
Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells

Abstract
:1. Introduction
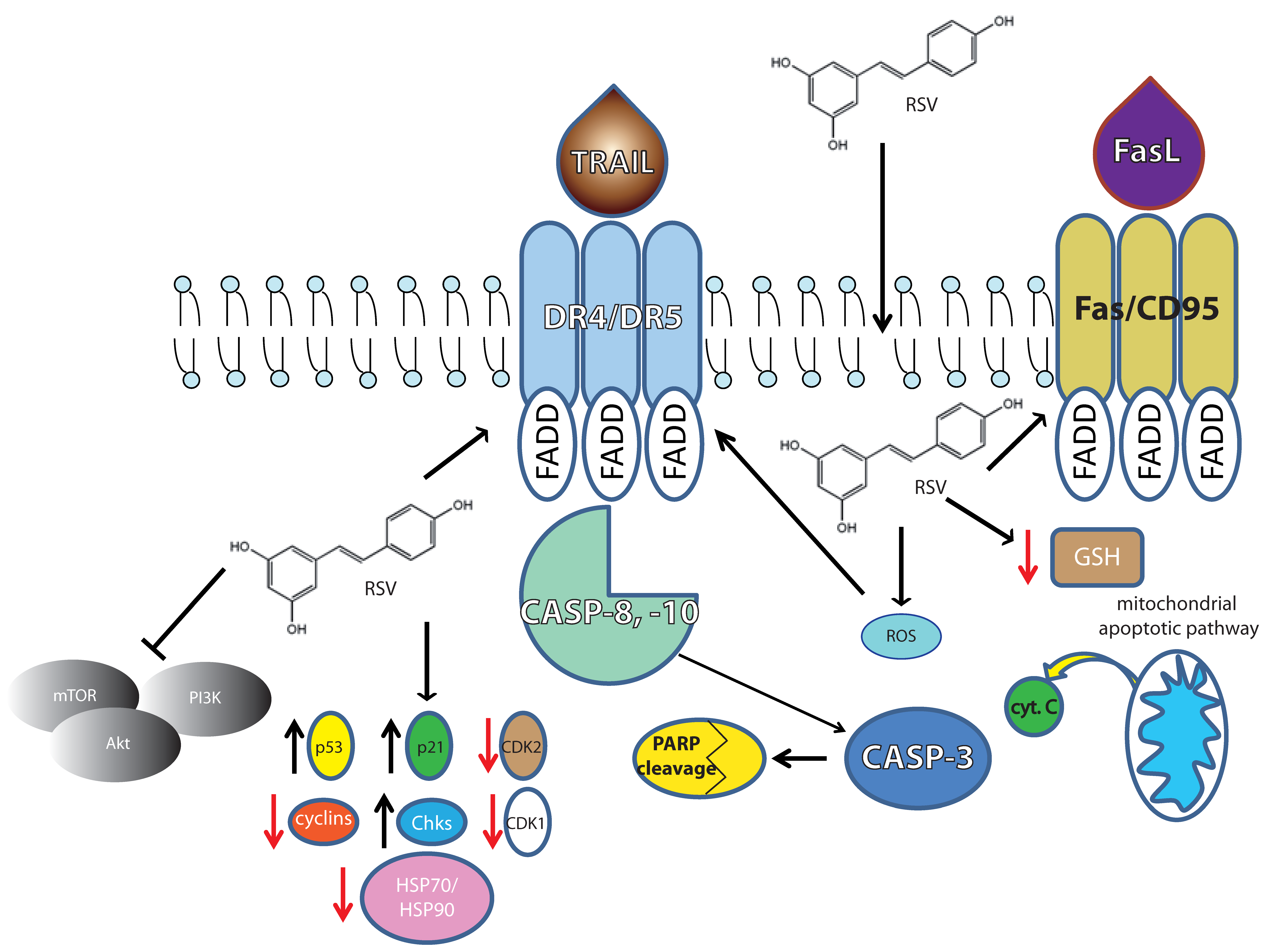
2. Molecular Targets during Apoptosis Onset in Lymphomas
2.1. The Intrinsic Apoptotic Pathway
2.2. The ER Stress
2.3. The Antioxidant and Pro-Oxidant Activities
2.4. Differentiation and Death Receptor Pathway
2.5. EBV+ Burkitt’s Lymphoma
2.6. Immunological Modulation
3. Cell Cycle Progression as a Target in Lymphomas
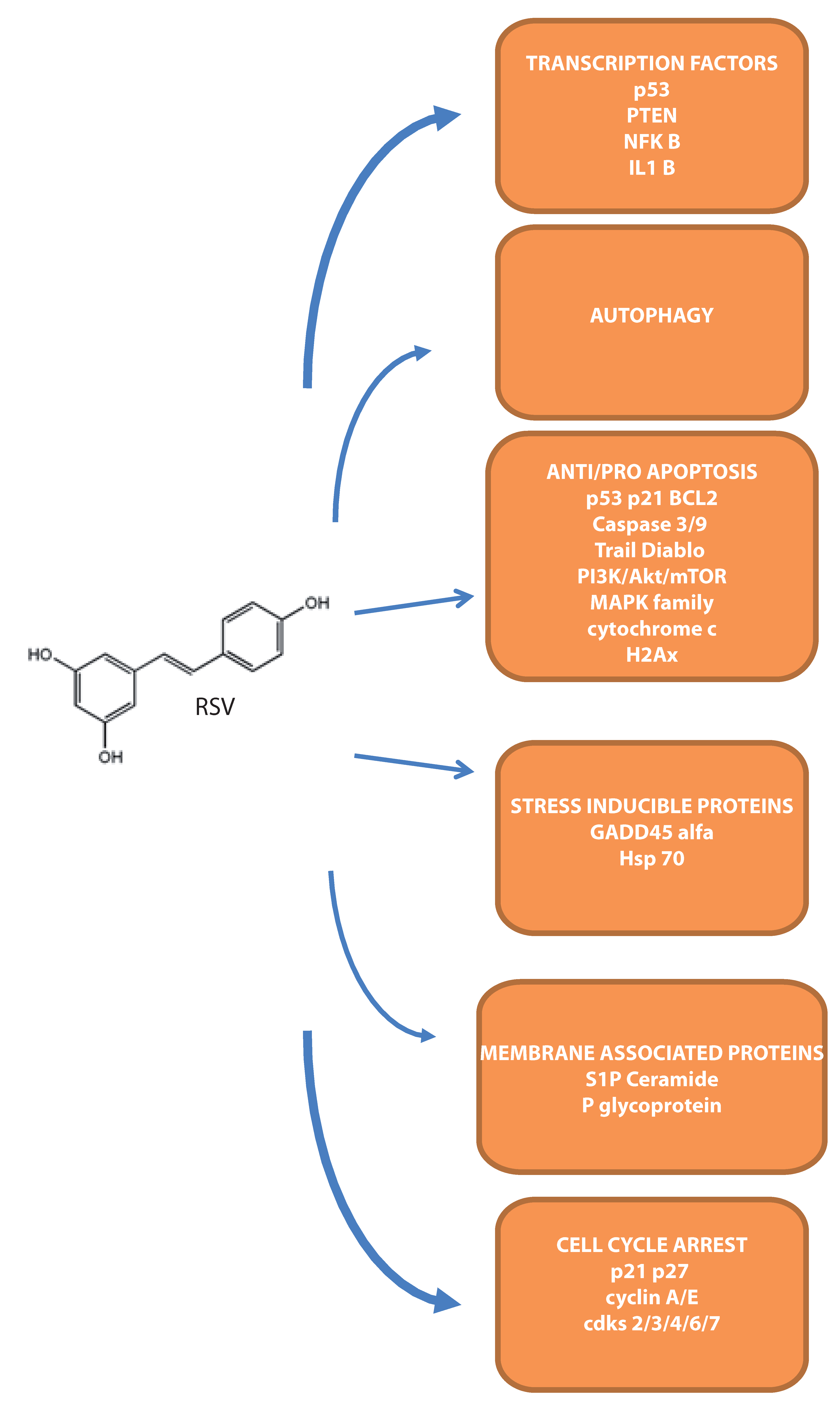
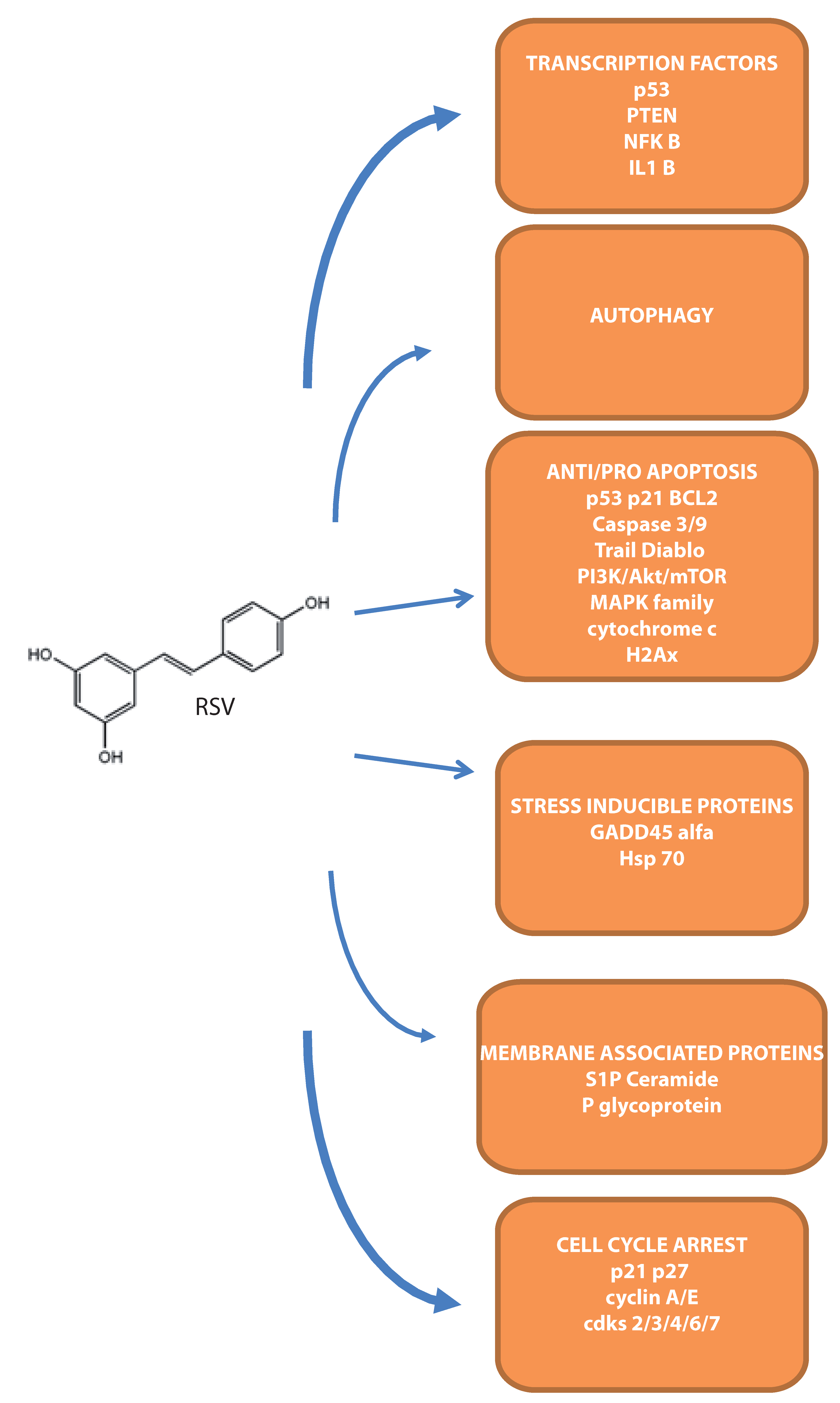
4. Molecular Targets during Apoptosis Onset in Leukemias
4.1. Intracellular Apoptotic Mediators
4.2. Transcription Factors Modulated by RSV
4.3. Stress Inducible Proteins Involved in RSV–Mediated Cell Death
4.4. The Involvement of Membrane-Associated Proteins
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Latruffe, N.; Lancon, A.; Frazzi, R.; Aires, V.; Delmas, D.; Michaille, J.J.; Djouadi, F.; Bastin, J.; Cherkaoui-Malki, M. Exploring new ways of regulation by Resveratrol involving miRNAs, with emphasis on inflammation. Ann. N. Y. Acad. Sci. 2015, 1348, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lancon, A.; Frazzi, R.; Latruffe, N. Anti-Oxidant, Anti-Inflammatory and Anti-Angiogenic Properties of Resveratrol in Ocular Diseases. Molecules 2016, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of Resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Gusman, J.; Malonne, H.; Atassi, G. A reappraisal of the potential chemopreventive and chemotherapeutic properties of Resveratrol. Carcinogenesis 2001, 22, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Mustafi, S.B.; Ganguly, S.; Chatterjee, M.; Raha, S. Resveratrol induces apoptosis in K562 (chronic myelogenous leukemia) cells by targeting a key survival protein, heat shock protein 70. Cancer Sci. 2008, 99, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Grosso, S.; Jacquel, A.; Belhacene, N.; Colosetti, P.; Cassuto, J.P.; Auberger, P. Imatinib mesylate-resistant human chronic myelogenous leukemia cell lines exhibit high sensitivity to the phytoalexin Resveratrol. FASEB J. 2008, 22, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Multiple molecular targets of Resveratrol: Anti-carcinogenic mechanisms. Arch. Biochem. Biophys. 2009, 486, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Frazzi, R.; Valli, R.; Tamagnini, I.; Casali, B.; Latruffe, N.; Merli, F. Resveratrol-mediated apoptosis of hodgkin lymphoma cells involves SIRT1 inhibition and FOXO3a hyperacetylation. Int. J. Cancer 2013, 132, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Frazzi, R.; Tigano, M. The multiple mechanisms of cell death triggered by Resveratrol in lymphoma and leukemia. Int. J. Mol. Sci. 2014, 15, 4977–4993. