1. Introduction
One of the outstanding challenges in virtual screening is the development of a fast and robust algorithm to compare many compounds and identify subsets with biological activity. It is often desired that such subsets are diverse with respect to their basic molecular scaffolds. Such scaffold hopping can be used to break out of the protected “patent space” or to find molecules with different, more desirable pharmacological properties.
All the above have prompted the development and application of ligand-based techniques that rely on a set of active/inactive ligands [
1]. One highly popular idea is to represent a compound as a vector with the presence (1) or absence (0) of a set of features describing chemical structure and properties. This method is very efficient as both vector generation (fingerprint) and screening task (vector comparison) are computationally inexpensive. Unfortunately, fingerprint representations have very limited capacity, do not scale well with increasing compound size and complexity, depend strongly on a set of predefined features, and have poor scaffold-hopping performance [
2,
3].
As the affinity of a ligand-receptor interaction depends on atomic interactions in all three dimensions, alternative approaches incorporating 3D features have emerged. Unfortunately they introduce ligand flexibility into the equation, which is computationally much more expensive and error-prone primarily due to insufficient conformational sampling. Methods to overcome this problem have been proposed. One idea was to represent the 3D molecule as a set of absolute, invariant molecular fragments generated from 2D topologies [
4]. This approach, focusing more on representation consistency rather than realism, was successfully applied for ligand alignments in CoMFA (Comparative Molecular Field Analysis) [
5].
Other methods have been proposed, with shape-based representations of small molecules [
6,
7], molecular interaction field-based representations [
8] or surface-based representations [
9], among others. Although the computation time is decreased compared to the simple atomic distance-based methods, they still require generating multiple conformations (5–50 per ligand) for all compounds in the dataset [
10].
One way to avoid conformational search, yet still describe the shape of a compound, is to represent the molecule’s connectivity, rather than its particular conformation. Such representations are closely related to molecular graphs, typically used to visualize chemical compounds. Among the examples of graph-based approaches are reduced graphs [
11] and extended reduced graphs (ErG) [
12]. However, these methods use graphs only as an intermediate stage and calculates a set of descriptors characterizing their composition and topology, which are then used to compare molecules.
In this work, we went a step further and developed a method that utilizes a graph as a final representation of a molecule. Although such an approach is computationally more challenging, graphs were successfully used to compare molecules, using both 2D and 3D structures [
13,
14], 3D pharmacophores [
15], or even protein binding sites [
16]. 3D methods, however, relay on pre-generated ligand conformations, which are computationally expensive and can influence results significantly.
In this work, we use a graph to describe the 2D structure of a molecule. As features, we utilize the pharmacophoric properties of atoms, which express the binding potential of a molecule and are more general than functional groups. We complemented this representation with a graph alignment procedure that allows for comparing and combining of multiple models. Our approach was implemented as a customizable Python module called DeCAF (Discrimination, Comparison, Alignment tool for 2D PHarmacophores) that can be easily combined with Open Babel or RDKit to facilitate ligand-based drug design.
2. Materials and Methods
DeCAF source code, documentation and installation instructions are publicly available at
http://bitbucket.org/marta-sd/decaf. All data and results presented in this study were created and analyzed using Python (v. 2.7) [
17].
To generate and compare different fingerprints in the SEA benchmark (see
