2. Results and Discussion
Chromatographic separation of the ethyl acetate extract of
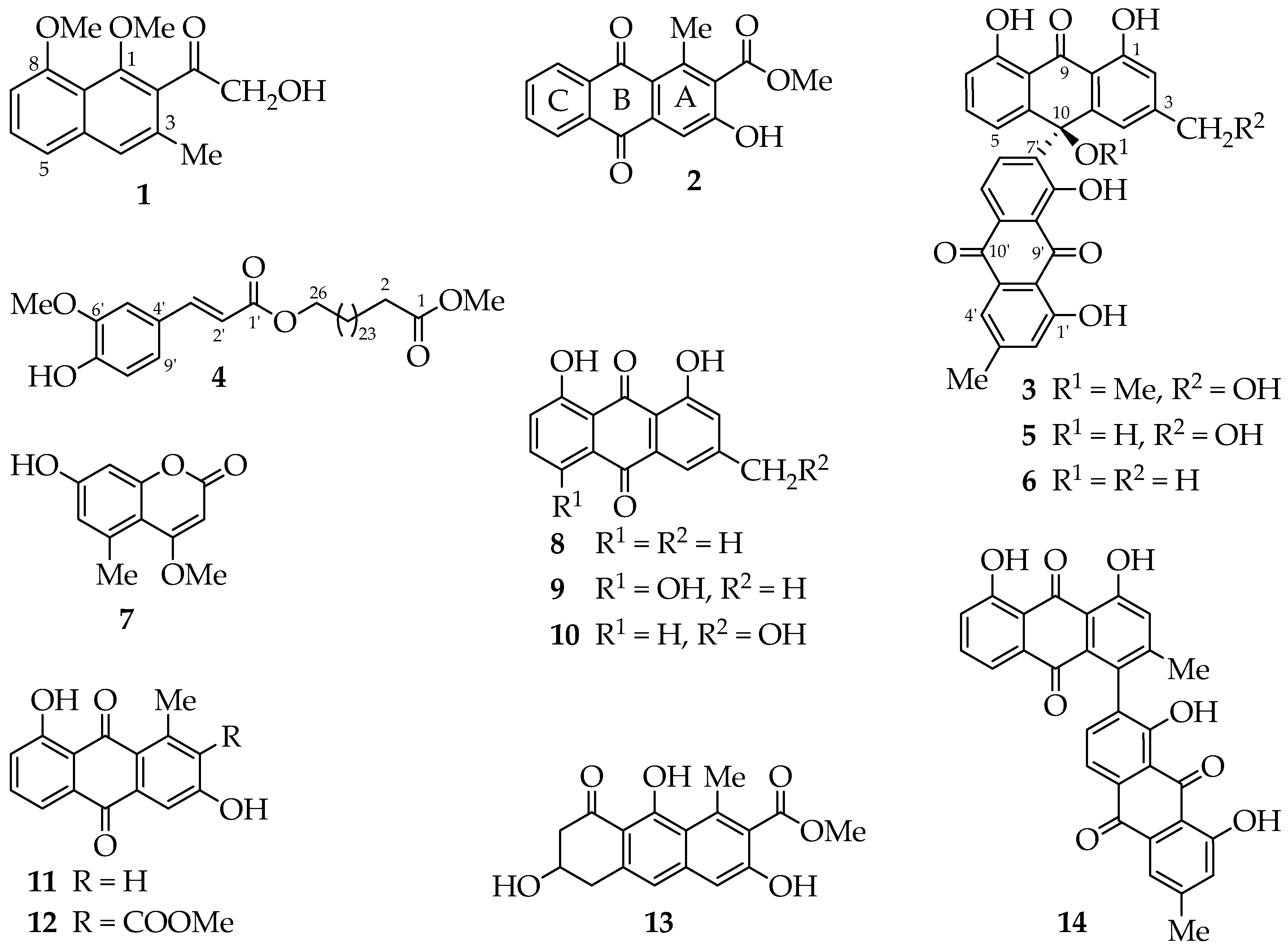
A. megalacantha roots furnished fourteen compounds (
Figure 1). All spectroscopic data, including UV, IR and NMR data, were in good agreement with the reported data for the known compounds chrysalodin (
5) [
14], 10-(chrysophanol-7′-yl)-10-hydroxychrysophanol-9-anthrone(
6) [
9,
15], 7-hydroxy-4-methoxy-5-methylcoumarin (
7) [
16], chrysophanol (
8) [
17], helminthosporin (
9) [
18], aloeemodin (
10) [
19], aloesaponarin II (
11) [
20], aloesaponarin I (
12) [
21], aloesaponol I (
13) [
20,
21] and asphodelin (
14) [
22]. It is worth to point out that this is the first report of 7-hydroxy-4-methoxy-5-methylcoumarin and its kind from the genus and most probably from the family of Asphodelaceae, having been previously reported from stem bark of
Toona ciliate [
16].