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Rebe, C.; Micheau, O.; Athias, A.; Gambert, P.; Grazide, S.; Laurent, G.; Latruffe, N.; Solary, E. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of Resveratrol and death receptor ligands in colon carcinoma cells. Oncogene 2004, 23, 8979–8986. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.T.; Yang, S.H.; Chang, T.C.; Changou, C.A.; Lai, H.Y.; Fu, E.; HuangFu, W.C.; Davis, P.J.; Lin, H.Y.; Liu, L.F. Mechanisms of dihydrotestosterone action on Resveratrol-induced anti-proliferation in breast cancer cells with different ERalpha status. Oncotarget 2015, 6, 35866–35879. [Google Scholar] [PubMed]
- Van Ginkel, P.R.; Yan, M.B.; Bhattacharya, S.; Polans, A.S.; Kenealey, J.D. Natural products induce a G protein-mediated calcium pathway activating p53 in cancer cells. Toxicol. Appl. Pharmacol. 2015, 288, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Berger, S.M.; Holz, M.K. Resveratrol as a novel treatment for diseases with mTOR pathway hyperactivation. Ann. N. Y. Acad. Sci. 2015, 1348, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Rebe, C.; Lacour, S.; Filomenko, R.; Athias, A.; Gambert, P.; Cherkaoui-Malki, M.; Jannin, B.; Dubrez-Daloz, L.; Latruffe, N.; et al. Resveratrol-induced apoptosis is associated with Fas redistribution in the rafts and the formation of a death-inducing signaling complex in colon cancer cells. J. Biol. Chem. 2003, 278, 41482–41490. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Chen, G.; Xu, Z.; Yang, G.; Li, B.; Wu, X.; Xiao, W.; Xie, B.; Hu, L.; Sun, X.; et al. Pterostilbene induces apoptosis and cell cycle arrest in diffuse large B-cell lymphoma cells. Sci. Rep. 2016, 6, 37417. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.Y.; Chen, C.Y.; Shyu, H.W.; Hong, S.; Chen, H.M.; Chiou, Y.H.; Lin, K.H.; Chou, M.C.; Wang, L.Y.; Wang, Y.F. Resveratrol induces cell death and inhibits human herpesvirus 8 replication in primary effusion lymphoma cells. Chem. Biol. Interact. 2015, 242, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Al-Rasheed, M.; Manogaran, P.S.; Al-Hussein, K.A.; Platanias, L.C.; al Kuraya, K.; Uddin, S. Curcumin induces apoptosis via inhibition of PI3'-kinase/AKT pathway in acute T cell leukemias. Apoptosis 2006, 11, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Uddin, S.; Bu, R.; Khan, O.S.; Ahmed, S.O.; Ahmed, M.; Al-Kuraya, K.S. Resveratrol suppresses constitutive activation of AKT via generation of ROS and induces apoptosis in diffuse large B cell lymphoma cell lines. PLoS ONE 2011, 6, e24703. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Dufort, F.J.; Blair, D.; Wagner, D.; Roberts, M.F.; Chiles, T.C. Inhibition of phosphatidylinositol 3-kinase-mediated glucose metabolism coincides with Resveratrol-induced cell cycle arrest in human diffuse large B-cell lymphomas. Biochem. Pharmacol. 