On the Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and O-alkylhydroxylamines: Unexpected Degradation of P-C-P Bridge

 and
and
Abstract
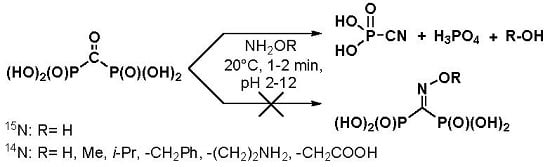
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
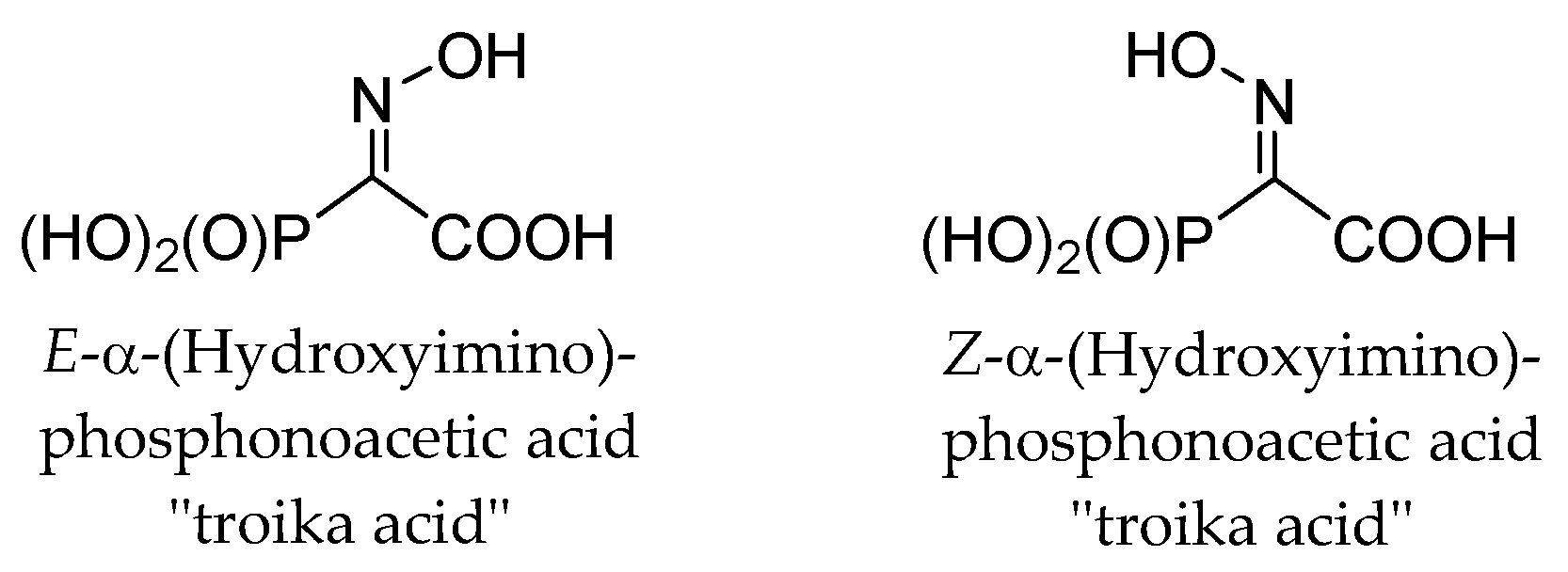{kind=link}
{kind=link}
{kind=link}
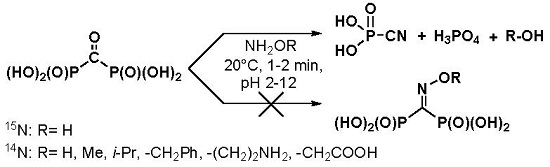
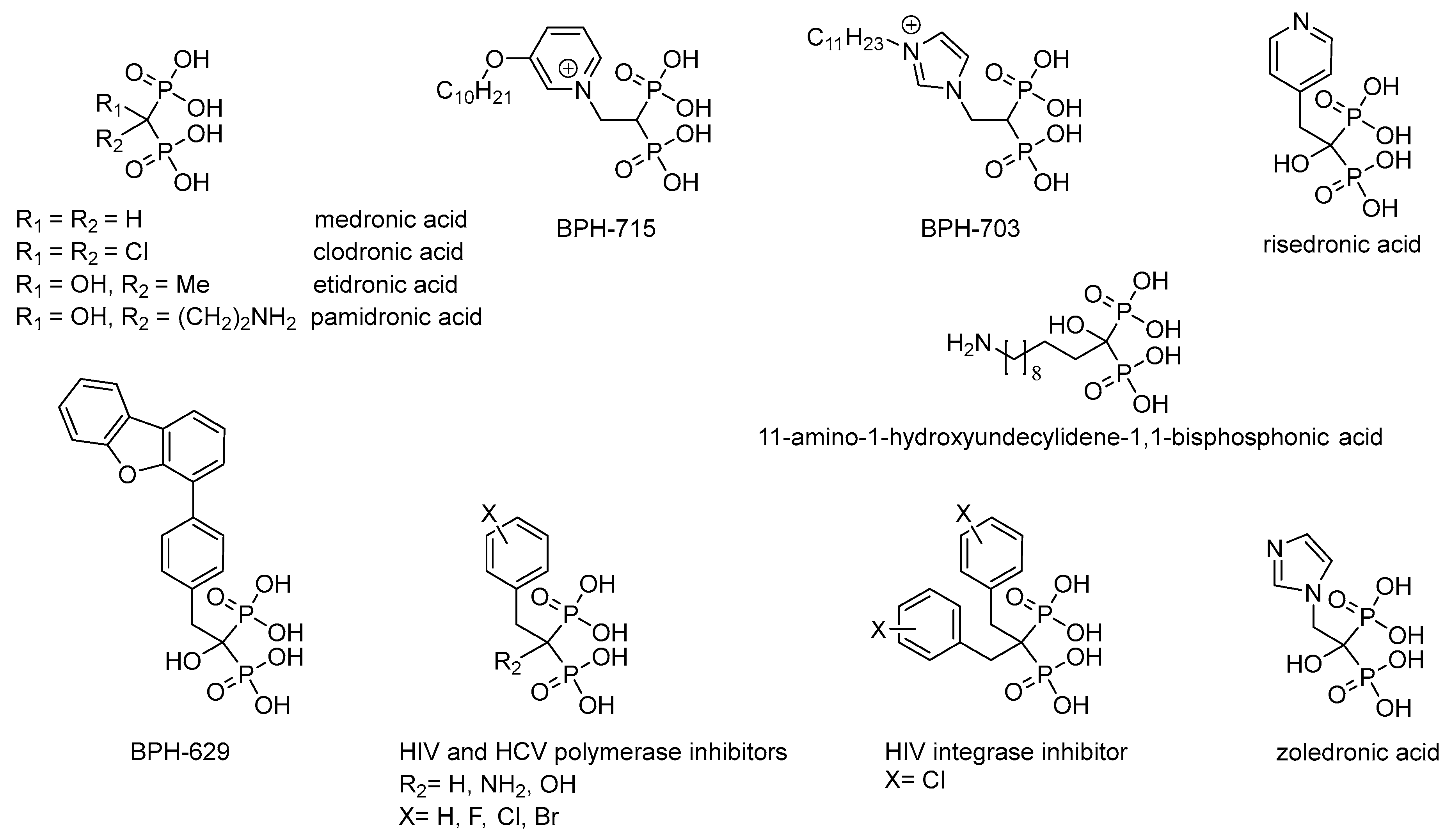
1. Introduction
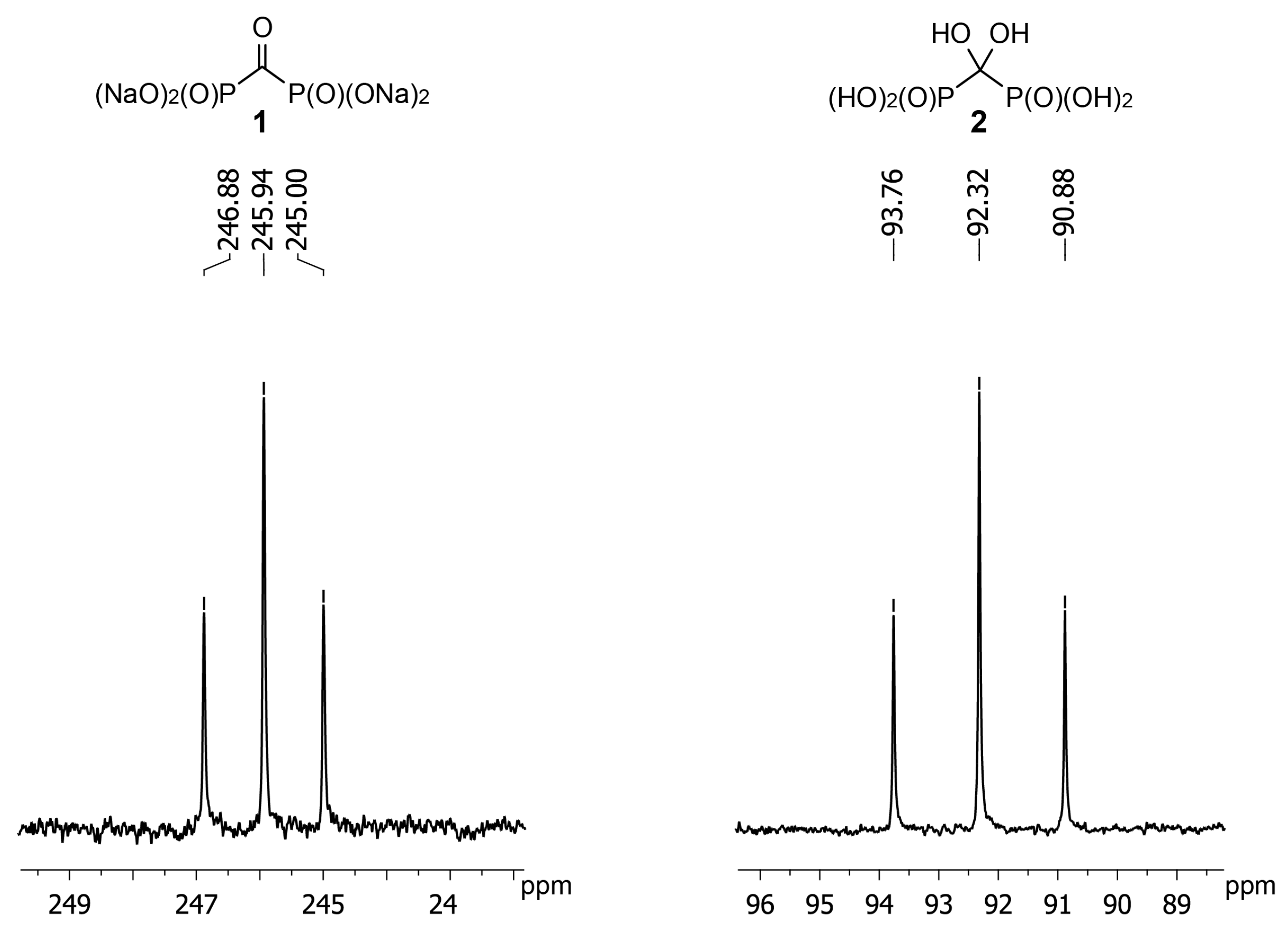
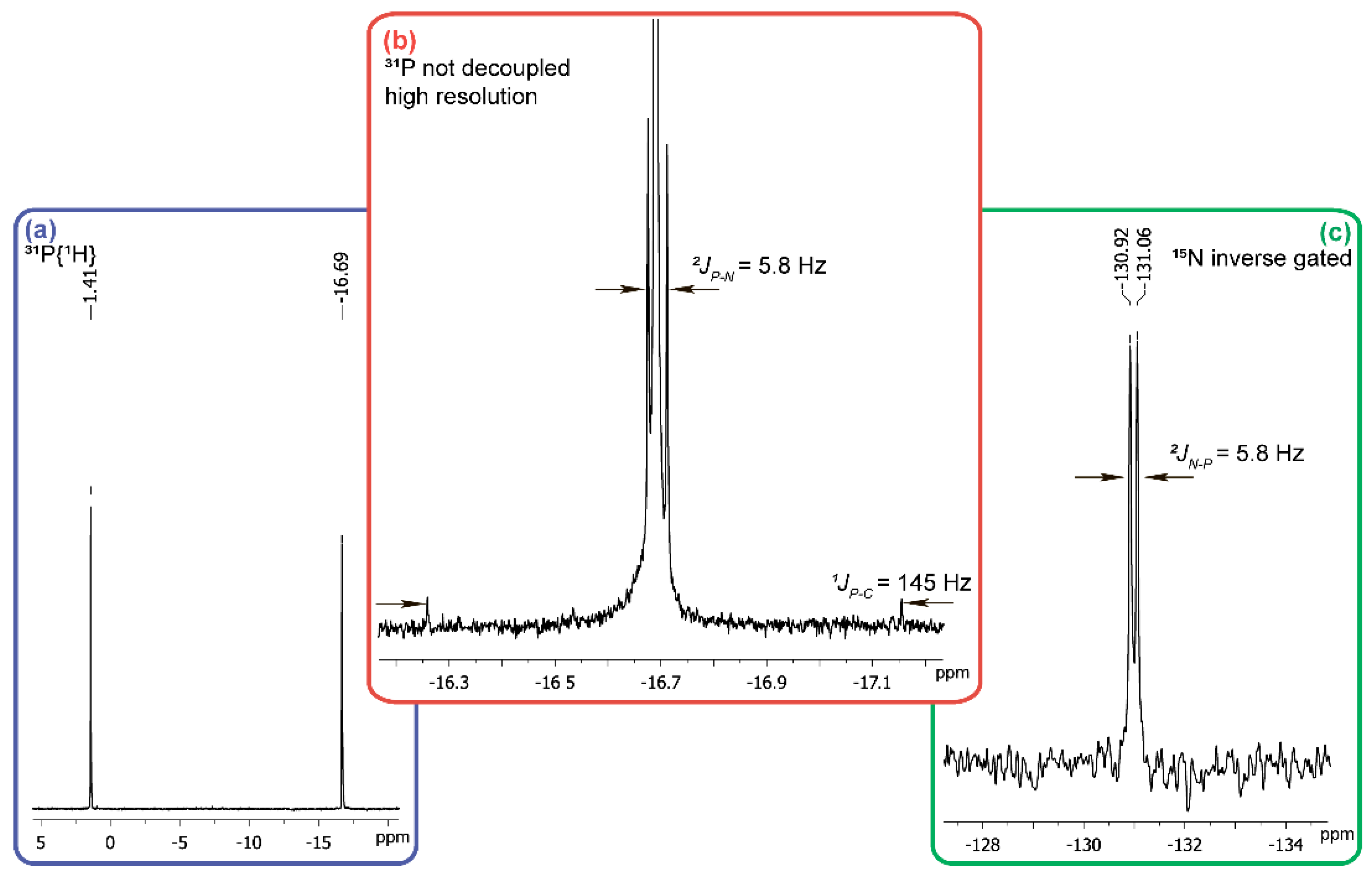
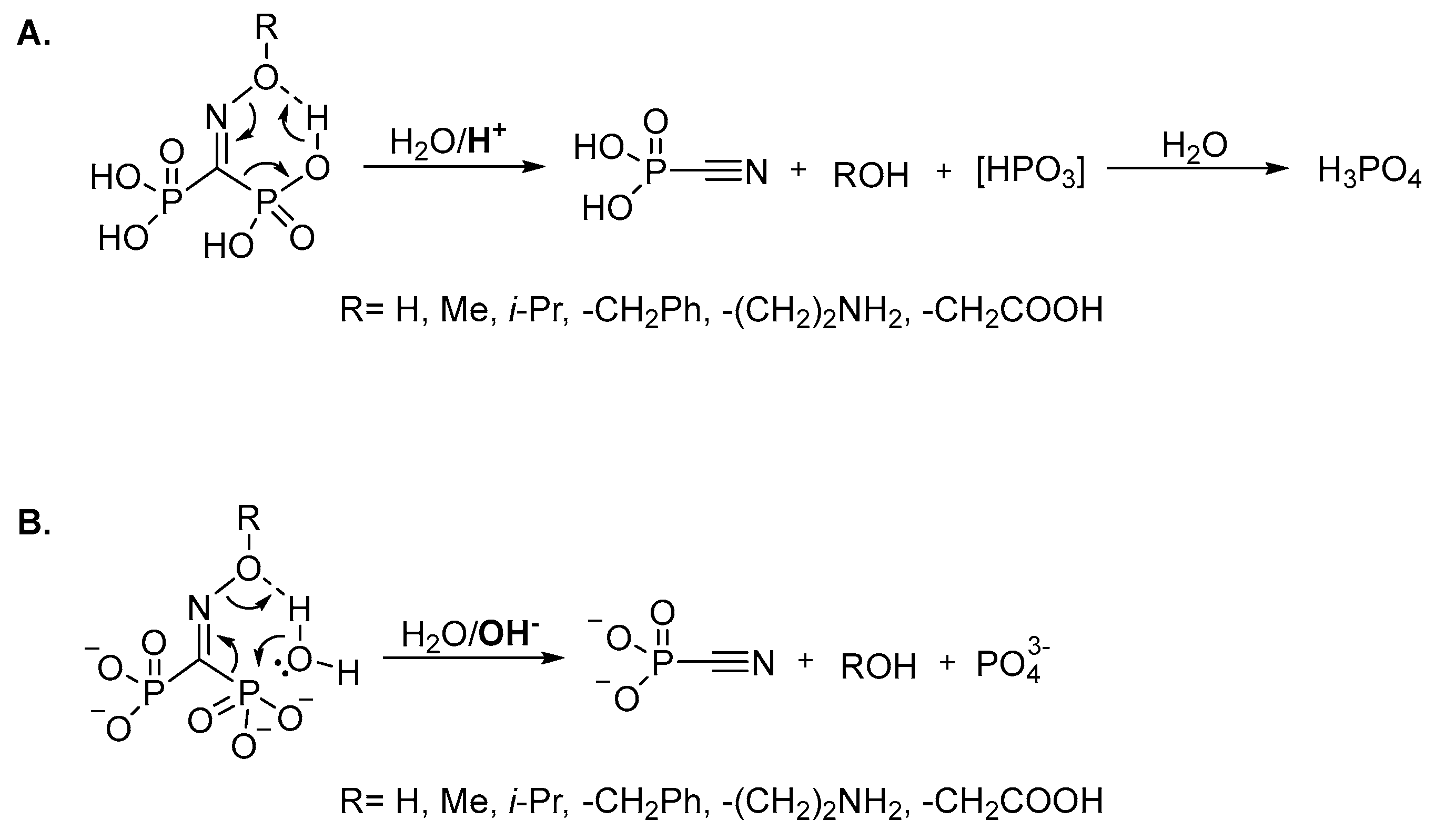
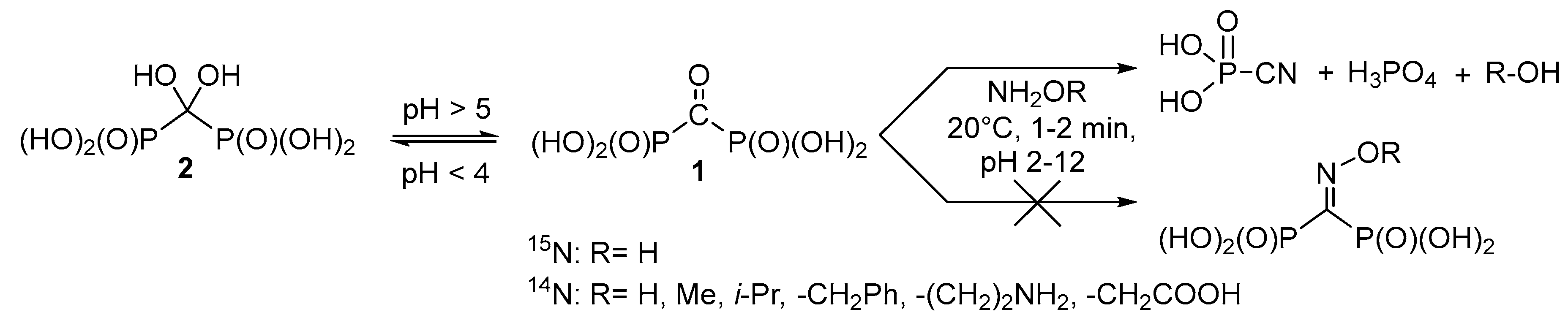
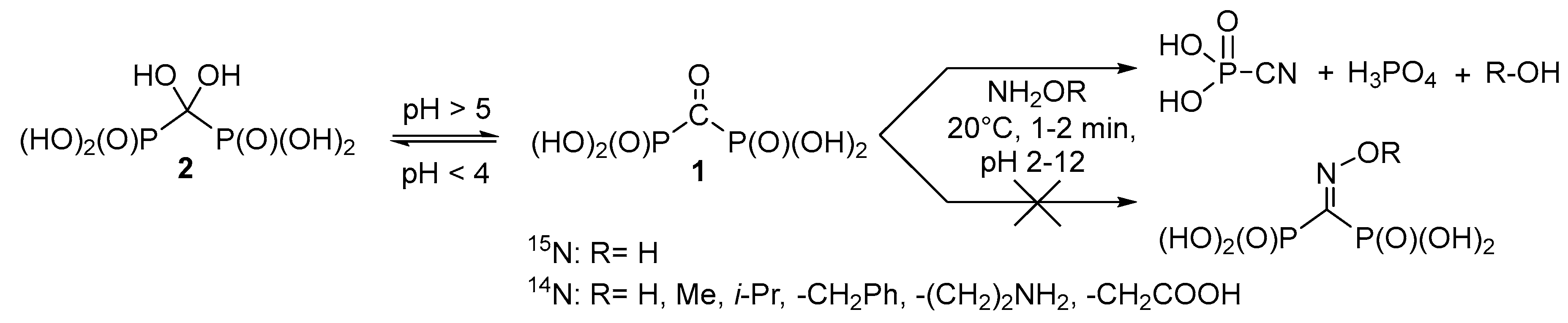
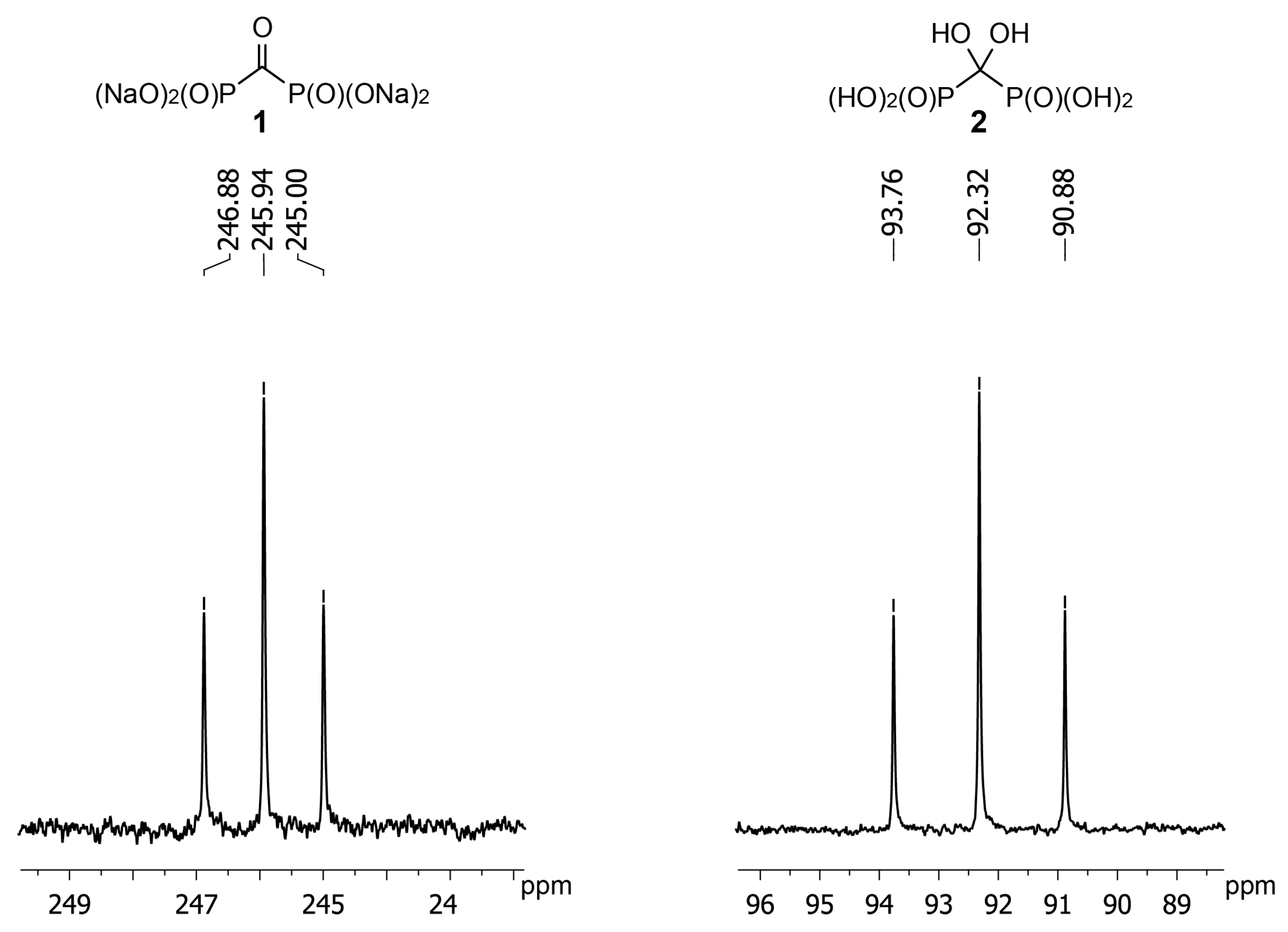
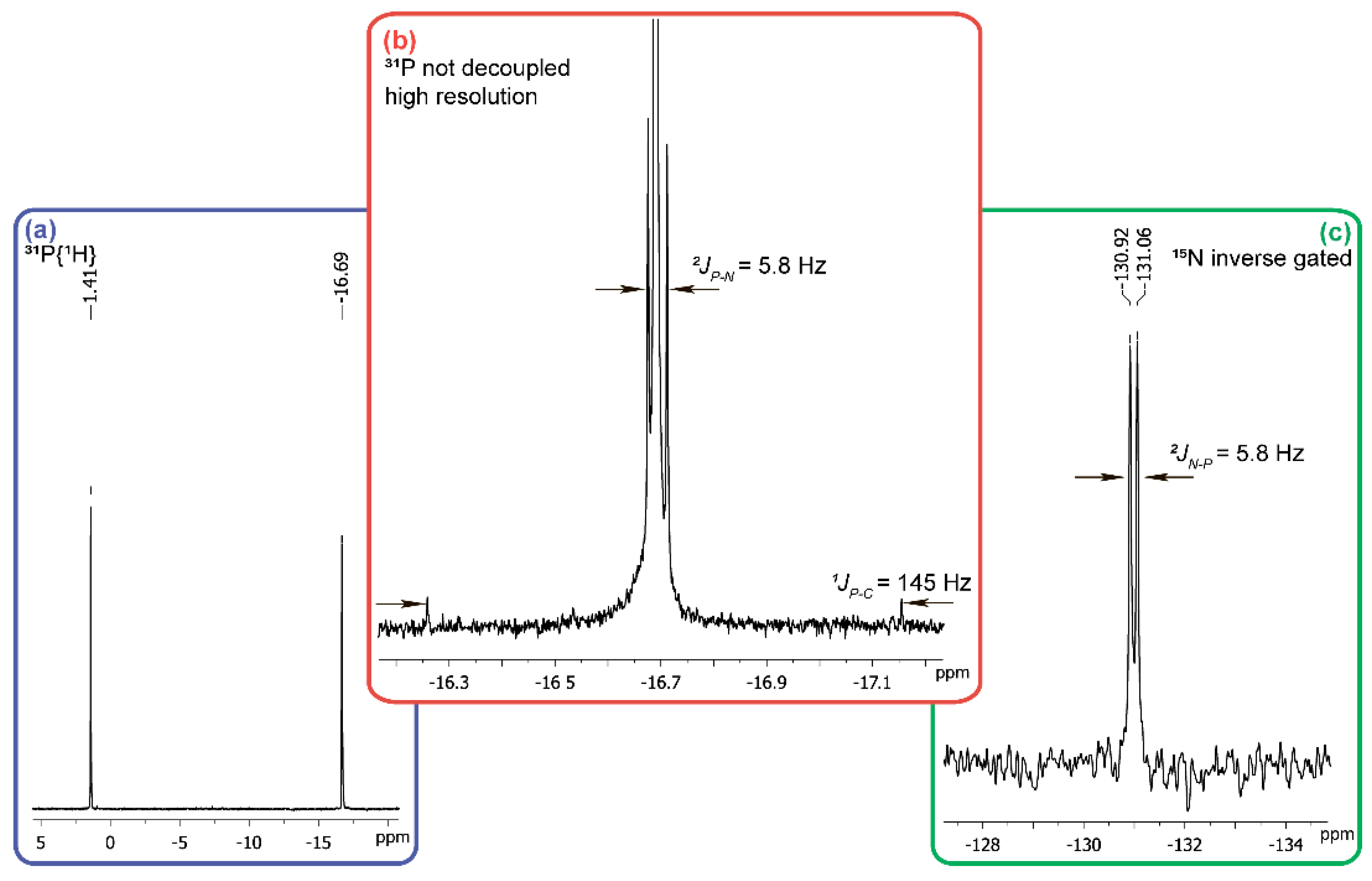
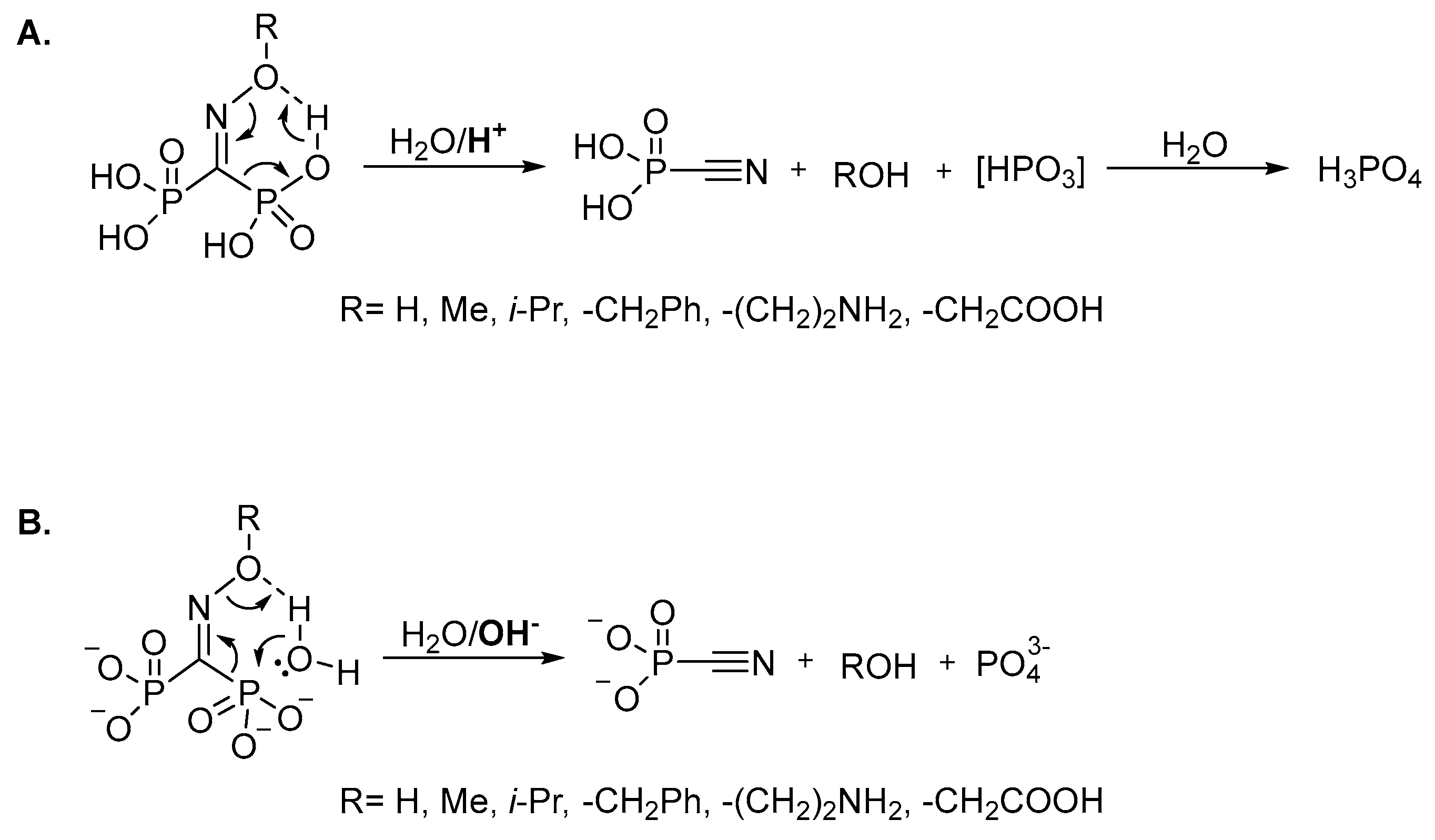
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Tetraethyl 1,1-Dichloromethylene Bisphosphonate
3.2. Synthesis of Carbonyl Diphosphonic Acid (1)
3.3. Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and Its Esters, General Procedure
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Russell, R.G. Bisphosphonates: From bench to bedside. Ann. N. Y. Acad. Sci. 2006, 1068, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Maiuolo, L.; Delso, I.; Algieri, V.; de