Compound
1 was isolated as colourless amorphous solid and the molecular formula C
15H
16O
4, deduced from its HR-ESI-MS ([M + Na]
+ m/
z 283.0916), indicated eight degrees of unsaturation. The UV (λ
max 252, 276 nm) and IR (ν
max 1701, 1618, 1571, 1346 cm
−1) spectra revealed absorptions for conjugated ketone and aromatic moieties. The
13C-NMR spectrum (
Table 1) revealed ten
sp2 hybridized carbon atoms of a naphthalene skeleton. The presence of a hydroxylmethylketone (δ
H 4.55, δ
C 70.5; δ
C 207.5), a methyl (δ
H 2.30, δ
C 19.0) and two methoxy (δ
H 3.73, δ
C 64.1; δ
H 3.99, δ
C 56.3) substituents were also evident. The
1H-NMR spectrum (
Table 1) showed three mutually
ortho/
meta-coupled aromatic protons at δ
H 6.97 (dd,
J = 7.6, 1.2 Hz, 1H), 7.44 (t,
J = 7.8 Hz, 1H) and 7.40 (dd,
J = 7.8, 1.4 Hz, 1H), corresponding to H-5, H-6, and H-7, respectively.
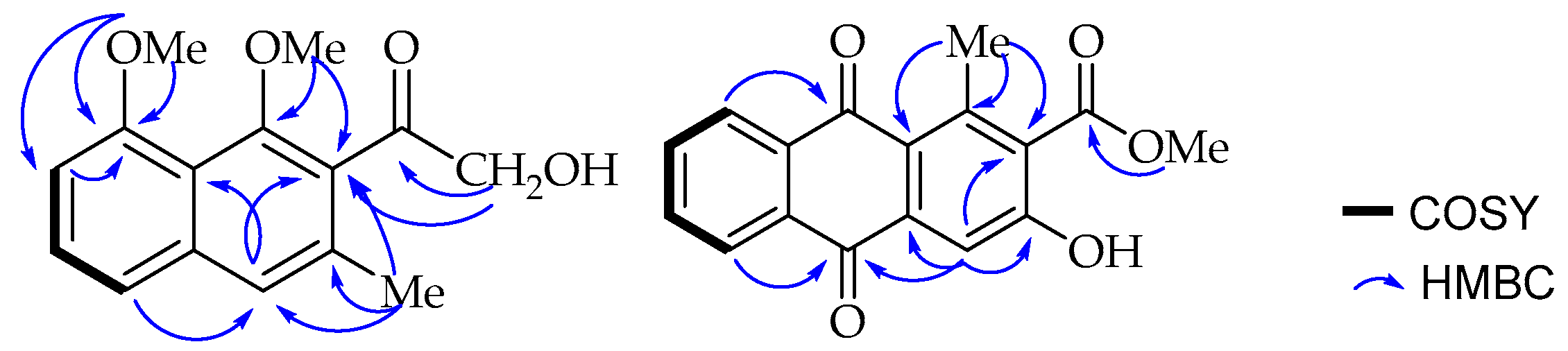
One of the methoxy groups (δ
H 3.99, δ
C 56.3) is present at C-8 (δ
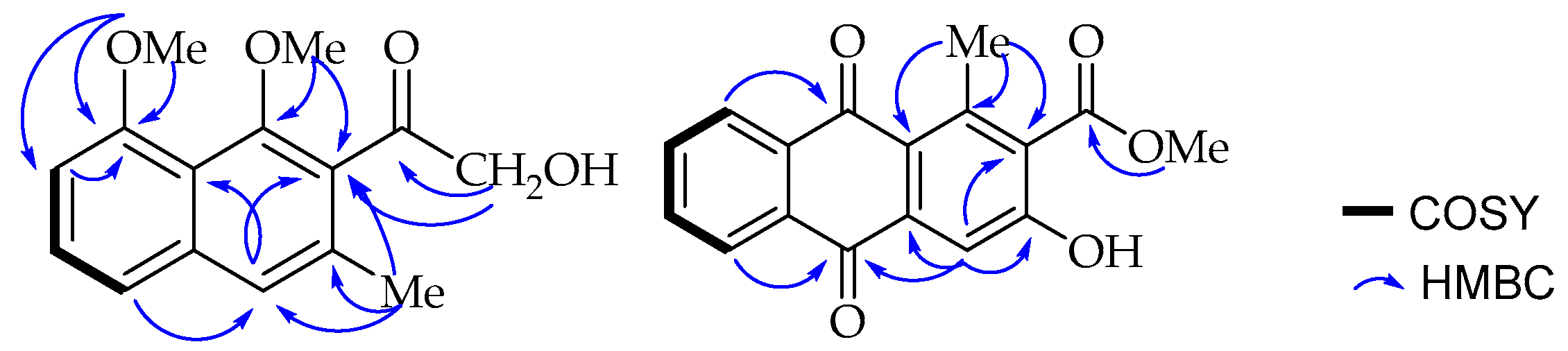
C 157.0) according to its long range HMBC correlation (
Figure 2) with the neighbouring carbons, C-7 (δ
C 106.7) and C-8 (δ
C 157.0). A broad singlet aromatic proton at δ
H 7.48 (H-4) showed a long-range NOE interaction in the NOESY spectrum with H-5 (δ
H 6.97) and the methyl group (δ
H 2.30; δ
C 19.0) located as expected according to biosynthetic considerations at C-3 (δ
C 133.6). The substituents at C-1 (δ
C 154.8) and C-2 (δ
C 132.0) were, therefore, established to be the methoxy (δ
H 3.73, δ
C 64.1) and the hydroxymethylketone unit (δ
H 4.55, δ
C 70.5; δ
C 207.5) respectively, based on a
3JC,H HMBC correlation of both H-4 (δ
H 7.48) and the hydroxymethyl protons (δ
H 4.55) with C-2 (δ
C 132.0). These placements were further confirmed by the long range HMBC correlations observed between methoxy protons (δ
H 3.73) and its nearby C-1 and C-2 carbons. Interestingly, the down-field shifted signal at δ
C 207.5 for the aryl ketone in the
13C-NMR spectrum is consistent with a group being
ortho-disubstituted, resulting in a deviation from planarity as a result of steric repulsion. Therefore, based on the above spectroscopic evidence, the new compound was characterized as 2-hydroxymethylacetyl-1,8-dimethoxy-3-methylnaphthalene, which was given the trivial name 1,8-dimethoxynepodinol (
1).
Compound
2 was isolated as yellow solid. The HR-ESI-MS provided a pseudomolecular ion peak at
m/
z 297.0753 [M + H]
+, corresponding to the molecular formula of C
17H
12O
5. The UV spectrum (λ
max 264, 298, 336, 371 nm) and NMR spectra suggested an anthraquinone skeleton [
23]. The
13C-NMR spectrum in total contains seventeen carbon signals (
Table 1), among them three carbonyl signals (δ