2006, 72, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Hussain, A.R.; Siraj, A.K.; Manogaran, P.S.; Al-Jomah, N.A.; Moorji, A.; Atizado, V.; Al-Dayel, F.; Belgaumi, A.; El-Solh, H.; et al. Role of phosphatidylinositol 3'-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood 2006, 108, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Gao, Y.Y.; Liu, B.Q.; Niu, X.F.; Zhuang, Y.; Wang, H.Q. Resveratrol-induced cytotoxicity in human Burkitt’s lymphoma cells is coupled to the unfolded protein response. BMC Cancer 2010, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Dey, A.; Sen, R.; Chatterjee, M.; Chattopadhyay, S.; Bandyopadhyay, S.K. Intracellular GSH depletion triggered mitochondrial Bax translocation to accomplish Resveratrol-induced apoptosis in the U937 cell line. J. Pharmacol. Exp. Ther. 2011, 336, 206–214. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Arena, G.; Stecca, C.; Raciti, M.; Mattia, E. Resveratrol inhibits proliferation and survival of Epstein Barr virus-infected Burkitt's lymphoma cells depending on viral latency program. Mol. Cancer Res. 2011, 9, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Arena, G.; Lacanna, E.; Oliviero, G.; Colavita, F.; Mattia, E. Resveratrol inhibits Epstein Barr Virus lytic cycle in Burkitt’s lymphoma cells by affecting multiple molecular targets. Antivir. Res. 2012, 96, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Radwan, F.F.; Zhang, L.; Hossain, A.; Doonan, B.P.; God, J.M.; Haque, A. Mechanisms regulating enhanced human leukocyte antigen class II-mediated CD4 + T cell recognition of human B-cell lymphoma by resveratrol. Leuk. Lymphoma 2012, 53, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Chiles, T.C. Resveratrol induces apoptosis in transformed follicular lymphoma OCI-LY8 cells: Evidence for a novel mechanism involving inhibition of BCL6 signaling. Int. J. Oncol. 2006, 29, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.A.; Pozo-Guisado, E.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.M.; Castellon, E.A. Mechanisms involved in Resveratrol-induced apoptosis and cell cycle arrest in prostate cancer-derived cell lines. J. Androl. 2007, 28, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.; Xia, Y.; Simayi, M.; Ikramullah, S.; He, Y.; Cui, S.; Li, S.; Wushouer, Q. Resveratrol induces cell cycle arrest and apoptosis in human eosinophils from asthmatic individuals. Mol. Med. Rep. 2016, 14, 5231–5236. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.J.; Wu, K.; Wang, C.; Wan, D.M. Polydatin-induced cell apoptosis and cell cycle arrest are potentiated by Janus kinase 2 inhibition in leukemia cells. Mol. Med. Rep. 2016, 13, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Wenzel, U.; Daniel, H.; Planas, J.M. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J. Agric. Food Chem. 2008, 56, 4813–4818. [Google Scholar] [CrossRef] [PubMed]
