Labradorins with Antibacterial Activity Produced by Pseudomonas sp.

Abstract
:1. Introduction
2. Results and Discussion
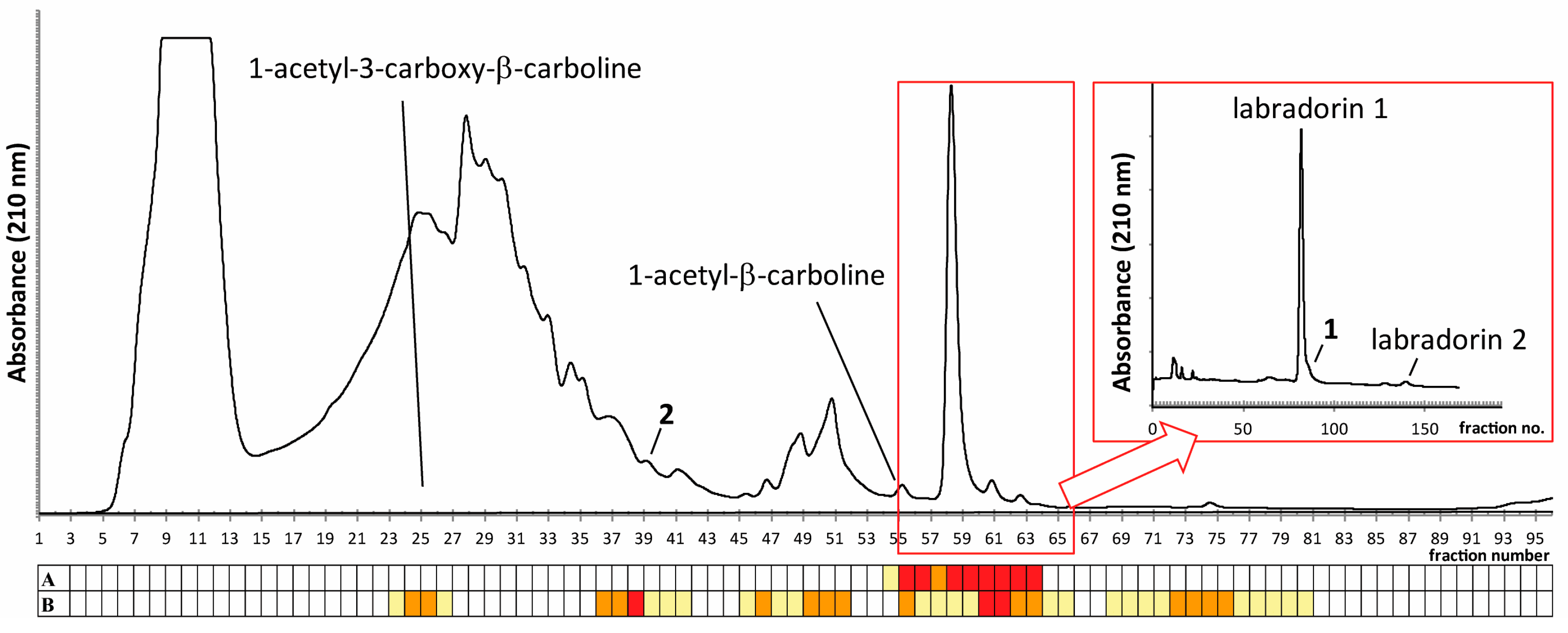
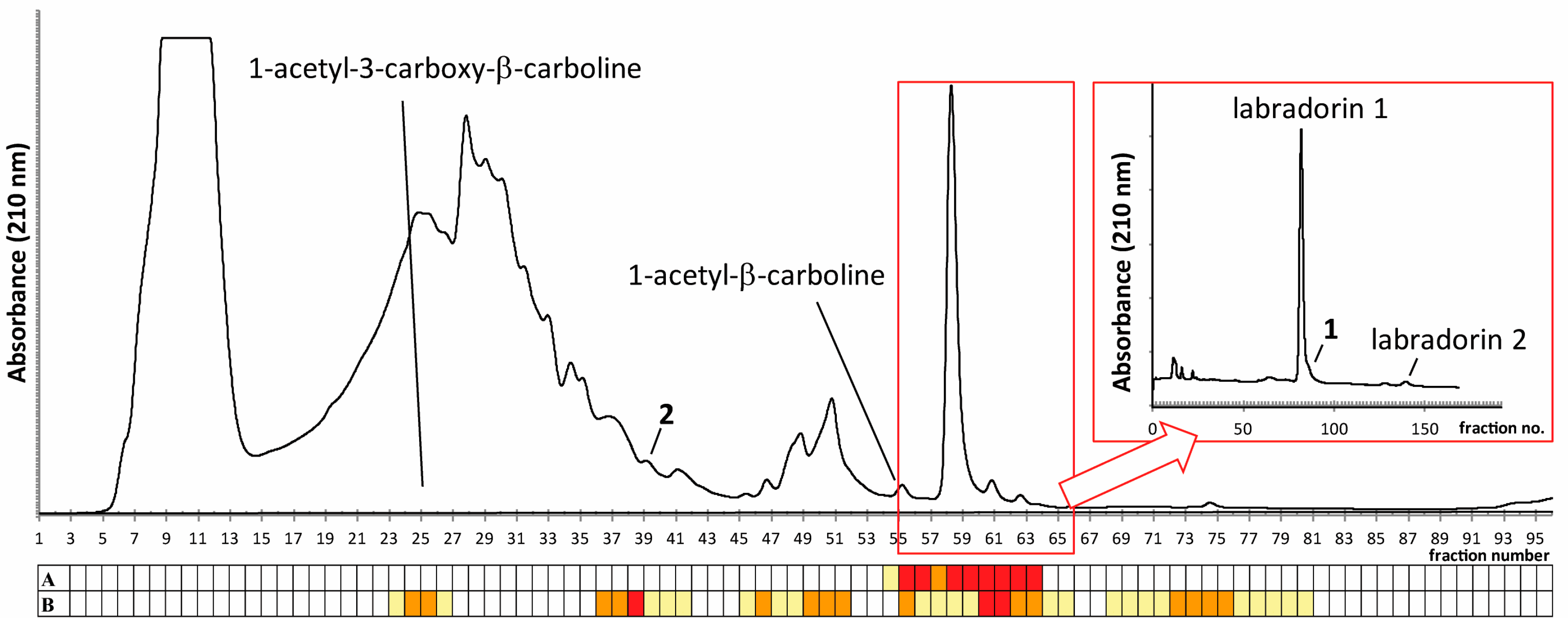
2.1. Isolation of Compounds
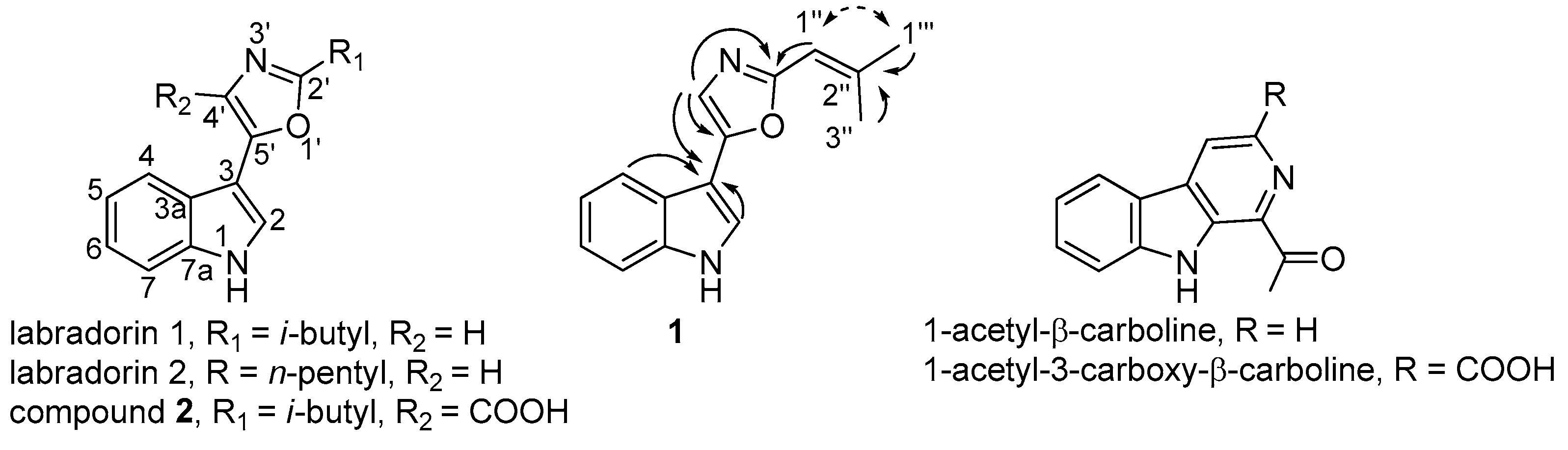
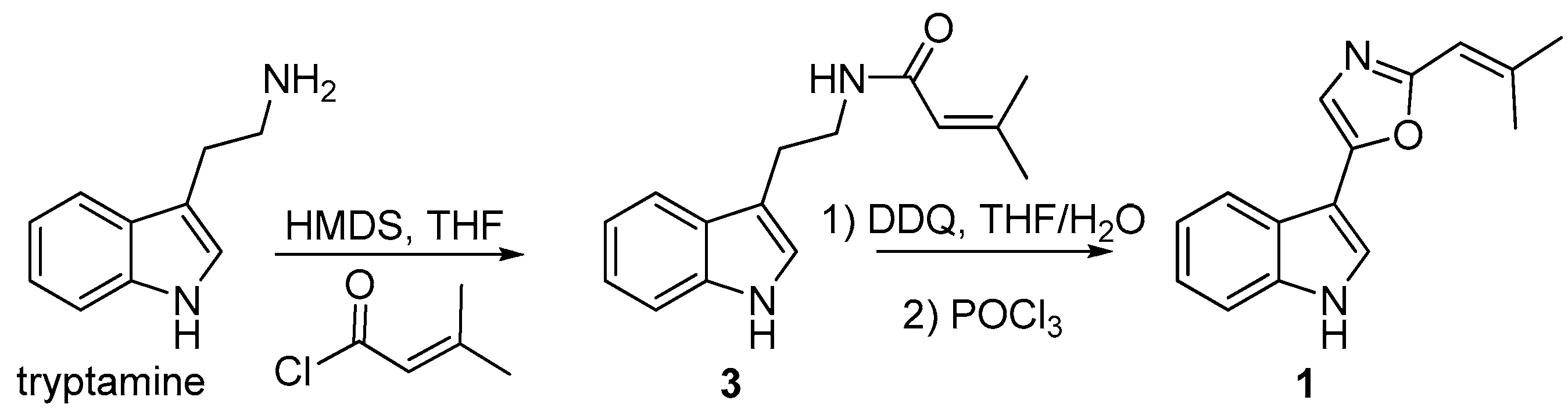
2.2. Identification of Compound 1