Section 3.2), we used Open Babel (development version from 30.08.2015) [
18]. Open Babel was also used to read molecules and automatically convert them to DeCAF pharmacophore models. Ligand-based virtual screening benchmark (see
Section 3.1) was downloaded from Github as a git repository [
19] and run with RDKit (version 2016.03.2) [
20]. All other data and Python scripts used in our study can be downloaded from
http://bitbucket.org/marta-sd/decaf-supplementary. The repository also contains instructions on how to prepare a Python environment with all packages needed to reproduce our results and repeat our analysis.
2.1. Representation
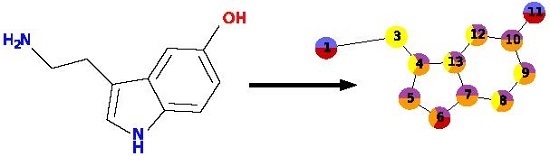
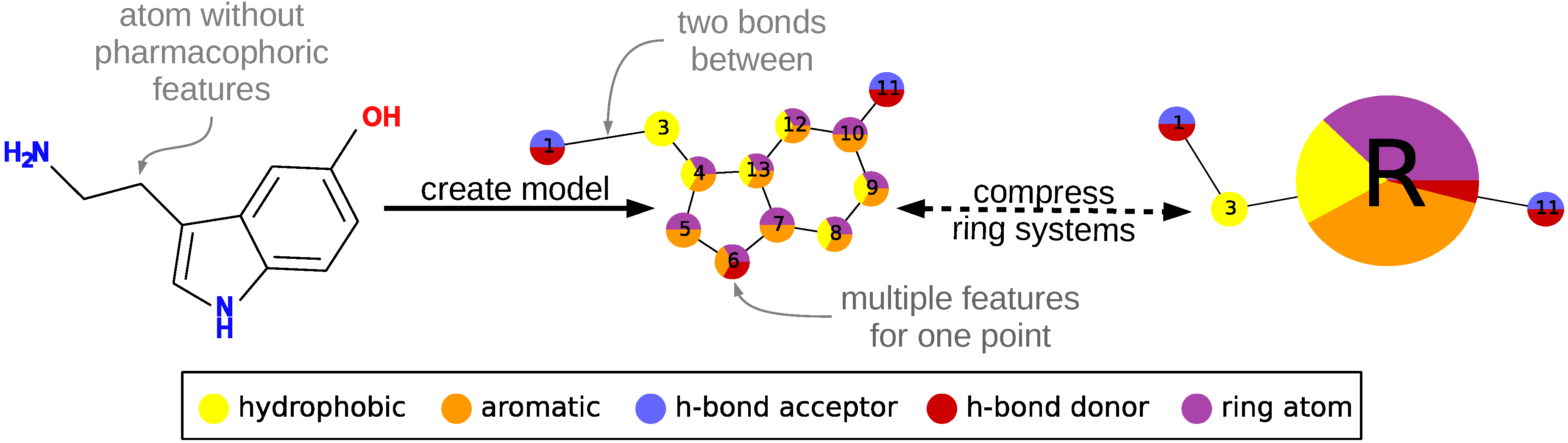
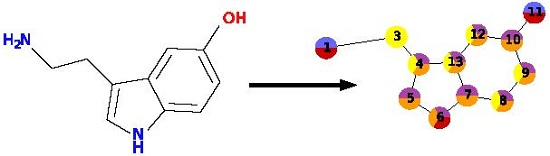
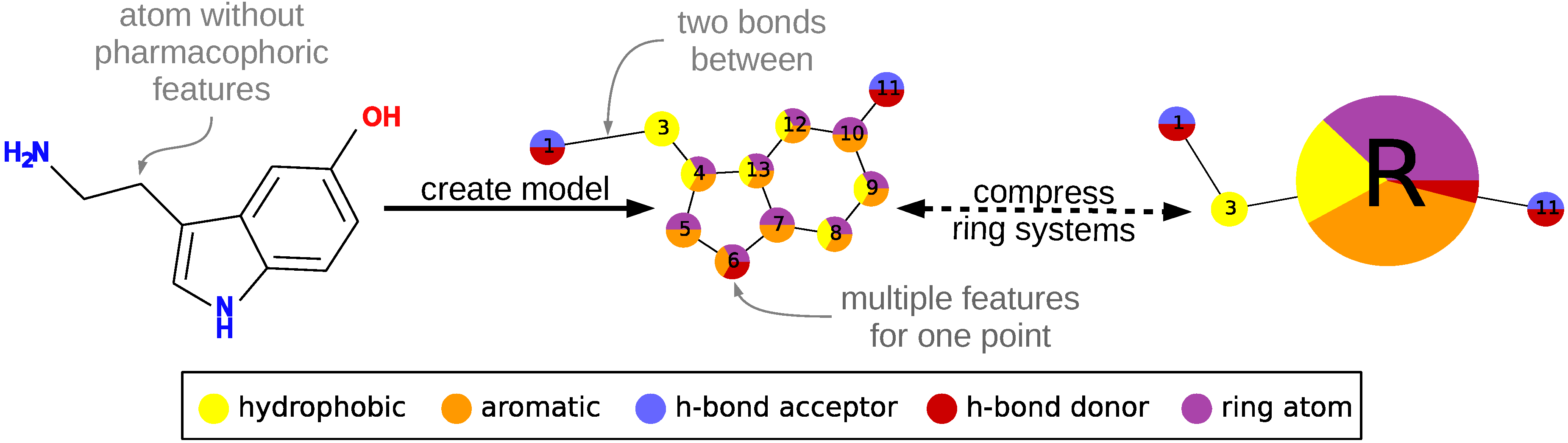
To describe a molecule, DeCAF substitutes its functional groups with pharmacophoric points (see
Figure 1), hence the “F” in the program’s name. Anything you can define with a SMARTS (SMiles ARbitrary Target Specification) pattern can be a feature and there are 5 elementary pharmacophoric points already implemented in DeCAF:(i) hydrophobic (“HH”); (ii) aromatic (“AR”); (iii) hydrogen bond donor (“HD”); (iv) hydrogen bond acceptor (“HA”); and (v) ring atoms (“R”) . The ring feature allows for the simplification of the alignment procedure (see next section and
Supplementary Methods) and provides a natural penalty for aligning rings to aliphatic fragments of a molecule.
Each point and each of its features has a weight (the default is 1.0), which expresses the importance of that part of the model or its frequency, if the model was generated from multiple molecules. Nodes are organized into an undirected graph, in which atoms are connected to their direct neighbors or to the next atom in the molecule, if a neighbor is not present in the graph.
No calibration of the weighting scheme was performed as DeCAF aims to be applicable to a broad spectrum of chemical space. However, the pharmacophores can be manually tuned to incorporate the expert knowledge into the model. Weights can be changed to differentiate between crucial and less significant features. Edges can be added or removed to modify the distance constraints. In this study, DeCAF was tested by screening multiple targets without any target-specific adjustments. For some of the targets, we show how incorporating external knowledge can improve screening results (see
Section 4).
2.2. Alignment
To compare two models, DeCAF aligns them by finding their maximal common subgraph. This is a common problem in cheminformatics, to which a solution was presented by Barrow and Burstall [
21]. Similarly to their standard solution, DeCAF generates a modular product of two graphs and then searches for the best maximal clique in this product, which corresponds to the highest-scoring common part of two models.
The maximal common subgraph problem is NP-hard, and no polynomial algorithm for solving it is known. DeCAF uses additional simplifications and constraints to develop a fast, heuristic algorithm for finding a common part of two models. In this section, we will sketch the algorithm, and a more detailed description can be found in the
Supplementary Methods.
The alignment procedure is divided into two phases. During the first phase, a coarse-grained version of each model is generated, in which rings and ring systems are compressed to single nodes using the pharmacophore “R” feature described above. Then, DeCAF generates their modular product and finds all maximal cliques within it using the Bron–Kerbosch algorithm with pivoting [
22,
23]. We select a clique with the highest similarity score, which corresponds to the best alignment of the reduced models.
One important modification to the Barrow and Burstall algorithm is that we introduce an additional constraint when adding edges to the modular product: we connect only those pairs whose corresponding nodes are in similar distance in both models. This decreases the number of edges in the modular product and therefore reduces the number of cliques. It also prevents DeCAF from producing alignments that do not preserve distances between features.