Nino, A.; Tejero, T. Chemical approaches to inhibitors of isoprenoid biosynthesis: Targeting farnesyl and geranylgeranyl pyrophosphate synthases. RSC Adv. 2017, 7, 10947–10967. [Google Scholar] [CrossRef]
- Russell, R.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Hosking, D.; Lyles, K.; Brown, J.P.; Fraser, W.D.; Miller, P.; Curiel, M.D.; Devogelaer, J.P.; Hooper, M.; Su, G.; Zelenakas, K.; et al. Long-term control of bone turnover in paget’s disease with zoledronic acid and risedronate. J. Bone Miner. Res. 2007, 22, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Saji, H. Advances in drug design of radiometal-based imaging agents for bone disorders. Int. J. Mol. Imaging 2011, 2011, 537687. [Google Scholar] [CrossRef] [PubMed]
- Turhanen, P.A.; Vepsalainen, J.J.; Peraniemi, S. Advanced material and approach for metal ions removal from aqueous solutions. Sci. Rep. 2015, 5, 8992. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Lipton, A. Antitumor effects and anticancer applications of bisphosphonates. Semin. Oncol. 2010, 37, S30–S40. [Google Scholar] [CrossRef] [PubMed]
- No, J.H.; de Macedo Dossin, F.; Zhang, Y.; Liu, Y.L.; Zhu, W.; Feng, X.; Yoo, J.A.; Lee, E.; Wang, K.; Hui, R.; et al. Lipophilic analogs of zoledronate and risedronate inhibit plasmodium geranylgeranyl diphosphate synthase (ggpps) and exhibit potent antimalarial activity. Proc. Natl. Acad. Sci. USA 2012, 109, 4058–4063. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.; Feng, X.; Ko, T.P.; Huang, C.H.; Hu, Y.; Zheng, Y.; Bogue, S.; Nakano, C.; Hoshino, T.; Zhang, L.; et al. Structure and inhibition of tuberculosinol synthase and decaprenyl diphosphate synthase from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2014, 136, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, E.; Burzynska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, L.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Yanvarev, D.V.; Korovina, A.N.; Usanov, N.N.; Khomich, O.A.; Vepsalainen, J.; Puljula, E.; Kukhanova, M.K.; Kochetkov, S.N. Methylene bisphosphonates as the inhibitors of HIV RT phosphorolytic activity. Biochimie 2016, 127, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Agapkina, J.; Yanvarev, D.; Anisenko, A.; Korolev, S.; Vepsalainen, J.; Kochetkov, S.; Gottikh, M. Specific features of