- Zunino, S.J.; Storms, D.H. Resveratrol alters proliferative responses and apoptosis in human activated B lymphocytes in vitro. J. Nutr. 2009, 139, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Verras, M.; McNeil, B.; Koong, A.C.; Denko, N.C. Plant stilbenes induce endoplasmic reticulum stress and their anti-cancer activity can be enhanced by inhibitors of autophagy. Exp. Cell Res. 2015, 339, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.J.; Lee, T.J.; Lee, S.H.; Lee, J.M.; Seo, J.H.; Jeong, Y.J.; Park, J.W.; Kwon, T.K. Elevated gadd153/chop expression during Resveratrol-induced apoptosis in human colon cancer cells. Biochem. Pharmacol. 2007, 73, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kucinska, M.; Piotrowska, H.; Luczak, M.W.; Mikula-Pietrasik, J.; Ksiazek, K.; Wozniak, M.; Wierzchowski, M.; Dudka, J.; Jager, W.; Murias, M. Effects of hydroxylated Resveratrol analogs on oxidative stress and cancer cells death in human acute T cell leukemia cell line: prooxidative potential of hydroxylated Resveratrol analogs. Chem. Biol. Interact. 2014, 209, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. Ursolic acid, a pentacyclin triterpene, potentiates TRAIL-induced apoptosis through p53-independent up-regulation of death receptors: Evidence for the role of reactive oxygen species and JNK. J. Biol. Chem. 2011, 286, 5546–5557. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Yadav, V.R.; Ravindran, J.; Aggarwal, B.B. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2011, 71, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Panayiotidis, M.I.; Cidlowski, J.A. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J. Biol. Chem. 2007, 282, 30452–30465. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.C.; Chang, C.L.; Chien, H.F.; Wu, C.H.; Lin, L.I. Resveratrol enhances the expression of death receptor Fas/CD95 and induces differentiation and apoptosis in anaplastic large-cell lymphoma cells. Cancer Lett. 2011, 309, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.V.; Hirpara, J.L.; Chawdhury, S.H.; Pervaiz, S. Chemopreventive agent Resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood 1998, 92, 996–1002. [Google Scholar] [PubMed]
- Passoni, L.; Scardino, A.; Bertazzoli, C.; Gallo, B.; Coluccia, A.M.; Lemonnier, F.A.; Kosmatopoulos, K.; Gambacorti-Passerini, C. ALK as a novel lymphoma-associated tumor antigen: Identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood 2002, 99, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Burrows, S.R.; Thomson, S.A.; Moss, D.J.; Cresswell, P.; Poulsen, L.M.; Cooper, L. Class I processing-defective Burkitt’s lymphoma cells are recognized efficiently by CD4+ EBV-specific CTLs. J. Immunol. 1997, 158, 3619–3625. [Google Scholar] [PubMed]