By default, DeCAF does not make any assumptions about the data. It requires a strict match between the distances in the compared molecules. With additional knowledge of the receptor and its ligands, it is possible to increase the accepted distance to better describe the target’s preferences. However, for a study with diverse targets, it might lead to false positive results. Therefore, only default parameters have been used. We also show an example of the target with less strict distance preference in the
Section 4.
The result of the first phase of the alignment procedure is a coarse-grained alignment, which is an approximation of the actual comparison of the two original models. In the second phase of the algorithm, nodes that form rings are aligned with respect to the already aligned parts of the pharmacophores, resulting in a fine-grained alignment. This final alignment can then be used to calculate the similarity between the two models. DeCAF can also combine them into a single pharmacophore model using shared nodes as a core of the new model. To assess similarity between the models, DeCAF uses Dice similarity score for non-binary. The similarity score of models P and Q is given as:
where:
is a score for an alignment A,
is a similarity between nodes i and j, and
is a weights of node i.
For each pair of aligned nodes, DeCAF computes , which is the ratio of their common features by the number of all features, multiplied by the sum of their weights (). is the sum over those values and serves as a score for a given clique during the alignment procedure. It is also used to compute the final similarity score : is divided by the sum of weights of all features present in the two models. is a counterpart of the doubled number of common features in the binary version of Dice’s similarity scores, while the sum of all weights is an analogue to the number of all features.
2.3. Benchmark Preparation and Dataset Development
Although there are ready-to-use datasets available, most of them are designed for structure-based screening. For example, in the DUD-E (Directory of Useful Decoys, Enhanced) database, “decoys” (compounds assumed to be inactive) are chosen to have similar physicochemical properties to active compounds, but dissimilar structures, so that binding to the target would be highly unlikely. This makes them very easy to distinguish with ligand-based methods, which use structural similarity. Hence, we tested our method on two ligand-based benchmarks: screening benchmark for fingerprints provided by Riniker and Landrum [