HIV-1 integrase inhibition by bisphosphonate derivatives. Eur. J. Med. Chem. 2014, 73, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yanvarev, D.V.; Korovina, A.N.; Usanov, N.N.; Kochetkov, S.N. Non-hydrolysable analogues of inorganic pyrophosphate as inhibitors of hepatitis C virus RNA-dependent RNA-polymerase. Russ. J. Bioorg. Chem. 2012, 38, 224–229. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Cao, R.; Yin, F.; Hudock, M.P.; Guo, R.T.; Krysiak, K.; Mukherjee, S.; Gao, Y.G.; Robinson, H.; Song, Y.; et al. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: An X-ray and NMR investigation. J. Am. Chem. Soc. 2009, 131, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- Lacbay, C.M.; Mancuso, J.; Lin, Y.S.; Bennett, N.; Gotte, M.; Tsantrizos, Y.S. Modular assembly of purine-like bisphosphonates as inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2014, 57, 7435–7449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Reilly, J.E.; Loerch, K.A.; Hohl, R.J.; Wiemer, D.F. Synthesis of isoprenoid bisphosphonate ethers through c-p bond formations: Potential inhibitors of geranylgeranyl diphosphate synthase. Beilstein J. Org. Chem. 2014, 10, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Grün, A.; Garadnay, S.; Greiner, I.; Keglevich, G. Synthesis of hydroxymethylenebisphosphonic acid derivatives in different solvents. Molecules 2016, 21, 1046. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, E.; Kafarski, P. Physiologic activity of bisphosphonates—Recent advances. Open Pharm. Sci. J. 2016, 3, 56–78. [Google Scholar] [CrossRef]
- Demkowicz, S.; Rachon, J.; Daśko, M.; Kozak, W. Selected organophosphorus compounds with biological activity. Applications in medicine. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Abdou, W.M.; Ganoub, N.A.; Fahmy, A.F.; Shaddy, A.A. Symmetrical and asymmetrical bisphosphonate esters. Synthesis, selective hydrolysis, and isomerization. Monatsh. Chem. 2006, 137, 105–116. [Google Scholar] [CrossRef]
- Mckenna, C.E.; Higa, M.T.; Cheung, N.H.; Mckenna, M.C. Facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 1977, 155–158. [Google Scholar] [CrossRef]
- McKenna, C.E.; Kashemirov, B.A. Recent progress in carbonylphosphonate chemistry. Top. Curr. Chem. 2002, 220, 201–238. [Google Scholar]