- Adhami, V.M.; Afaq, F.; Ahmad, N. Involvement of the retinoblastoma (pRb)-E2F/DP pathway during antiproliferative effects of Resveratrol in human epidermoid carcinoma (A431) cells. Biochem. Biophys. Res. Commun. 2001, 288, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Adhami, V.M.; Afaq, F.; Feyes, D.K.; Mukhtar, H. Resveratrol causes WAF-1/p21-mediated G(1)-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin. Cancer Res. 2001, 7, 1466–1473. [Google Scholar] [PubMed]
- Joe, A.K.; Liu, H.; Suzui, M.; Vural, M.E.; Xiao, D.; Weinstein, I.B. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin. Cancer Res. 2002, 8, 893–903. [Google Scholar] [PubMed]
- Estrov, Z.; Shishodia, S.; Faderl, S.; Harris, D.; Van, Q.; Kantarjian, H.M.; Talpaz, M.; Aggarwal, B.B. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-ĸB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood 2003, 102, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Tsai, S.H.; Chen, L.; Lin-Shiau, S.Y.; Lin, J.K. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem. Pharmacol. 2003, 65, 1053–1060. [Google Scholar] [CrossRef]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakazato, T.; Xian, M.J.; Sagawa, M.; Ikeda, Y.; Kizaki, M. Resveratrol induces apoptosis of human malignant B cells by activation of caspase-3 and p38 MAP kinase pathways. Biochem. Pharmacol. 2006, 71, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Sui, T.; Ma, L.; Bai, X.; Li, Q.; Xu, X. Resveratrol inhibits the phosphatidylinositide 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in the human chronic myeloid leukemia K562 cell line. Oncol. Lett. 2014, 7, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, D.; Tinhofer, I.; Tonko, M.; Hubl, H.; Ausserlechner, M.J.; Greil, R.; Kofler, R.; Csordas, A. Resveratrol causes arrest in the S-phase prior to Fas-independent apoptosis in CEM-C7H2 acute leukemia cells. Cell Death Differ. 2000, 7, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.F.; White, J.E.; Maheshwari, J.G.; Chikkappa, G. Anti-leukemia effect of resveratrol. Leuk. Lymphoma 2002, 43, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Frojdo, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Dorrie, J.; Gerauer, H.; Wachter, Y.; Zunino, S.J. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 2001, 61, 4731–4739. [Google Scholar] [PubMed]
- Wang, B.; Liu, J.; Gong, Z. Resveratrol induces apoptosis in K562 cells via the regulation of mitochondrial signaling pathways. Int. J. Clin. Exp. Med. 2015, 8, 16926–16933. [Google Scholar] [PubMed]