24] and a benchmark using SEA as the screening method based on a Lounkine et al. study [
25].
The dataset used in the first benchmark was provided by the authors and consisted of 88 targets from MUV (Maximum Unbiased Validation), ChEMBL, and DUD (Directory of Useful Decoys) databases. The data, together with scripts running the benchmark, were downloaded from Github [
19]. Scripts were adapted for DeCAF: molecule representation and similarity function were added to
fingerprint_lib.py and
scoring_functions.py, respectively. In addition, the main script—
calculate_scored_lists.py—was parallelized.
The dataset for the second benchmark was not available, thus we reconstructed it using the description and supplementary files from the original work by Lounkine et al. For each of 73 targets, we created an extensive set of its potentially bioactive ligands by retrieving activity data from the ChEMBL database [
26] and selecting ligands with
,
,
or
values below 1000 nM (accessed 10.2015).
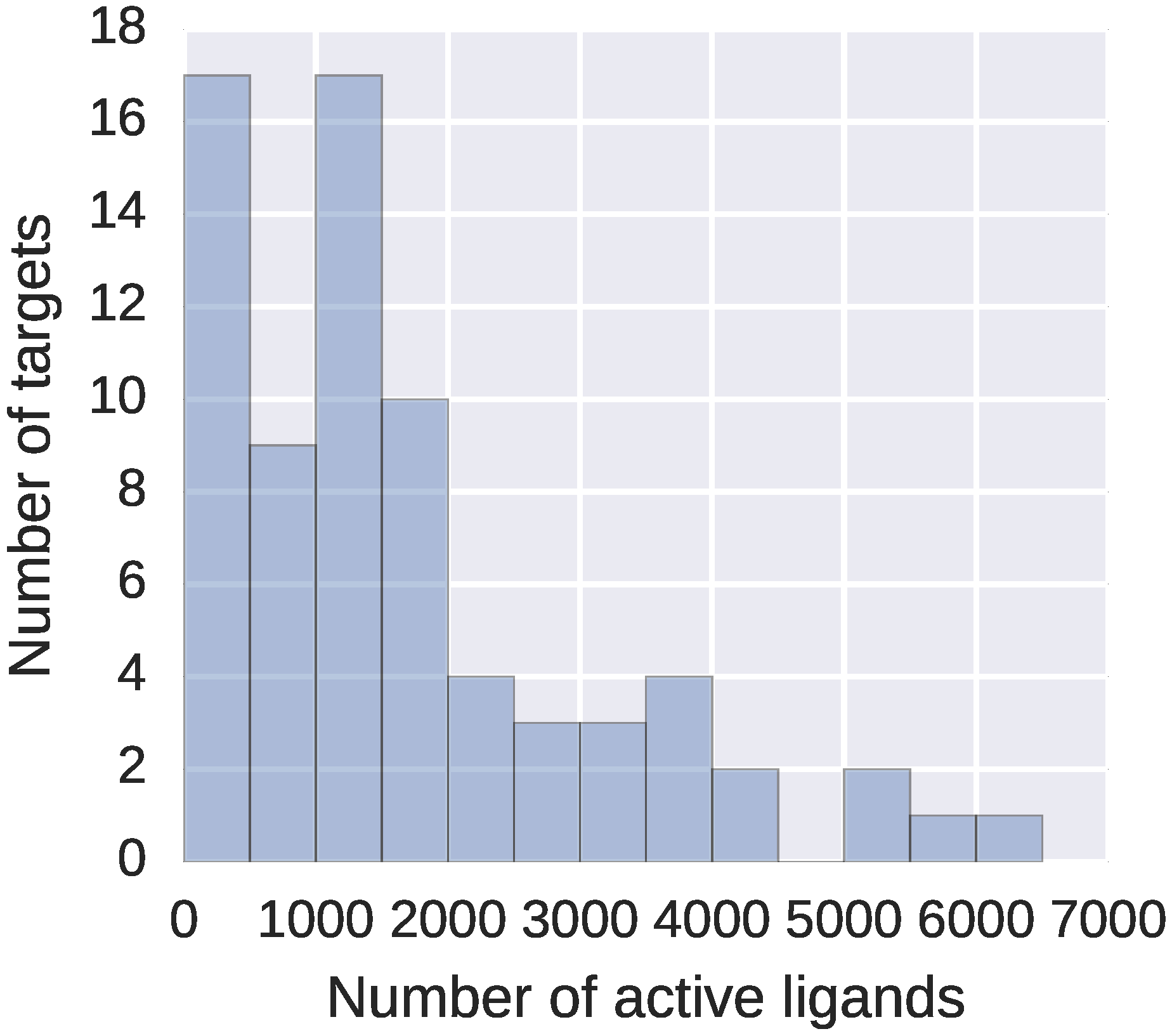
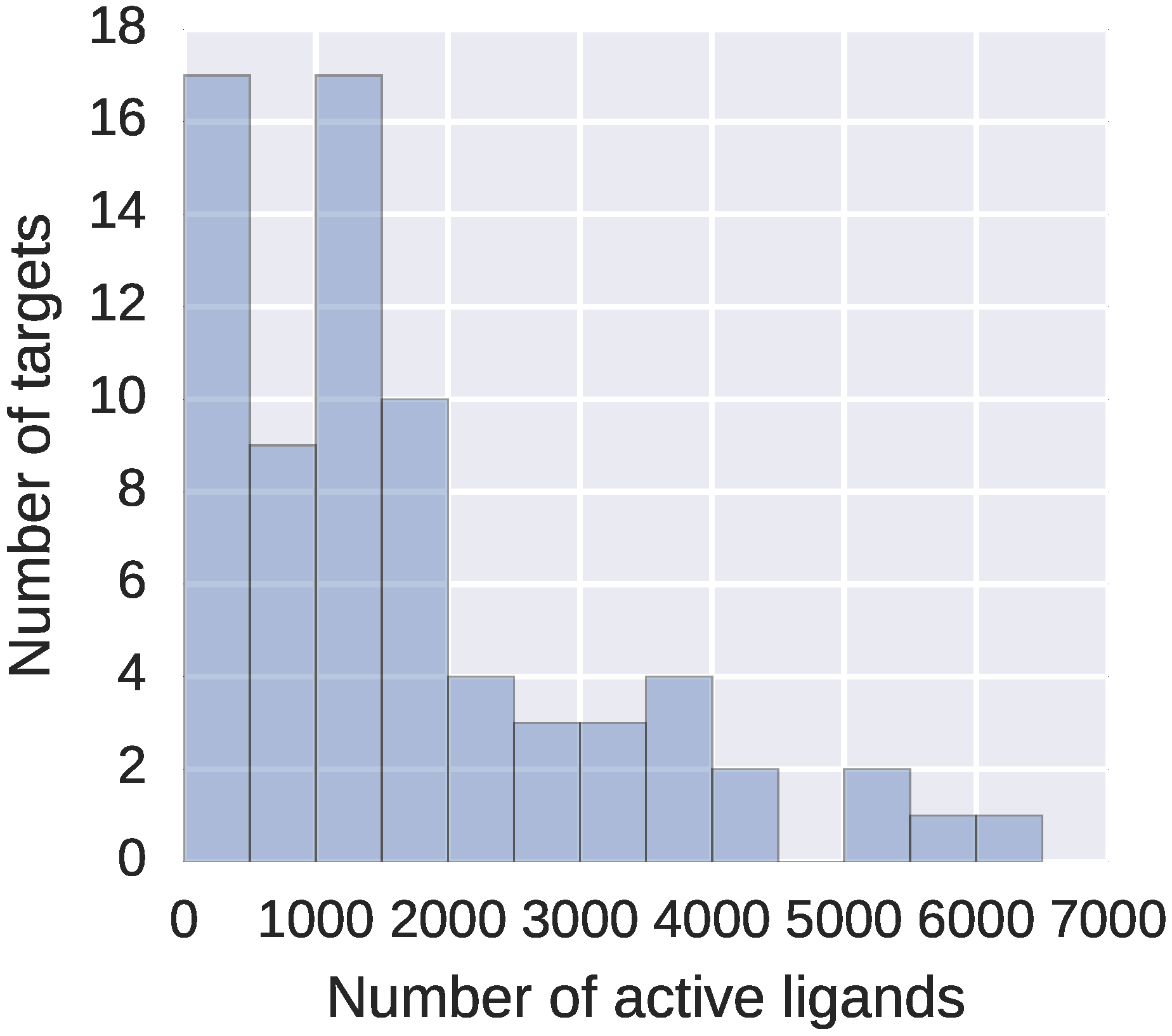
From this dataset, we filtered out all peptides and peptide-like compounds with more than 20 amino acids. Including longer peptides or other polymers, for which the overall 3D conformation defines their properties, would only slow down the computations but not improve the activity predictions – neither DeCAF, nor fingerprints would represent them properly. Final refined dataset consisted of 73 targets, with 59 to 6229 high affinity ligands describing each target (see
Figure 2).
To test the models, data regarding interactions between 656 drugs tested by Lounkine et al. and the 73 targets were constructed. The validation set consists of: (i) low affinity (<30,000 nM) drug-target pairs from ChEMBL; (ii) those not present in ChEMBL, but reported by Lounkine et al. in their study; and (iii) present in the DrugBank database. All drug-target pairs with even lower affinity or without any data about interaction were assumed to be inactive.
For detailed description of each drug and target, see
Supplementary file 1 and the original work by Lounkine et al.
