | ||
| 11a | 33.4, CH2 | 1.89–1.91 c | 8, 10, 13, 14, 16 |
| 11b | 1.25–1.37 c | 10, 14, 16 | |
| 12 | 42.3, CH | 2.13, t (5.7) | 8, 9, 15, 17 |
| 13a | 31.9, CH2 | 1.70–1.72 c | 9, 16 |
| 13b | 0.99, dd (11.5, 7.0) | 11, 16 | |
| 14a | 25.5, CH2 | 1.97–2.12 c | 8, 10, 11, 16 |
| 14b | 1.71–1.76 c | 8, 10, 16 | |
| 15a | 29.3, CH2 | 1.48, dt (12.0, 2.7) | 17 |
| 15b | 1.30, td (12.0, 5.0) | 9, 12 | |
| 16 | 74.3, qC | ||
| 17a | 68.3, CH2 | 3.37, dd (9.0, 5.0) | 12, 15 |
| 17b | 3.27, dd (9.0, 5.0) | 12, 15 | |
| 18a | 76.2, CH2 | 4.02, d (10.0) | 3, 1′ |
| 18b | 3.16, d (10.0) | 3, 5, 19, 1′ | |
| 19 | 17.2, CH3 | 0.76, s | 3, 5, 18 |
| 20 | 16.0, CH3 | 1.05, s | 1, 5, 9 |
| 1′ | 102.4, CH | 5.51, s | 3, 18, 3′, 7′ |
| 2′ | 140.6, qC | ||
| 3′ | 127.3, CH | 7.51, d (6.5) | 1′, 5′, 7′ |
| 4′ | 128.8, CH | 7.35, d (6.5) | 2′, 6′ |
| 5′ | 129.3, CH | 7.36, m | |
| 6′ | 128.8, CH | 7.35, d (6.5) | 2′, 4′ |
| 7′ | 127.3, CH | 7.51, d (6.5) | 1′, 3′, 5′ |
| OH-16 | 3.07, s | ||
| OH-17 | 3.50, t (5.0) |
| Compound | IC50 (µM) | ||||
|---|---|---|---|---|---|
| A549 a | CNE1-LMP1 b | A375 c | MCF-7 d | HaCaT e | |
| 1 | 68.7 | 35.4 | 39.8 | 35.2 | 42.6 |
| 4 | 16.2 | 3.12 | 37.3 | 42.3 | 10.4 |
| 5 | 5.20 | 4.00 | 40.5 | 66.2 | 8.00 |
| 6 | >100 | >100 | >100 | >100 | >100 |
| 7 and 8 | 0.44 | 0.20 | 0.10 | 0.17 | >47.5 |
| paclitaxel | 3.0 × 10−2 | 4.2 × 10−3 | 8.9 × 10−3 | 1.4 × 10−2 | 0.024 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Niu, S.; Ding, Z.; Wang, R.; Dai, Q.; Wei, W.; Luo, R.; Liu, L. Isolation and Characterization of Aphidicolin Derivatives from Tolypocladium inflatum. Molecules 2017, 22, 1168. https://doi.org/10.3390/molecules22071168
Lin J, Niu S, Ding Z, Wang R, Dai Q, Wei W, Luo R, Liu L. Isolation and Characterization of Aphidicolin Derivatives from Tolypocladium inflatum. Molecules. 2017; 22(7):1168. https://doi.org/10.3390/molecules22071168
Chicago/Turabian StyleLin, Jie, Shubin Niu, Zhengfeng Ding, Renlei Wang, Qun Dai, Wei Wei, Rongrong Luo, and Ling Liu. 2017. "Isolation and Characterization of Aphidicolin Derivatives from Tolypocladium inflatum" Molecules 22, no. 7: 1168. https://doi.org/10.3390/molecules22071168
APA StyleLin, J., Niu, S., Ding, Z., Wang, R., Dai, Q., Wei, W., Luo, R., & Liu, L. (2017). Isolation and Characterization of Aphidicolin Derivatives from Tolypocladium inflatum. Molecules, 22(7), 1168. https://doi.org/10.3390/molecules22071168