- Li, G.; He, S.; Chang, L.; Lu, H.; Zhang, H.; Zhang, H.; Chiu, J. GADD45α and annexin A1 are involved in the apoptosis of HL-60 induced by resveratrol. Phytomedicine 2011, 18, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Liu, Y.; Li, Q.; Guo, X.; Gu, L.; Ma, Z.G.; Zhu, Y.P. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed. Environ. Sci. 2013, 26, 902–911. [Google Scholar] [PubMed]
- Wang, L.; Wang, C.; Jia, Y.; Liu, Z.; Shu, X.; Liu, K. Resveratrol Increases Anti-Proliferative Activity of Bestatin Through Downregulating P-Glycoprotein Expression Via Inhibiting PI3K/Akt/mTOR Pathway in K562/ADR Cells. J. Cell Biochem. 2016, 117, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.P.; Xiong, M.; Xu, C.S.; Duan, L.N.; Dong, Y.Q.; Luo, Y.; Niu, T.H.; Lu, C.R. Resveratrol induces apoptosis of human chronic myelogenous leukemia cells in vitro through p38 and JNK-regulated H2AX phosphorylation. Acta Pharmacol. Sin. 2015, 36, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Trung, L.Q.; Espinoza, J.L.; Takami, A.; Nakao, S. Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS ONE 2013, 8, e55183. [Google Scholar]
- Tian, H.; Yu, Z. Resveratrol induces apoptosis of leukemia cell line K562 by modulation of sphingosine kinase-1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 2755–2762. [Google Scholar] [PubMed]
- Mustafi, S.B.; Chakraborty, P.K.; Raha, S. Modulation of Akt and ERK1/2 pathways by resveratrol in chronic myelogenous leukemia (CML) cells results in the downregulation of Hsp70. PLoS ONE 2010, 5, e8719. [Google Scholar]
- De la Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Clement, M.V.; Pervaiz, S. Pro-oxidant activity of low doses of resveratrol inhibits hydrogen peroxide-induced apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kuršvietienė, I.S.L.; Mongirdienė, A.; Bernatonienė, J. Multiplicity of effects and health benefits of resveratrol. Medicina 2016, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Ragione, F.D.; Cucciolla, V.; Borriello, A.; Pietra, V.D.; Racioppi, L.; Soldati, G.; Manna, C.; Galletti, P.; Zappia, V. Resveratrol arrests the cell division cycle at S/G2 phase transition. Biochem. Biophys. Res. Commun. 1998, 250, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Karawajew, L.; Wuchter, C.; Ruppert, V.; Drexler, H.; Gruss, H.J.; Dorken, B.; Ludwig, W.D. Differential CD95 expression and function in T and B lineage acute lymphoblastic leukemia cells. Leukemia 1997, 11, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Halicka, D.; Klimek, P.; Kowal, M.; Chocholska, S.; Dmoszynska, A. Resveratrol increases rate of apoptosis caused by purine analogues in malignant lymphocytes of chronic lymphocytic leukemia. Ann. Hematol. 