2.3. Identification of Compound 2
2.4. Identification of Known β-Carbolines
2.5. Biological Activities of Compounds 1 and 2
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Isolate Origin, Identity and Maintenance
3.3. Culture Conditions and Metabolite Sampling
3.4. Sample Work-Up and Isolation Procedures
3.5. In Vitro Bioassay
3.6. Analysis by LC-HRMS
3.7. MIC Determination
3.8. Toxicity Determination
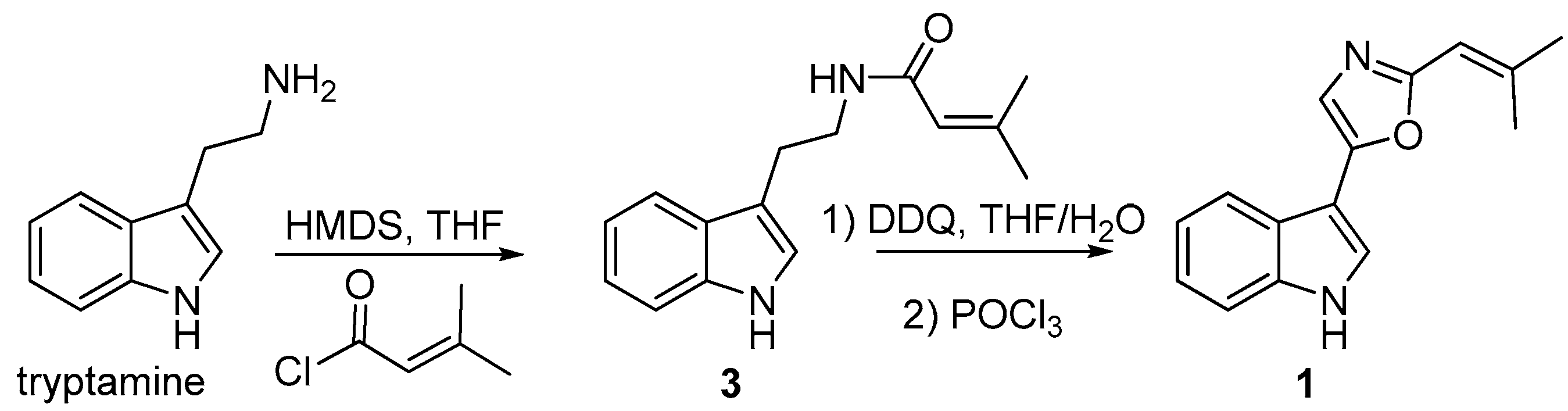
3.9. Synthesis of Compound 1
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Global Action Plan on Antimicrobial Resistance. Available online: http://www.who.int/drugresistance/global_action_plan/en/ (accessed on 20 March 2017).