- Quimby, O.T.; Prentice, J.B.; Nicholson, D.A. Tetrasodium carbonyldiphosphonate. Synthesis, reactions, and spectral properties. J. Org. Chem. 1967, 32, 4111–4114. [Google Scholar] [CrossRef]
- Mullins, N.D.; Maguire, N.M.; Ford, A.; Das, K.; Arnold, E.; Balzarini, J.; Maguire, A.R. Exploring the role of the alpha-carboxyphosphonate moiety in the HIV-RT activity of alpha-carboxy nucleoside phosphonates. Org. Biomol. Chem. 2016, 14, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Kashemirov, B.A.; Ju, J.Y.; Bau, R.; Mckenna, C.E. “Troika acids”: Synthesis, structure, and fragmentation pathways of novel alpha-(hydroxyimino)phosphonoacetic acids. J. Am. Chem. Soc. 1995, 117, 7285–7286. [Google Scholar] [CrossRef]
- Breuer, E. Fragmentations and rearrangements of alpha-hydroxyiminoalkylphosphonates. Curr. Org. Chem. 2006, 10, 2357–2370. [Google Scholar] [CrossRef]
- Dirksen, A.; Dawson, P.E. Rapid oxime and hydrazone ligations with aromatic aldehydes for biomolecular labeling. Bioconj. Chem. 2008, 19, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. Catalysis in Chemistry and Enzymology; Dover Publications, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Khomutov, A.R.; Vepsalainen, J.J.; Shvetsov, A.S.; Hyvonen, T.; Keinanen, T.A.; Pustobaev, V.N.; Eloranta, T.O.; Khomutov, R.M. Synthesis of hydroxylamine analogues of polyamines. Tetrahedron 1996, 52, 13751–13766. [Google Scholar] [CrossRef]