4. Discussion
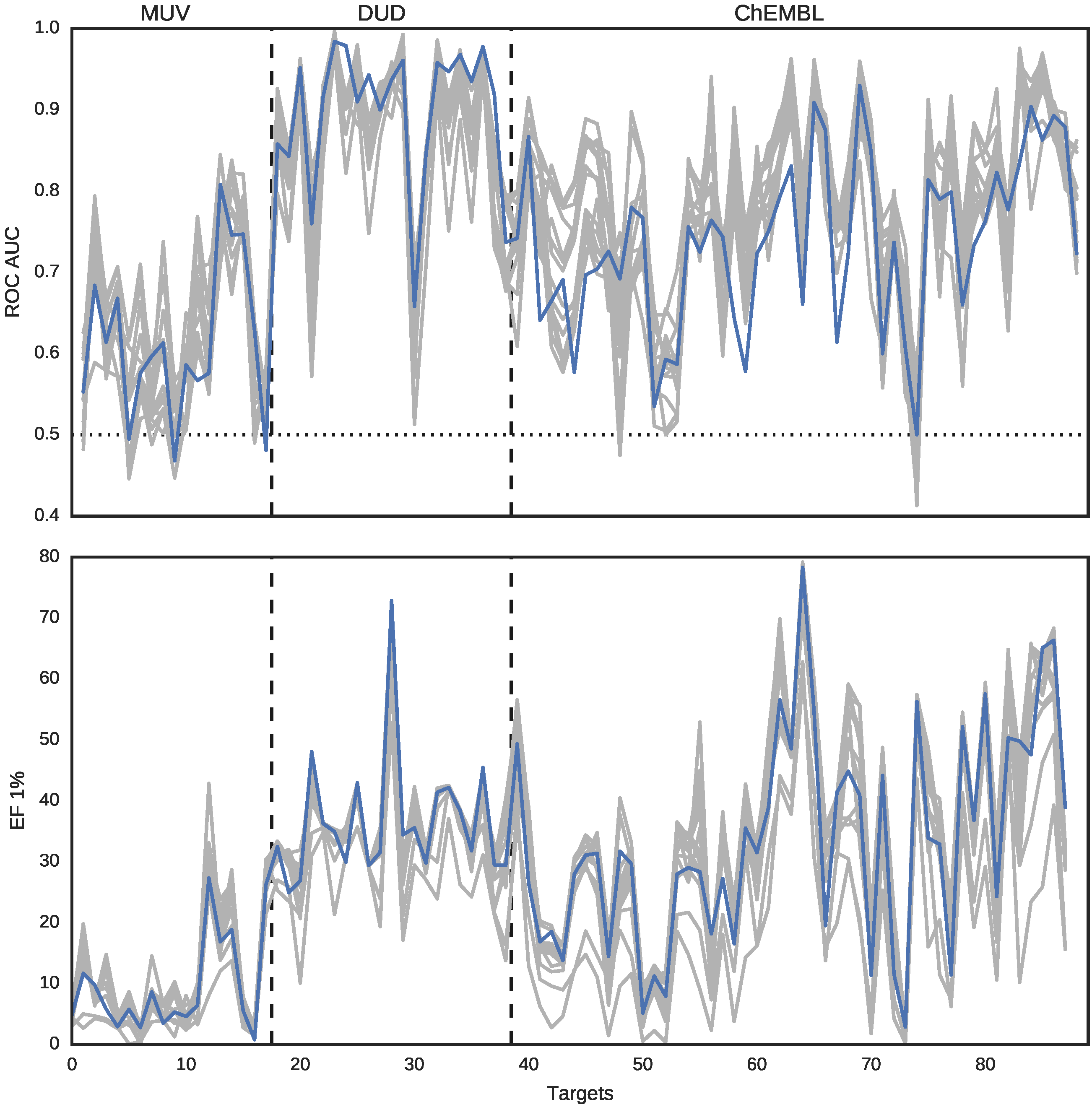
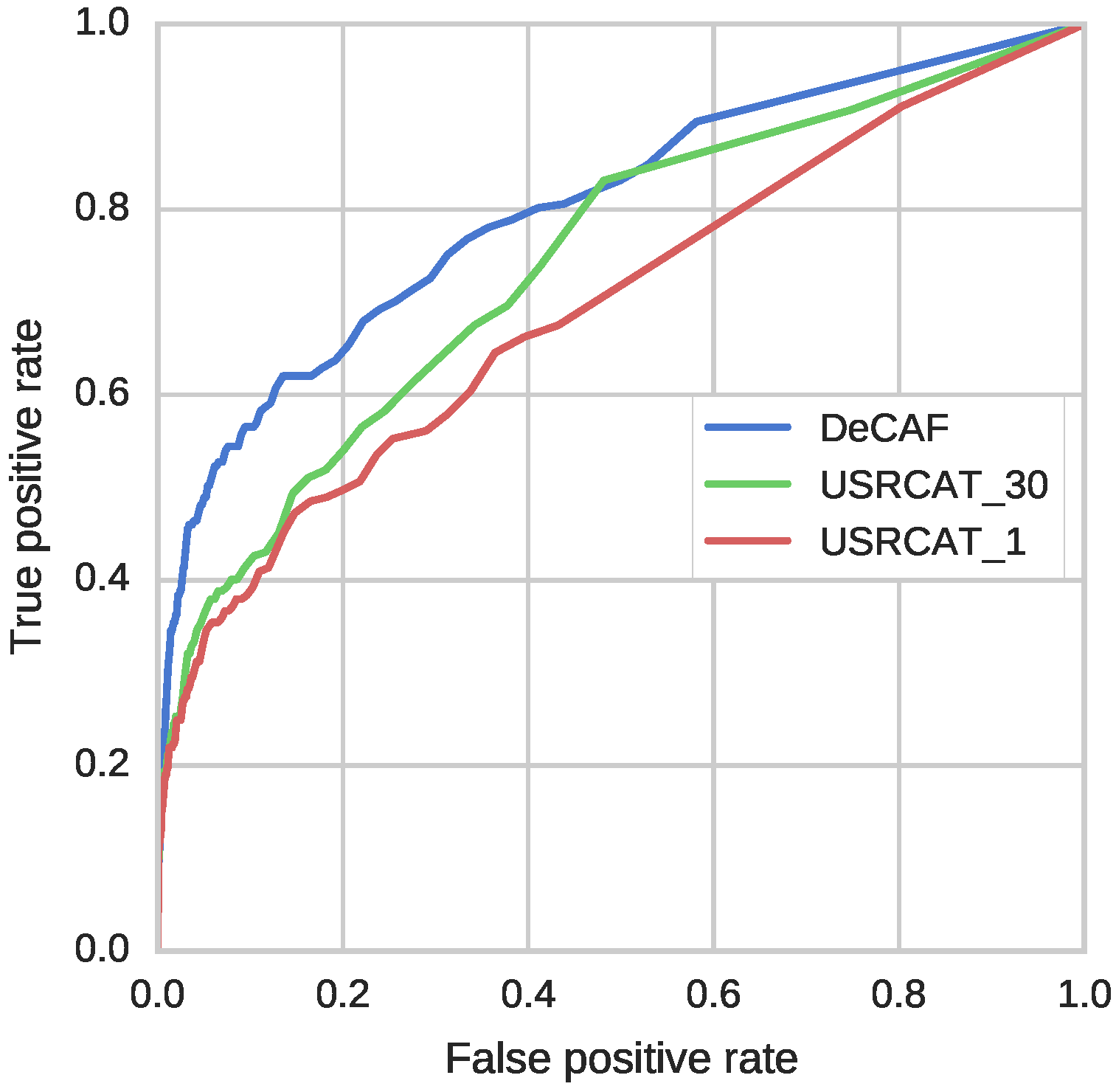
In this section, we focus on the results from the second benchmark, i.e., screening with an SEA classifier. This benchmark provides non-relative scores for activity prediction, with the same interpretation across different datasets and different representations of a molecule. This allows to directly compare activity predictions obtained with different methods.
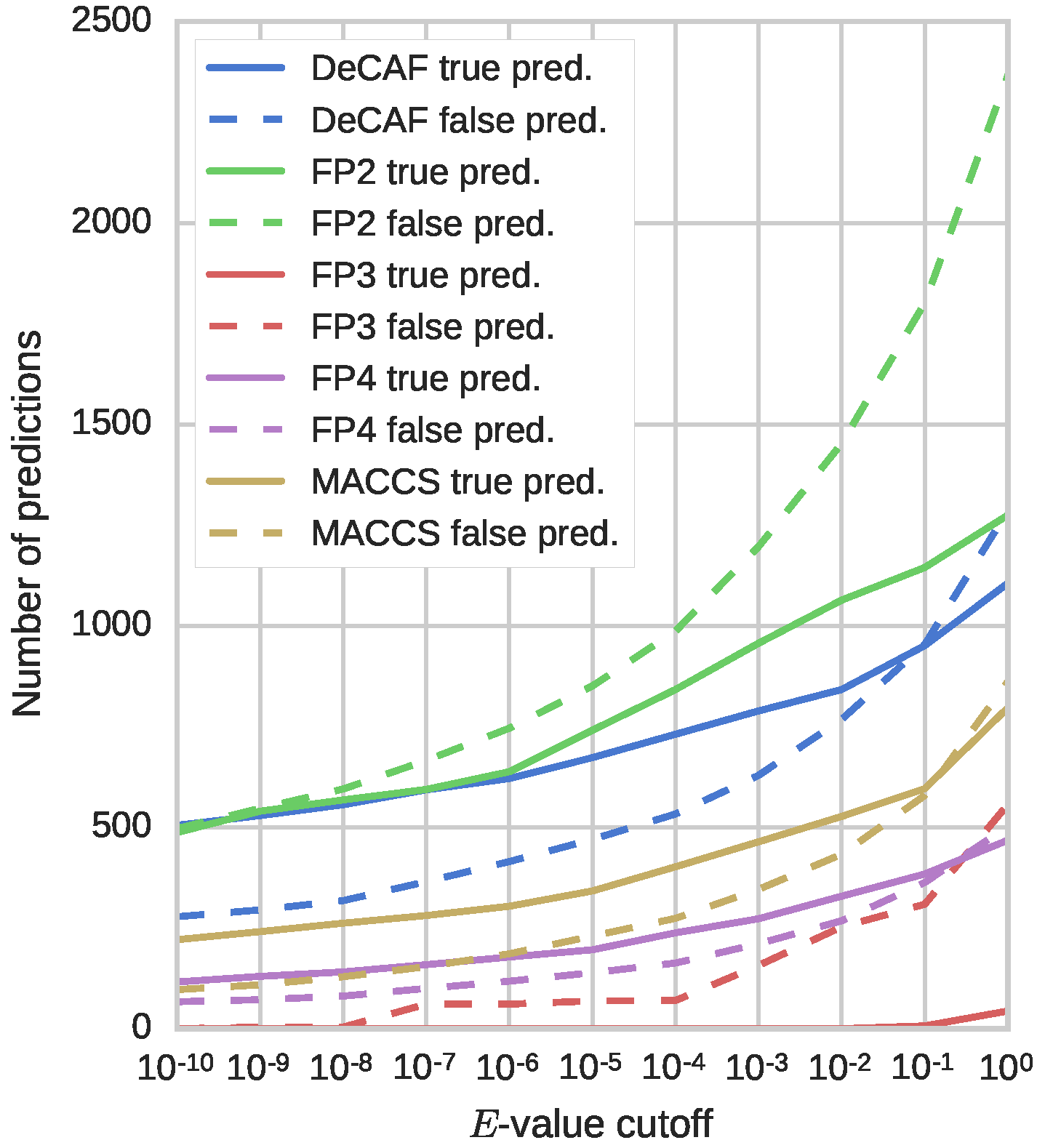
In most cases, DeCAF’s advantages over the four fingerprints tested on SEA benchmark can be explained by the limited capabilities of these fingerprints to represent complex substructures and spatial relationships between different parts of a molecule (see
Figure 4 in Supplementary Analysis). On the other hand, 3D methods such as USRCAT rely on the shape of a compound and therefore can suffer from using incorrect conformation(s). DeCAF tries to overcome those two contradicting approaches by converting a molecule into a 2D graph preserving the spatial relationships between pharmacophore groups. Although DeCAF performs generally better than the tested fingerprints, it sometimes fails to correctly predict activity when default parameters are used. Contrary to fingerprints, however, DeCAF allows to adjust feature importance and parameters of the alignment procedure. In this section, we show two examples that illustrate how a researcher can fine-tune DeCAF for a specific task. For more general assessment of the results, see
Supplementary Analysis.