2011, 90, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Bernhard, D.; Senfter, M.; Anether, G.; Loeffler, M.; Kroemer, G.; Kofler, R.; Csordas, A.; Greil, R. Resveratrol, a tumor-suppressive compound from grapes, induces apoptosis via a novel mitochondrial pathway controlled by Bcl-2. FASEB J. 2001, 15, 1613–1615. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Schilder, Y.D.; Dirsch, V.M. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J. Biol. Chem. 2007, 282, 26759–26766. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Jiang, S.; Zhang, L.; Bai, Y. Molecular mechanism of apoptosis induction by resveratrol, a natural cancer chemopreventive agent. Int. J. Vitam. Nutr. Res. 2008, 78, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, N.C.; Baek, S.J. The anticancer effects of resveratrol: modulation of transcription factors. Nutr. Cancer 2012, 64, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Zand, H.; Jeddi-Tehrani, M.; Koohdani, F.; Shidfar, F.; Keshavarz, S.A. PTEN over-expression by resveratrol in acute lymphoblastic leukemia cells along with suppression of AKT/PKB and ERK1/2 in genotoxic stress. J. Nat. Med. 2015, 69, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Csermely, P. Heat shock proteins in the regulation of apoptosis: New strategies in tumor therapy: A comprehensive review. Pharmacol. Ther. 2004, 101, 227–257. [Google Scholar] [CrossRef] [PubMed]
- Withey, J.M.; Harvey, A.J.; Crompton, M.R. RNA interference targeting of Bcr-Abl increases chronic myeloid leukemia cell killing by 17-allylamino-17-demethoxygeldanamycin. Leuk. Res. 2006, 30, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Smyth, M.J.; Johnstone, R.W. HMBA induces activation of a caspase-independent cell death pathway to overcome P-glycoprotein-mediated multidrug resistance. Blood 2000, 95, 2378–2385. [Google Scholar] [PubMed]
- Chiarini, F.; del Sole, M.; Mongiorgi, S.; Gaboardi, G.C.; Cappellini, A.; Mantovani, I.; Follo, M.Y.; McCubrey, J.A.; Martelli, A.M. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia 2008, 22, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Pan, S.F.; Feng, Y.; Jin, B.H.; Liu, X.; Zhou, L.H.; Hou, F.G.; Wang, W.H.; Fu, X.L.; Han, Z.F.; et al. Zuo Jin Wan reverses P-gp-mediated drug-resistance by inhibiting activation of the PI3K/Akt/NF-ĸB pathway. BMC Complement. Altern. Med. 2014, 14, 279. [Google Scholar] [CrossRef] [PubMed]
- Vang, O.; Ahmad, N.; Baile, C.A.; Baur, J.A.; Brown, K.; Csiszar, A.; Das, D.K.; Delmas, D.; Gottfried, C.; Lin, H.Y.; et al. What is new for an old molecule? Systematic review and recommendations on the use of resveratrol. PLoS ONE 2011, 6, e19881. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Pathway Affected | Cell Type | Concentration Ranges | References |
|---|---|---|---|
| Apoptosis | |||