- Butler, M.S.; Buss, A.D. Natural products-The future scaffolds for novel antibiotics? Biochem. Pharmacol. 2006, 71, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Jones, O.A.H.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.L.; Wishart, D.S. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, B.O.; Van Lanen, S.G.; Baltz, R.H. Microbial genome mining for accelerated natural products discovery: Is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 2014, 41, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes-a review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput In Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Hökeberg, M.; Gerhardson, B.; Johnsson, L. Biological control of cereal seed-borne diseases by seed bacterization with greenhouse-selected bacteria. Eur. J. Plant Pathol. 1997, 103, 25–33. [Google Scholar] [CrossRef]
- Johansson, P.M.; Wright, S.A.I. Low-Temperature Isolation of Disease-Suppressive Bacteria and Characterization of a Distinctive Group of Pseudomonads. Appl. Environ. Microbiol. 2003, 69, 6464–6474. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Knight, J.C.; Herald, D.L.; Davenport, R.; Pettit, R.K.; Tucker, B.E.; Schmidt, J.M. Isolation of Labradorins 1 and 2 from Pseudomonas syringae pv. coronafaciens. J. Nat. Prod. 2002, 65, 1793–1797. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, F.; Dill, V.; Dowling, A.; Thanwisai, A.; Bode, E.; Chantratita, N.; ffrench-Constant, R.; Bode, H.B. Identification and isolation of insecticidal oxazoles from Pseudomonas sp. Beilstein. J. Org. Chem. 2012, 8, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Omer, Z.S.; Tombolini, R.; Gerhardson, B. Plant colonization by pink-pigmented facultative methylotrophic bacteria (PPFMs). FEMS Microbiol. Ecol. 2004, 47, 319–326. [Google Scholar] [CrossRef]
- Joshi, B.S.; Kamat, V.N.; Gawad, D.H. Some β-Carboline Alkaloids of Ailanthus malabarica DC. Heterocycles 1977, 7, 193–200. [Google Scholar] [CrossRef]
- Faini, F.; Torres, R.; Delle Monache, F.; Marini-Bettólo, G.B.; Castillo, M. 1-Acetyl-3-Carboxy-β-Carboline, a New Acid and Other Constituents of Vestia Lycioides. Planta Med. 1980, 38, 128–132. [Google Scholar] [CrossRef]
- Faini, F.; Castillo, M.; Torres, R. A new β-carboline alkaloid from Vestia Lycioides. Phytochemistry 1978, 17, 338. [Google Scholar] [CrossRef]
- Cao, L.H.; Zhang, W.; Luo, J.G.; Kong, L.Y. Five New β-Carboline-Type Alkaloids from Stellaria dichotoma var. lanceolata. Helv. Chim. Acta 2012, 95, 1018–1025. [Google Scholar] [CrossRef]
- Zhou, T.S.; Ye, W.C.; Wang, Z.T.; Che, C.T.; Zhou, R.H.; Xu, G.J.; Xu, L.S. β-Carboline alkaloids from Hypodematium squamuloso-pilosum. Phytochemistry 1998, 49, 1807–1809. [Google Scholar] [CrossRef]
- Lan, W.J.; Fu, S.J.; Xu, M.Y.; Liang, W.L.; Lam, C.K.; Zhong, G.H.; Xu, J.; Yang, D.P.; Li, H.J. Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri. Mar. Drugs 2016, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.L.; Cardellina, J.H., II. Aromatic Secondary Metabolites from the Sponge Tedania ignis. J. Nat. Prod. 1991, 54, 1056–1061. [Google Scholar] [CrossRef]
- Proksa, B.; Uhrín, D.; Šurdíková, M.; Fuska, J. 1-Acetyl-β-carboline, a new metabolite of Streptomyces kasugaensis. Acta Biotechnol. 1990, 10, 337–340. [Google Scholar] [CrossRef]