C 184.0, 183.5 and 168.3). The former two signals were attributed to a quinone system and the latter to the carboxylic acid methyl ester. In the
1H-NMR spectrum (
Table 1), four mutually coupled aromatic protons of AA′BB′ spin pattern centered at δ
H 8.19 (dd,
J = 7.7, 1.4 Hz, 1H), 7.86 (td,
J = 7.5, 1.4 Hz, 1H), 7.91 (td,
J = 7.5, 1.5 Hz, 1H) and 8.24 (dd,
J = 7.7, 1.5 Hz, 1H) were assigned to H-5, H-6, H-7, and H-8, respectively, located at the disubstituted ring C. A singlet at δ
H 7.75 was assigned to H-4 in ring A, which otherwise is fully substituted with a methyl group shifted downfield to
δH 2.71 due to the deshielding effect of the neighbouring carbonyl group attached to C-1 (δ
C 142.3). Furthermore, a carboxylic acid methyl ester (δ
H 3.95; δ
C 52.9, δ
C 168.3) at C-2 (δ
C 130.8) (based on biosynthetic considerations) and a hydroxy group at C-3 (δ
C 159.0). These assignments were in agreement with the biosynthetic consideration that appear to have been formed through folding of the octaketide chain in an unusual way as in aloesaponarin II [
24] and further confirmed by the HMBC correlation (
Figure 2) of methyl protons (δ
H 2.71) with C-1, C-1a (δ
C 125.3) and C-2; and H-4 (δ
H 7.75) with C-1a, C-2 and C-3.
The
13C-NMR spectrum further showed the presence of five
sp2 methines (δ
C 112.8, 127.2, 128.0, 134.4, 135.5), one methyl (δ
C 20.0) and seven
sp2 quaternary carbons (δ
C 125.3, 130.8, 133.5, 136.2, 138.3, 142.3, 159.0). These data are consistent with the compound being methyl 3-hydroxy- 1-methyl-9,10-dioxo-9,10-dihydroanthracene-2-carboxylate (
Figure 1) which was given the trivial name aloesaponarin III. This is the first report on the natural occurrence of compound
2 that had previously been reported as a synthetic intermediate [
25].
Compound
3 was isolated as yellow amorphous powder and was assigned the molecular formula C
31H
22O
9 based on the HR-ESI-MS analysis (negative mode; [M − H]
− m/
z 537.1863) and
13C-NMR data (
Table 1). The positive mode of HR-MS did not show the molecular ion peak, instead the demethoxylated pseudo molecular ion peak [M – OCH
3]
+ (
m/
z = 507.1034) was observed similar to the related dimeric anthraquinones [
9,
12]. Its UV–vis spectrum showed characteristic absorptions (λ
max 215, 261, 384, 432 nm) consistent with an anthrone-anthraquinone dimer [
14]. The presence of four highly downfield shifted proton signals (δ
H 12.36, 12.39, 12.10, 11.76) corresponding to hydroxyl groups involved in hydrogen bonding and three carbonyl groups (δ
C 193.1, 192.5, 182.0), a methyl (δ
H 2.43; δ
C 22.4) and hydroxymethyl (δ
H 4.64; δ
C 64.5) groups (
Table 1) supported the suggestion that the compound is a dimer of 1,8-dihydroxyanthraquinone/anthrone derivative similar to chrysalodin (
5) [
14].