- Khomutov, M.A.; Mandal, S.; Weisell, J.; Saxena, N.; Simonian, A.R.; Vepsalainen, J.; Madhubala, R.; Kochetkov, S.N. Novel convenient synthesis of biologically active esters of hydroxylamine. Amino Acids 2010, 38, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Ilia, G.; Iliescu, S.; Macarie, L.; Popa, A. Synthesis of tervalent phosphorus esters in biphasic system using potassium phosphate as unique solid base. Heteroat. Chem. 2008, 19, 360–364. [Google Scholar] [CrossRef]
- Quimby, O.T.; Curry, J.D.; Nicholson, D.A.; Prentice, J.B.; Roy, C.H.J. Metalated methylenediphosphonate esters. Preparation, characterization and synthetic applications. J. Organomet. Chem. 1968, 13, 199–207. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
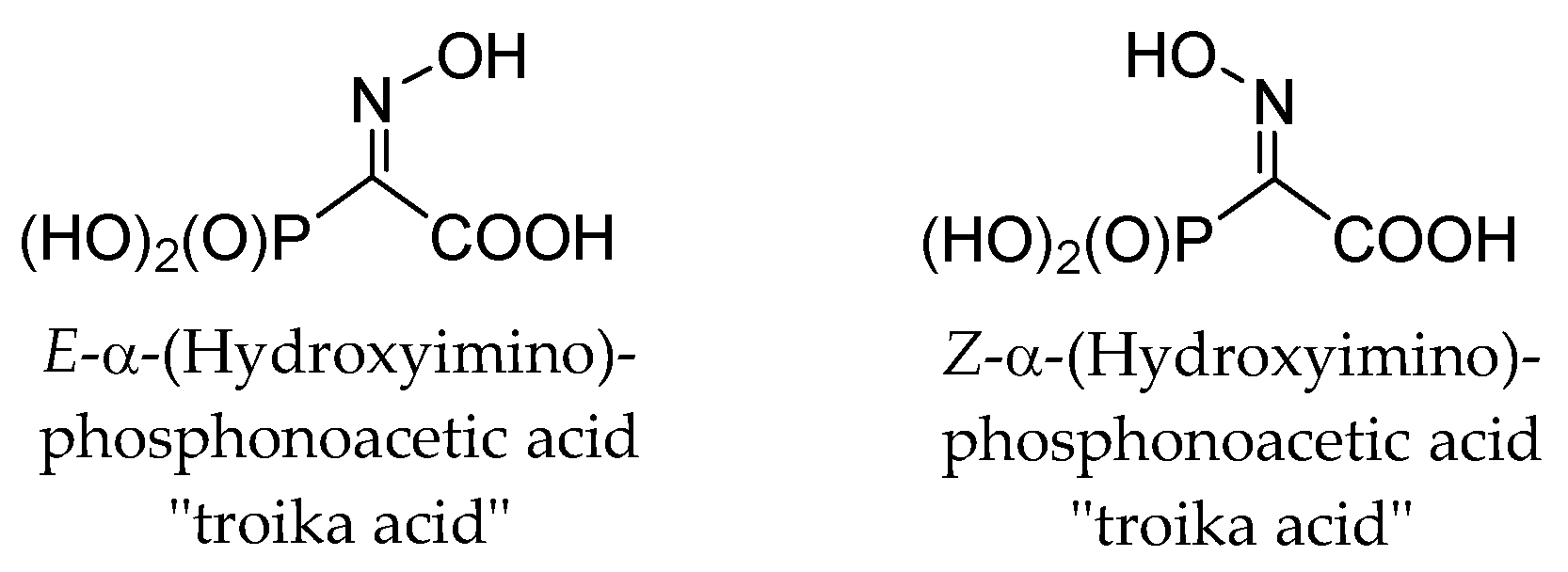
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khomich, O.A.; Yanvarev, D.V.; Novikov, R.A.; Kornev, A.B.; Puljulla, E.; Vepsäläinen, J.; Khomutov, A.R.; Kochetkov, S.N. On the Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and O-alkylhydroxylamines: Unexpected Degradation of P-C-P Bridge. Molecules 2017, 22, 1040. https://doi.org/10.3390/molecules22071040
Khomich OA, Yanvarev DV, Novikov RA, Kornev AB, Puljulla E, Vepsäläinen J, Khomutov AR, Kochetkov SN. On the Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and O-alkylhydroxylamines: Unexpected Degradation of P-C-P Bridge. Molecules. 2017; 22(7):1040. https://doi.org/10.3390/molecules22071040
Chicago/Turabian StyleKhomich, Olga A., Dmitry V. Yanvarev, Roman A. Novikov, Alexey B. Kornev, Elina Puljulla, Jouko Vepsäläinen, Alex R. Khomutov, and Sergey N. Kochetkov. 2017. "On the Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and O-alkylhydroxylamines: Unexpected Degradation of P-C-P Bridge" Molecules 22, no. 7: 1040. https://doi.org/10.3390/molecules22071040
APA StyleKhomich, O. A., Yanvarev, D. V., Novikov, R. A., Kornev, A. B., Puljulla, E., Vepsäläinen, J., Khomutov, A. R., & Kochetkov, S. N. (2017). On the Reaction of Carbonyl Diphosphonic Acid with Hydroxylamine and O-alkylhydroxylamines: Unexpected Degradation of P-C-P Bridge. Molecules, 22(7), 1040. https://doi.org/10.3390/molecules22071040