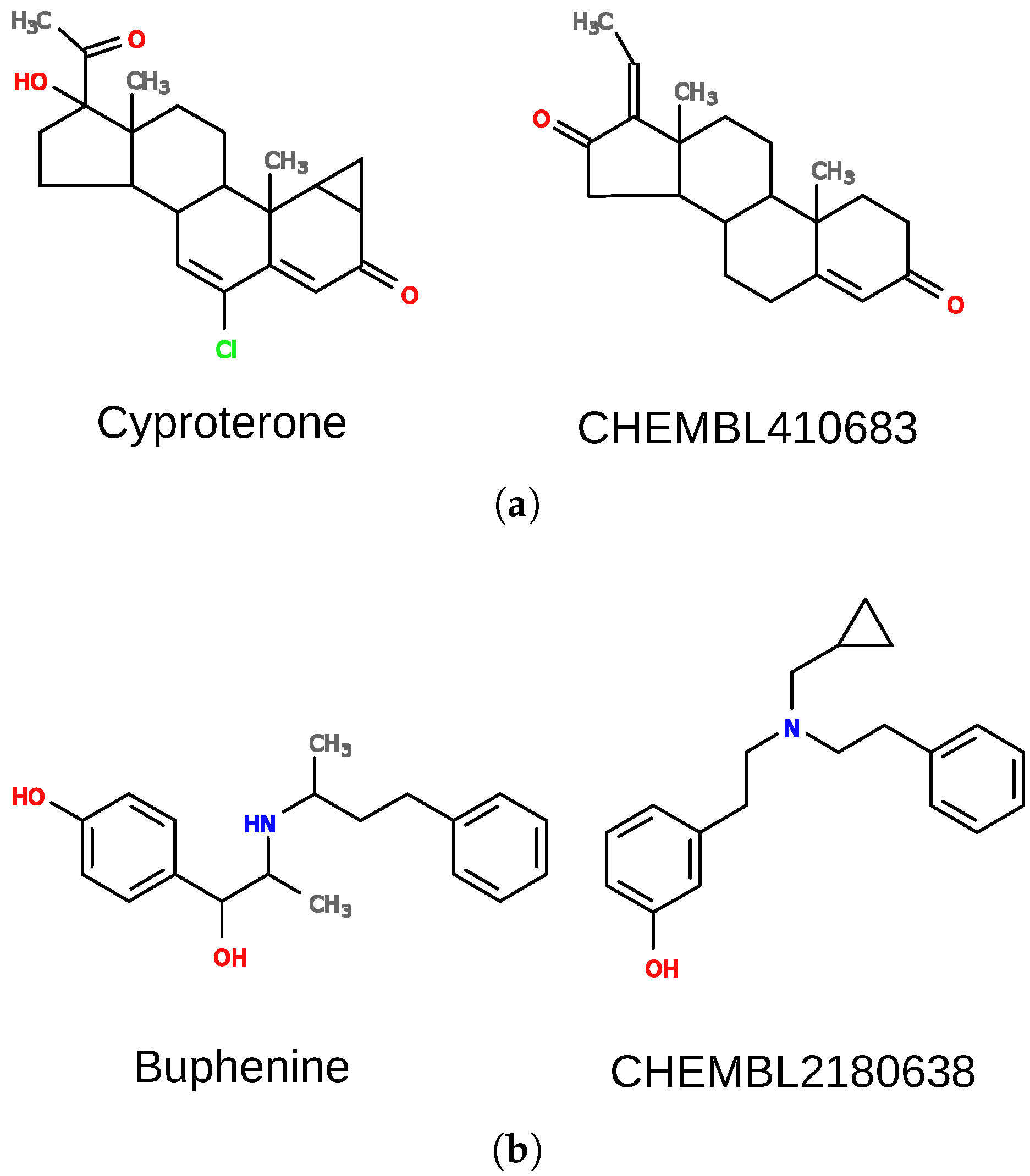
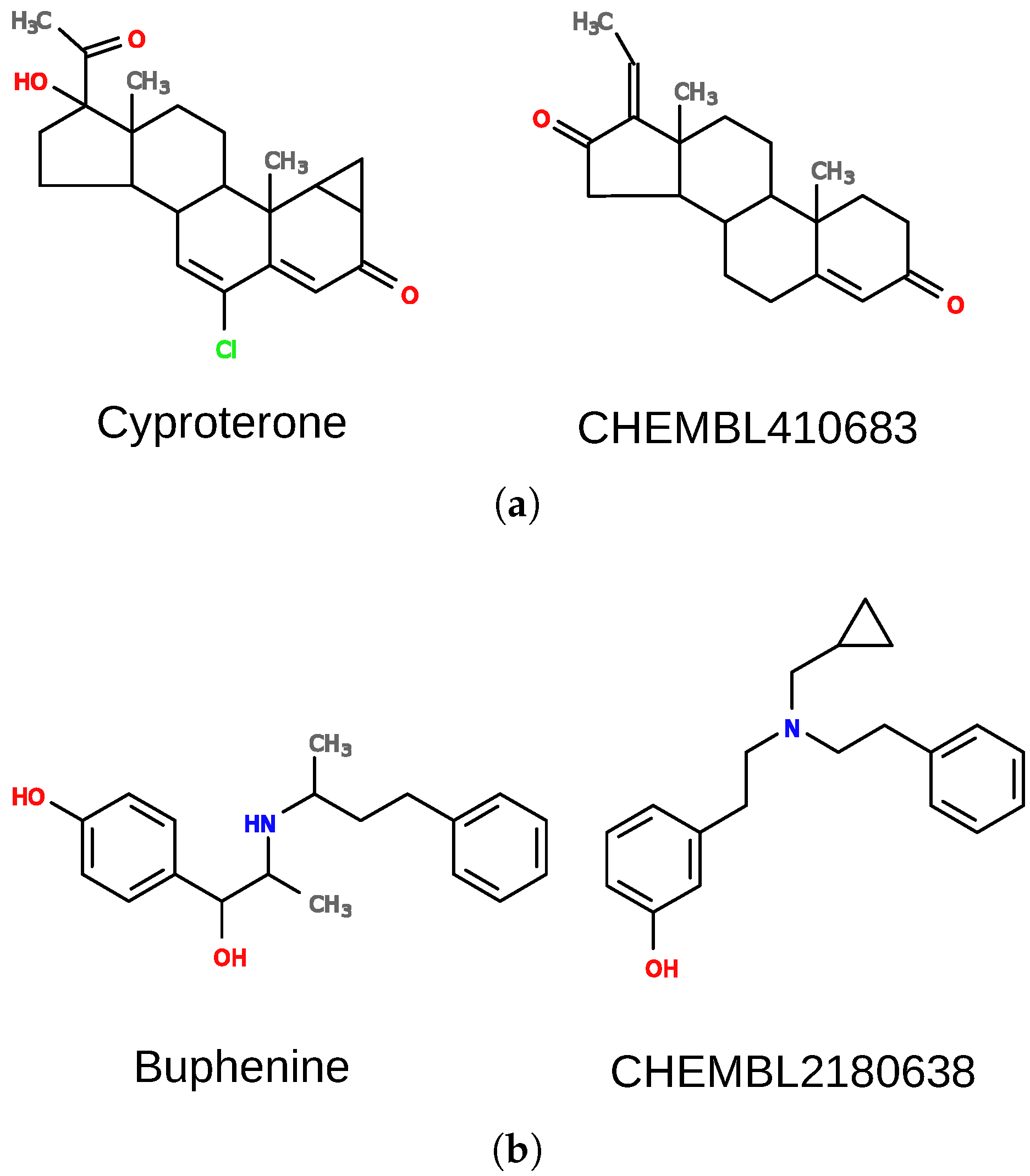
The first case is a false positive result for pregnane X receptor (nuclear receptor subfamily 1 group I member 2; NR1I2). There is no evidence that cyproterone (
Figure 6a) interacts with the receptor, yet it was predicted active, with high confidence (
E-value of
). Cyproterone shares a steroid scaffold with several pregnane X receptor ligands (e.g., CHEMBL410683), which is the biggest part of the molecule and dominates other features. There are, however, additional elements in the structure of cyproterone that probably prevent its interaction with the receptor, and they should also be important for the overall score. For such cases, the weights of some elements of the model can be changed to adjust their importance. When weights of nodes forming the ring system (with feature “R”) were decreased by a factor of 10 and other weights were increased by a factor of 10, the similarity of the two compounds dropped to 0.62.
Importantly, weights’ adjustment, as seen in the above example, may also be used in scaffold-hopping campaigns. In such cases, one would decrease the weight of specified scaffold atoms, and/or increase weights of ligands’ external pharmacophore groups responsible for target interactions. It is also possible to adjust the distance matching parameter, as shown below.
Another example is a case in which DeCAF fails to find true positives. Buphenine (
Figure 6b) is clearly similar to certain ligands of the kappa-type opioid receptor (OPRK1), yet it obtains an extremely poor
E-value of
for interaction with this receptor. DeCAF returns low similarity scores for buphenine and other ligands of the kappa-type opioid receptor due to different distances between the two rings in their structures. In such a case, it would be beneficial to use a less stringent distance matching value while aligning these compounds. In order to change this default behavior,
dist_tol parameter should be set to a value higher than 0. When