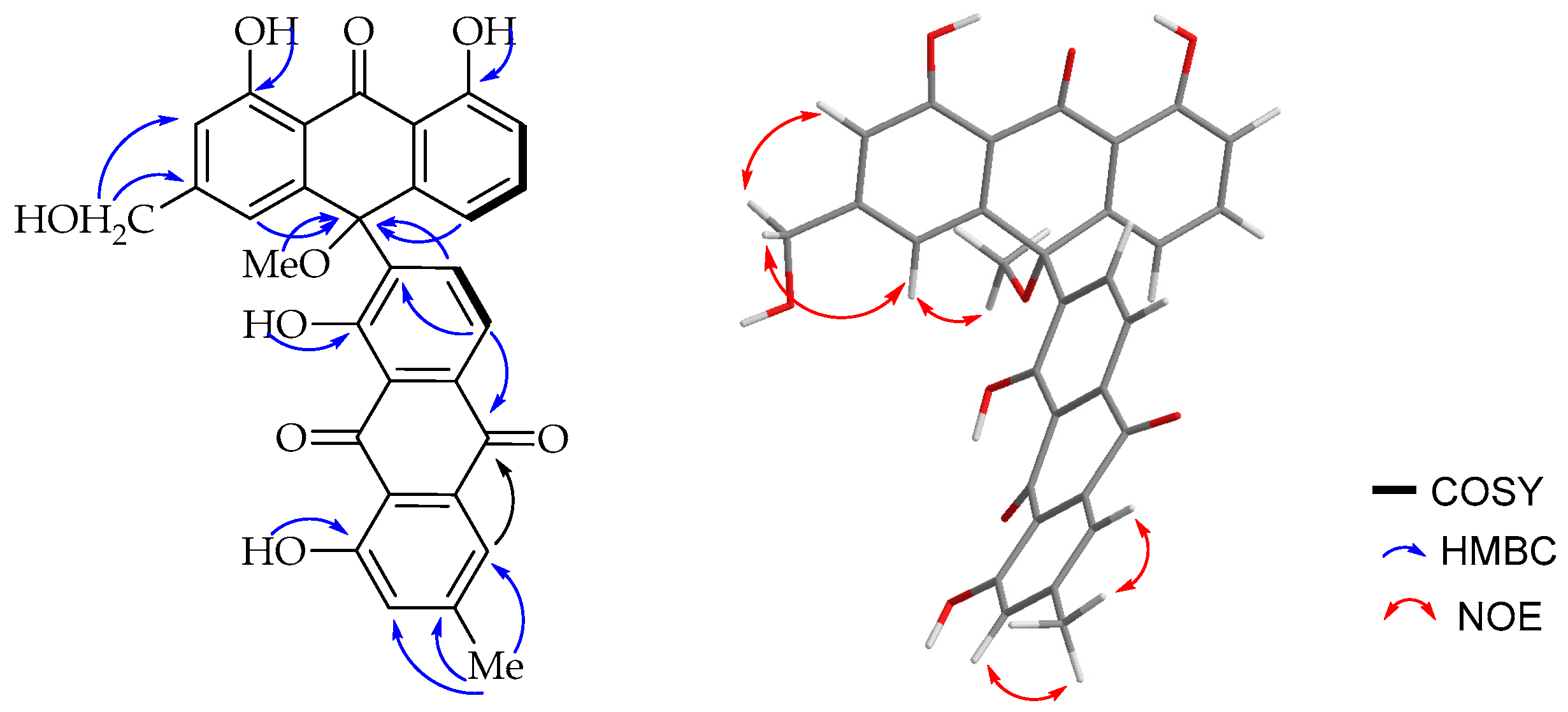
The
1H-NMR spectroscopic (
Table 1) features were virtually identical to those of chrysalodin (
5) with one-half of the molecule constituting aloeemodinanthrone, consisting of three mutually
ortho/
meta-coupled aromatic protons resonating at δ
H 6.77 (dd,
J = 8.0, 1.1Hz, 1H), 7.43 (t,
J = 8.2 Hz, 1H) and 6.95 (dd,
J = 8.5, 1.1 Hz, 1H) for H-5, H-6 and H-7 respectively,
meta-coupled protons at δ
H 6.76 (d,
J = 1.5 Hz, H-2) and 6.99 (d,
J = 1.5 Hz, H-4), and broad singlet proton signal at δ
H 4.64 for a hydroxymethyl group. The other half of the molecule constitutes a chrysophanol moiety, where the presence of
meta-coupled aromatic protons δ
H 7.60 (H-2′) and 7.02 (H-4′) with the biosynthetically expected methyl group (δ
H 2.43; δ
C 22.4) being located at C-3′ (δ
C 149.6) and a pair of downfield shifted
ortho-coupled protons at δ
H 7.89 (d,
J = 8.0 Hz, 1H) and δ
H 8.85 (d,
J = 8.0 Hz, 1H) for H-5′ and H-6′, respectively, clearly indicated the attachment of the aloeemodin moiety at C-7′ (δ
C 141.7) as in chrysalodin [
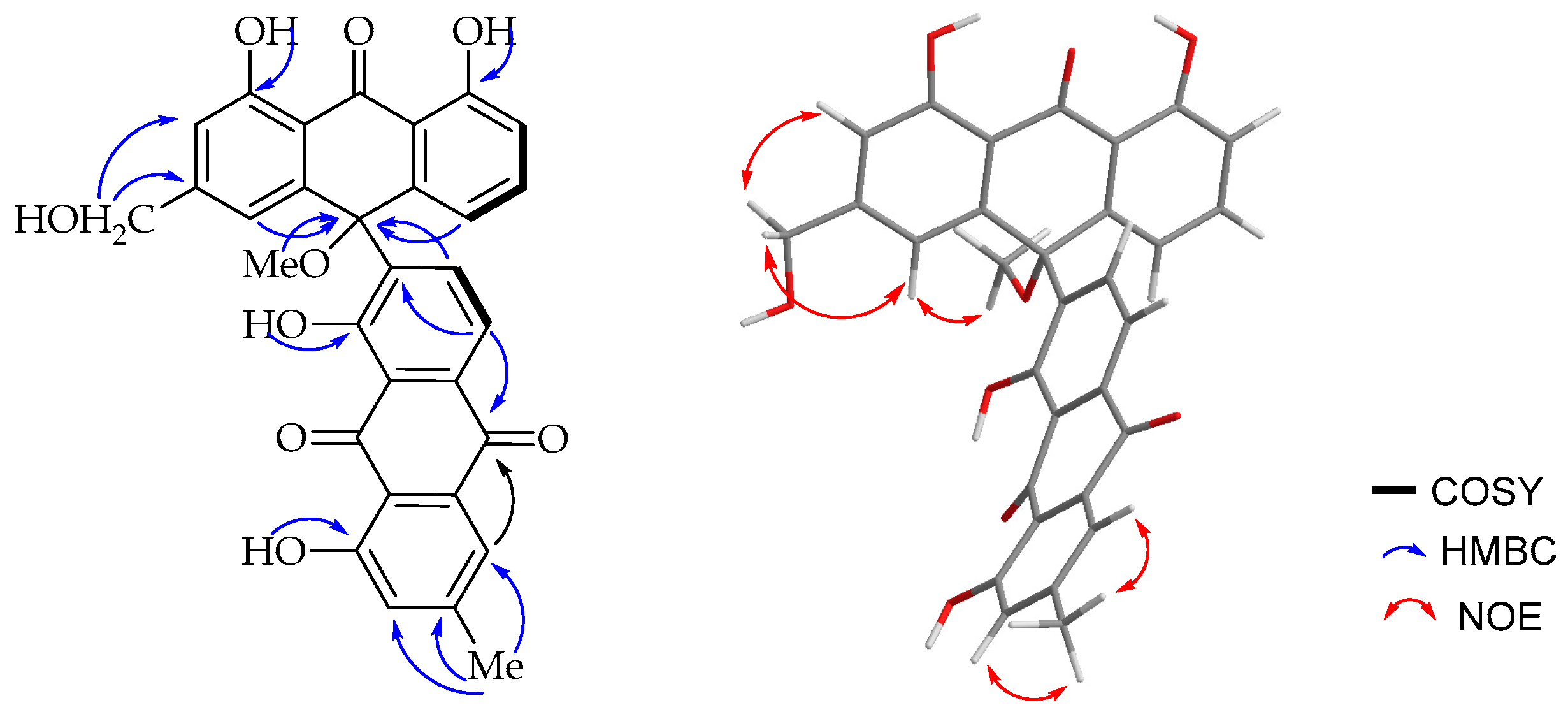
14]. The HMBC spectrum (
Figure 3) showed a
3JC,H correlation between H-6′ (δ
H 8.85) and C-10 (δ
C 75.6) supporting the suggestion that C-7′ is the point of attachment to the former half of the molecule. The only notable difference is the presence of a singlet integrating for three protons at δ
H 2.87 attached to the carbon resonating at δ
C 50.7 showing a HMBC correlation to C-10 (δ
C 75.6). The upfield chemical shift value of the methoxy group (δ
H 2.87) may be ascribed to anisotropy. It also showed an NOE (
Figure 3) correlation with H-4. The optical activity (
= +46 (
c 0.5, CH
2Cl
2)) and the CD spectrum confirms the chirality of compound
3. The absolute configuration at the chiral center was then established in comparison with a related dimeric anthraquinone, abiyquinone B [
26]. Compound
3 has opposite optical activity and a strong negative Cotton effect at shorter wavelength close to 235 nm in the CD spectrum. Based on the above spectroscopic evidence, the third compound is (
S)-10-(chrysophanol-7′-yl)-10-methoxy-aloeemodin-9-anthrone that was given the trivial name 10-
O-methylchrysalodin (
3).