- Levenfors, J.J.; Hedman, R.; Thaning, C.; Gerhardson, B.; Welch, C.J. Broad-spectrum antifungal metabolites produced by the soil bacterium Serratia plymuthica A 153. Soil Biol. Biochem. 2004, 36, 677–685. [Google Scholar] [CrossRef]
- Stanier, R.Y.; Palleroni, N.J.; Doudoroff, M.J. The aerobic pseudomonads: A taxonomic study. Gen. Microbiol. 1966, 43, 159–271. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, A.; Broberg, A.; Johansson, M.; Kenne, L.; Levenfors, J. Pseudotrienic acid A and B, two novel bioactive metabolites from Pseudomonas sp. MF381-IODS. J. Nat. Prod. 2005, 68, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Thaning, C.; Welch, C.J.; Borowicz, J.J.; Hedman, R.; Gerhardson, B. Suppression of Sclerotinia sclerotiorum apothecial formation by the soil bacterium. Serratia plymuthica: Identification of a chlorinated macrolide as one of the causal agents. Soil Biol. Biochem. 2001, 33, 1817–1826. [Google Scholar] [CrossRef]
- Weislow, O.S.; Kiser, R.; Fine, D.L.; Bader, J.; Shoemaker, R.H.; Boyd, M.R.J. New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J. Natl. Cancer Inst. 1989, 81, 577–586, Published erratum in J. Natl. Cancer Inst. 1989, 81, 963. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A.; et al. Design, synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Compound 2 | |||||
|---|---|---|---|---|---|---|
| Pos. | δC | δH | Mult. (Hz) | δC | δH | Mult. (Hz) |
| 1 | - | 8.38 | br. s | - | 8.61 | br. s |
| 2 | 122.3 | 7.56 | d (2.3) | 130.1 | 8.95 | d (2.8) |
| 3 | 105.7 | - | - | 104.0 | - | - |
| 3a | 124.3 | - | - | 124.9 | - | - |
| 4 | 120.3 | 7.86 | d (7.9) | 121.4 | 8.18 | d (7.5) |
| 5 | 121.6 | 7.27 | t (7.9) | 122.1 | 7.30 | t (7.5) |
| 6 | 123.7 | 7.31 | t (7.9) | 123.6 | 7.33 | t (7.5) |
| 7 | 112.1 | 7.46 | d (7.9) | 112.0 | 7.48 | d (7.5) |
| 7a | 136.4 | - | - | 136.0 | - | - |
| 1′ | - | - | - | - | - | - |
| 2′ | 159.6 | - | - | 160.2 | - | - |
| 3′ | - | - | - | - | - | - |
| 4′ | 119.4 | 7.33 | s | 122.2 | - | - |
| 5′ | 146.7 | - | - | 154.2 | - | - |
| 1′′ | 111.3 | 6.31 | br. s | 37.1 | 2.80 | d (7.3) |
| 2′′ | 147.5 | - | - | 27.8 | 2.31 | m |
| 3′′ | 21.3 | 2.32 | s | 22.6 | 1.10 | d (6.6) |
| 1′′′ | 28.1 | 2.04 | s | 22.6 | 1.10 | d (6.6) |
| COOH | - | - | 162.2 | n.d. a | - | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broberg, A.; Bjerketorp, J.; Andersson, P.; Sahlberg, C.; Levenfors, J.J. Labradorins with Antibacterial Activity Produced by Pseudomonas sp. Molecules 2017, 22, 1072. https://doi.org/10.3390/molecules22071072
Broberg A, Bjerketorp J, Andersson P, Sahlberg C, Levenfors JJ. Labradorins with Antibacterial Activity Produced by Pseudomonas sp. Molecules. 2017; 22(7):1072. https://doi.org/10.3390/molecules22071072
Chicago/Turabian StyleBroberg, Anders, Joakim Bjerketorp, Pierre Andersson, Christer Sahlberg, and Jolanta J. Levenfors. 2017. "Labradorins with Antibacterial Activity Produced by Pseudomonas sp." Molecules 22, no. 7: 1072. https://doi.org/10.3390/molecules22071072
APA StyleBroberg, A., Bjerketorp, J., Andersson, P., Sahlberg, C., & Levenfors, J. J. (2017). Labradorins with Antibacterial Activity Produced by Pseudomonas sp. Molecules, 22(7), 1072. https://doi.org/10.3390/molecules22071072