| Caspase 3, 8, 9 | NU-DUL-1; OCI-Ly8; U2932; SUDHL-4; DB; TMD8 | 24–32 μM; 20 μM | [16,17] |
| PARP | SUDHL-4; NU-DUL-1 | 24–32 μM | [16] |
| Bax/Bcl2 | SUDHL-4; NU-DUL-1 | 24–32 μM; 20 μM | [16,17] |
| Cytochrome c | SUDHL-4; HBL-1 | 25 and 50 μM | [18,19] |
| Cell survival | |||
| PI3K/Akt | SUDHL-4; SUDHL-5 | 25 and 50 μM | [20,21] |
| ER-stress | |||
| CHOP/GADD153 | Raji; Daudi | 10–200 μM | [22] |
| Pro-oxidant activity | |||
| ROS | BCBL-1; BC-1; P3HR1; BJAB | 20 μM | [17] |
| GSH depletion | U-937 | 50 μM | [23] |
| Death receptor pathway | |||
| DR5 | SUDHL-4; HBL-1 | 25 and 50 μM | [19] |
| EBV infection | |||
| EBV lytic antigens | Raji; Akata | 20–300 μM | [24,25] |
| Antigen presentation | |||
| DR and DM HLA class II | Nalm-6, Ramos, Daudi | 50 μM | [26] |
| Active cathepsins S, B, and D | Nalm-6, Ramos, Daudi | 50 μM | [26] |
| Cell cycle | |||
| S-phase | L-428 | 25 and 50 μM | [9] |
| p53; Bcl6; PI3K | OCI-Ly1; OCI-Ly18 | 25 μM | [20,27] |
| Cdk1 | Mouse lymphoma | 1–150 μM | [28] |
| Pathway Affected | Cell Type | Concentration Ranges | References |
|---|---|---|---|
| Apoptosis | |||
| CD95/caspase-3 | HL-60; K562 | 1–100 μM | [55,56] |
| Caspase-9 | SEM; RS4:11; MV4:11; REH; NALM-6; CEM; JURKAT; HL-60 | 50 μM | [55] |
| Cytochrome c | CEM; Molt-4 | 25 and 50 μM | [18,19] |
| PARP | OCI/AML3; OCIM2 | 5–75 μM | [47] |
| GADD45α | HL-60 | 12.5–200 μM | [57] |
| Autophagy | |||
| MDC (autophagosomes)/phagophore/LC3 | Molt-4; Jurkat; CEM | 25–250 μM | [58] |
| Cell survival | |||
| NF-kB | OCI/AML3; OCIM2 | 5–75 μM | [47] |
| IL-1 | OCI/AML3; OCIM2 | 5–75 μM | [47] |
| PI3K/Akt | Molt-4; Jurkat; CEM | 25–250 μM; 10 μM | [58,59] |
| Cell proliferation | |||
| p38; JNK (MAPK family) | K562 | 20–100 μM | [60] |
| Cell cycle | |||
| cyclins 2/6 | malignant NK | 12.5, 25 and 50 μM | [61] |
| Plasma membrane associated targets | |||
| Sphingosine kinases/sphingosine 1P | K562 | 20 and 40 μM | [62] |
| P-glycoprotein | K562 | 10 μM | [59] |
| Heat shock proteins | |||
| HSP70/HSP90 | K562 | 20–100 μM; 40 μM | [5,63] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frazzi, R.; Guardi, M. Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells. Molecules 2017, 22, 885. https://doi.org/10.3390/molecules22060885
Frazzi R, Guardi M. Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells. Molecules. 2017; 22(6):885. https://doi.org/10.3390/molecules22060885
Chicago/Turabian StyleFrazzi, Raffaele, and Manuela Guardi. 2017. "Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells" Molecules 22, no. 6: 885. https://doi.org/10.3390/molecules22060885
APA StyleFrazzi, R., & Guardi, M. (2017). Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells. Molecules, 22(6), 885. https://doi.org/10.3390/molecules22060885