Compound
4 was obtained as a colourless solid. The ESI-MS (
m/
z 625 (for [M + Na]
+) and HR-MS (
m/
z 603.5395 (for [M + H]
+) along with the NMR data was compatible with the molecular formula C
37H
62O
6. It has a UV absorption at λ
max 213, 278 nm. Its IR spectrum indicated the presence of hydroxyl (3410 cm
−1) and conjugated carbonyl (1637 cm
−1) functionalities. The
1H-NMR spectrum (in CDCl
3) exhibited signals corresponding to a ferulic acid moiety with a pair of doublets at δ
H 6.29 and 7.61 with a vicinal coupling constant
3JHH = 15.9 Hz. This indicated a
trans configuration and the signals were assigned to H-2′ and H-3′, respectively. These protons showed HMBC correlations with the ester carbonyl carbon at δ
C 167.5, suggesting that these protons were part of an α,β-unsaturated ester moiety. The three aromatic proton signals at δ
H 7.03 (
JHH = 1.9 Hz), 6.92 (
JHH = 8.2 Hz) and 7.07 (
JHH = 8.2, 1.9 Hz) were assigned to H-5′, H-8′, and H-9′, respectively based on their HH-COSY interactions and coupling constants. These assignments were further supported by the HMBC correlations of the downfield shifted proton H-3′ (δ
H 7.61) with C-5′ (δ
C 109.4) and C-9′ (δ
C 123.2) that in turn have HMQC correlations with H-5′ and H-9′, respectively. The
1H-NMR spectrum further showed the presence of a singlet at δ
H 5.84 corresponding to a phenolic hydroxy group, two singlets integrating for three protons each at δ
H 3.66 and 3.93 for two methoxy groups, two triplets at δ
H 4.19 and 2.30 for methylene groups (attached to oxygen and carbonyl respectively, deduced from their chemical shift) and multiplets corresponding to long methylene chain. The
13C-NMR spectrum contains signals corresponding to two ester carbonyl carbons (δ
C 174.5, 167.5), five aromatic/olefinic methine groups (δ
C 144.8, 123.2, 115.8, 114.8, 109.4), two oxygenated aromatic quaternary carbons (δ
C 148.0, 146.9), one oxymethylene (δ
C 64.8), two methoxy (δ
C 56.1, 51.6) and a number of methylene (34.3–24.9) carbons. The position of one methoxy group (δ
H 3.93; δ
C 56.1) was established at C-6′ (δ
C 148.0) according to its HMBC correlation to C-6′, while the hydroxy group at δ
H 5.84 showed a correlation to C-7′ (δ
C 146.9). The other methoxy group (δ
H 3.66; δ
C 51.6) corresponds to a methyl ester deduced from its upfield shifted (δ
C 51.6) signal [
12] and from its HMBC correlation with the ester carbonyl (δ
C 174.5). Comparison of these spectroscopic data with the literature revealed that the compound was very similar to ω-feruloyloxyacid [
27] and ethyl 24-(feruloyloxy)docosanoate [
28]. The only difference was the absence of a methoxy group and the length of the aliphatic chain. The NMR spectra and ESI-MS (
m/
z 625 [M + Na]
+, 601 [M−H]
−) were in accordance with the presence of a ω-oxygenated C
26 fatty acid chain, like ω-feruloyloxyacids with along aliphatic chain (C
19–C
27), reported from peat soil [
27]. Based on the spectroscopic data and the literature information, the structure of compound
4 was concluded to be methyl (
E)-26-((3-(4-hydroxy-3-methoxyphenyl)acryloyl)oxy)hexacosanoate that was given the trivial name methyl 26-
O-feruloyl-oxyhexacosanoate.
The isolates were evaluated for their cytotoxic activities against the human cervix cancer cell line KB-3-1, with cryptophycin-52 (IC
50 = 1.3 × 10
−5 µM) and griseofulvin (IC
50 = 19.0 µM) as positive controls, as described in previous reports [
29]. Two compounds, aloesaponarin II (
11) and aloesaponarin I (
12) showed good cytotoxic activity with IC
50 values of 0.98 µM and 16.00 µM, respectively whereas the dimeric anthraquinone asphodelin (
14) showed weak activity (>60.00 µM). The other compounds showed little or no inhibitory activities. The activity of aloesaponarin II is sixteen times higher compared to aloesaponarin I; however, both possess similar quinone nuclei with a methyl and hydroxyl substitutions at similar position. The only difference is the presence of an electron withdrawing methyl ester group in aloesaponarin I, which may negatively influence the cytotoxic activity of the compound.