dist_tol was set to 1.0, it was possible to match both rings in the structures, and obtain a similarity score of 0.73 for the molecules showed in
Figure 6b. As a result, we were able to predict buphenine interaction with the receptor with an
E-value =
. Please note that modifying this parameter can be beneficial for specific receptors as they can obviously differ in ligand promiscuity and specificity. However, changing them globally for all receptors may result in higher false positives rates.
5. Conclusions
The results presented in this work demonstrate that DeCAF performance in a ligand-based screening benchmark was comparable or in most cases even better than the tested fingerprints and USRCAT.
When describing a molecule, DeCAF emphasizes ligand physicochemical properties and their relative arrangements. Such a model, by capturing the spatial relationships needed for receptor interaction, should allow for the identification of molecules with different structures but similar interaction patterns. Furthermore, DeCAF’s models and alignment procedure can be further adjusted and tailored for a particular research problem, dealing with specific ligand groups independently, as seen with the examples in the
Section 4. To our knowledge, it is a unique and highly needed feature, which allows researchers to incorporate expert knowledge and gain insight into the computational model.
It is worth emphasizing that other applications besides this presented in this work are also possible. DeCAF can be used to create complex pharmacophore models by combining multiple chemical structures. Such models can also be adjusted to differentiate between crucial and insignificant features. Last but not least, DeCAF was implemented as a free, open-source Python package freely available at
http://bitbucket.org/marta-sd/decaf. We also provide all data used in our study and Python scripts used to create and analyze them as a git repository at
http://bitbucket.org/marta-sd/decaf-supplementary.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}