It is worth mentioning that several cancer chemotherapeutic agents such as doxorubicin, mitomycin C, and mitoxantrone contain a common structural quinone nucleus. This chemical structure allows them to be involved in multiple biological oxidative processes [
30]. The reduction of the quinone leads to toxic species (semiquinone anion radical and hydroquinone) which act selectively in hypoxic tissues like tumor cells [
31].
3. Materials and Methods
3.1. General Information
Melting points were recorded on B-540 melting point apparatus (Büchi, Flawil, Switzerland). Column chromatography was carried out on silica gel (0.06–0.2 mm, Merck, Darmstadt, Germany) deactivated with 3% aq. oxalic acid. Gel filtration was carried out on Sephadex LH-20 (GE Healthcare, Uppsala, Sweden). Analytical TLC was performed on Merck pre-coated silica gel 60 F254 plates (Merck, Darmstadt, Germany). UV spectra were recorded on a UV-3100PC spectrophotometer (VWR international, Darmstadt, Germany). High Resolution ESI-MS was done on a Micromass AC-TOF micro mass spectrometer (Micromass, Agilent Technologies 1200 series, Waldbronn, Germany). Optical rotations were measured on a P-1020 polarimeter (JASCO, Tokyo, Japan). CD spectra were measured on a JASCO J-810 CD spectrometer (JASCO, Tokyo, Japan). IR spectra were recorded on a Nicolet 380 FT-IR spectrometer (Thermo Electron Corporation, Madison, WI, USA). 1D NMR and 2D (COSY, HSQC, HMBC, NOESY) NMR spectra were recorded on an Avance 500 MHz spectrometer (Bruker, Rheinstetten, Germany) at 500 MHz (1H) and 125 MHz (13C) at 298 K using the residual solvent peaks as a reference.
3.2. Plant Materials
The roots of A. megalacantha were collected from West Arsinegele (7°22′26.2″ N 38°40′02.6″ E), Ethiopia, 240 Km away from Addis Ababa on the way to Shashemene in September 2016. The plant material was identified by professional botanist at Jimma University (Dr. Kitessa Hundera) and the voucher specimen (voucher number NA-07/16) has been deposited in the Jimma University Herbarium.
3.3. Extraction and Isolation
The air dried and powdered roots of A. megalacantha (840 g) were extracted exhaustively with ethyl acetate (3 L) four times each for 24 h at room temperature. The extract was then concentrated under reduced pressure using a rotary evaporator to yield 34 g of brown extract. A portion of the extract (30 g) was subjected to column chromatography on oxalic acid deactivated silica gel (400 g) and the column was eluted with petroleum ether containing increasing amounts of ethyl acetate to give 37 fractions each of ca. 250 mL.
Fractions 6–8 (eluting at 5% v/v ethyl acetate in petroleum ether) were further separated by repeated gel chromatography on Sephadex LH-20 (eluting with CH2Cl2/MeOH, 1:1) to give chrysophanol (4.0 mg), helminthosporin (3.0 mg) and compound 4 (2.1 mg). Fractions 15–18 (eluting at 10% v/v ethyl acetate in petroleum ether) were combined based on their TLC profile. Purification by column chromatography (column size: 60 cm length and 3 cm diameter) with an increasing gradient of ethyl acetate in petroleum ether gave asphodelin (8.0 mg), compound 2 (2.8 mg) and 10-(chrysophanol-7′-yl)-10-hydroxychrysophanol-9-anthrone (6.1 mg). Fractions 19–24 (eluting at 24% v/v ethyl acetate in petroleum ether) containing mixtures of five compounds were combined and subsequently subjected to column chromatography (column size: 80 cm length and 4 cm diameter) on silica gel (300 g) impregnated with oxalic acid (eluent: increasing gradient of ethyl acetate in petroleum ether). This was followed by further purification of the fractions on Sephadex LH-20 (dichloromethane/methanol, 1:1) yielding aloesaponarin I (4.8 mg), aloesaponarin II (3.9 mg), 4,7-dihydroxy-5-methylcoumarin (8.7 mg), compound 1 (4.2 mg) and aloeemodin (10.2 mg). Fractions 26–28 (eluting at 35% v/v ethyl acetate in petroleum ether) were collected as a yellow coloured solution. Further purification by gel chromatography using Sephadex LH 20 (eluting with CH2Cl2/MeOH; 1:1) gave compound 3 (3.4 mg) and chrysalodin (4.2 mg). Fractions 30–32 (eluting at 60% v/v ethyl acetate in petroleum ether) gave a colourless precipitate that was filtered and washed with dichloromethane/ethyl acetate mixture yielding aloesaponol I (21.0 mg).
1,8-Dimethoxynepodinol (
1): Colourless amorphous solid. m.p. 132–134 °C. UV (CH
3CN): λ
max (logε) = 252 (2.62), 276 (2.54) nm. IR (CH
2Cl
2) ν
max cm
−1 3472, 1701, 1618, 1571, 1346.
1H- and
13C-NMR (
Table 1). ESI-MS (rel. int.):
m/
z = 543 (100, [2 M + Na]
+), 283 (57, [M + Na]
+). HR-ESI-MS
m/
z = 283.0916, [M + Na]
+ (calculated for C
15H
16O
4Na, 283.0946).
Aloesaponarin III (
2): Yellow solid. m.p. 257–259 °C. UV (CH
3CN): λ
max (logε) = 264 (2.80), 298 (2.73), 336 (2.63), 371 (2.44) nm. IR (CH
2Cl
2) ν
max cm
−1 3363, 2948, 1732, 1662, 1577, 1235, 712.
1H- and
13C-NMR (
Table 1). ESI-MS (rel. int.)
m/
z = 615 (21, [2 M + Na]
+), 319 (43, [M + Na]
+), 297 (100, [M + H]
+). HR-ESI-MS
m/
z = 297.0753, [M + H]
+(calculated for C
17H
13O
5, 297.0762).
10-O-Methylchrysalodin (
3): Yellow amorphous solid. m.p. 228–230 °C. UV (CH
2Cl
2): λ
max (logε) = 215 (3.18), 261 (3.14), 384 (2.94), 432 (2.66) nm.
+46° (c 0.5, CH
2Cl
2).
1H- and
13C-NMR (
Table 1). ESI-MS (rel. int.)
m/
z= 507 (18, [M–OMe]
+), 537 (12, [M − H]
−). HR-ESI-MS
m/
z = 537.1863, [M − H]
− (calculated for C
31H
21O
9, 537.1855).
Methyl 26-O-feruloyl-oxyhexacosanoate (4): Colourless solid. m.p. 187–189 °C. UV (CH2Cl2): λmax (logε) = 213 (2.91), 278 (2.87) nm. IR (CH2Cl2) νmax cm−1 3410, 1762, 118, 1637. 1H-NMR (CDCl3) δH 7.61 (1H, d, J = 15.9 Hz, H-3′), 7.07 (1H, dd, J = 1.9, 8.2 Hz, H-9′), 7.03 (1H, d, J = 1.9 Hz, H-5′), 6.92 (1H, d, J = 8.2 Hz, H-8′), 6.29 (1H, d, J = 15.9 Hz, H-2′), 5.84 (1H, s, 7′-OH), 4.19 (2H, t, J = 1.8 Hz, H-26), 3.93 (3H, s, 6′-OCH3), 3.66 (3H, s, 1-OCH3), 2.30 (2H, t, J = 1.4 Hz, H-2), 1.18–1.23 (46H, m, methylene chain). 13C-NMR (CDCl3) δC 174.5 (C-1), 167.5 (C-1′), 148.0 (C-6′), 147.9 (C-7′), 144.8 (C-3′), 127.2 (C-4′), 123.2 (C-9′), 118.8 (C-8′), 115.8 (C-2′), 109.4 (C-5′), 64.8(C-26), 56.1 (6′-OCH3), 51.6 (1-OCH3), 34.3 (C-2), 34.1–24.9 (methylene chain). ESI-MS (rel.int.) m/z = 625 (100, [M + Na]+). ESI-MS m/z = 601, [M − H]−. HR-ESI-MS m/z = 603.5395, [M + H]+ (calculated for C37H63O6, 603.5363).
3.4. Cytotoxicity Assay
The human cervix carcinoma cell line KB-3-1 was used in the cytotoxicity assay as previously described [
29]. The cell line was cultivated as a monolayer in DMEM (Dulbecco’s modified Eagle medium) with glucose (4.5 g/L),
l-glutamine, sodium pyruvate and phenol red, supplemented with 10% fetal bovine serum (FBS) and were maintained at 5.3% CO
2 and 37 °C in humidified air. The cells at 70% confluence were detached with trypsin-ethylenediamine tetraacetic acid solution (0.05%; 0.02% in DPBS) and placed in sterile 96-well plates in a density of 10,000 cells in 100 μL medium per well. The dilution series of the compounds was prepared from stock solutions in DMSO of concentrations of 100 mM, 50 mM or 25 mM and the stock solutions were diluted with culture medium (10% FBS) down to pM concentrations. The dilution prepared from stock solution was added to the wells and each concentration was tested in at least six replicates. The control contained the same concentration of DMSO as the first dilution. After incubation for 72 h at 37 °C and 5.3% CO
2-humidified air, 30 μL of an aqueous resazurin solution (175 μM) was added to each well. The cells were incubated at the same conditions for 5 h. Subsequently, the fluorescence was recorded at a wavelength of 588 nm. The IC
50 values were calculated as a sigmoidal dose response curve using Graphpad Prism 4.03 (Graphpad software Inc., San Diego, CA, USA).



{kind=link}
{kind=link}
{kind=link}