
Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Screening of Potential Redox Regulators
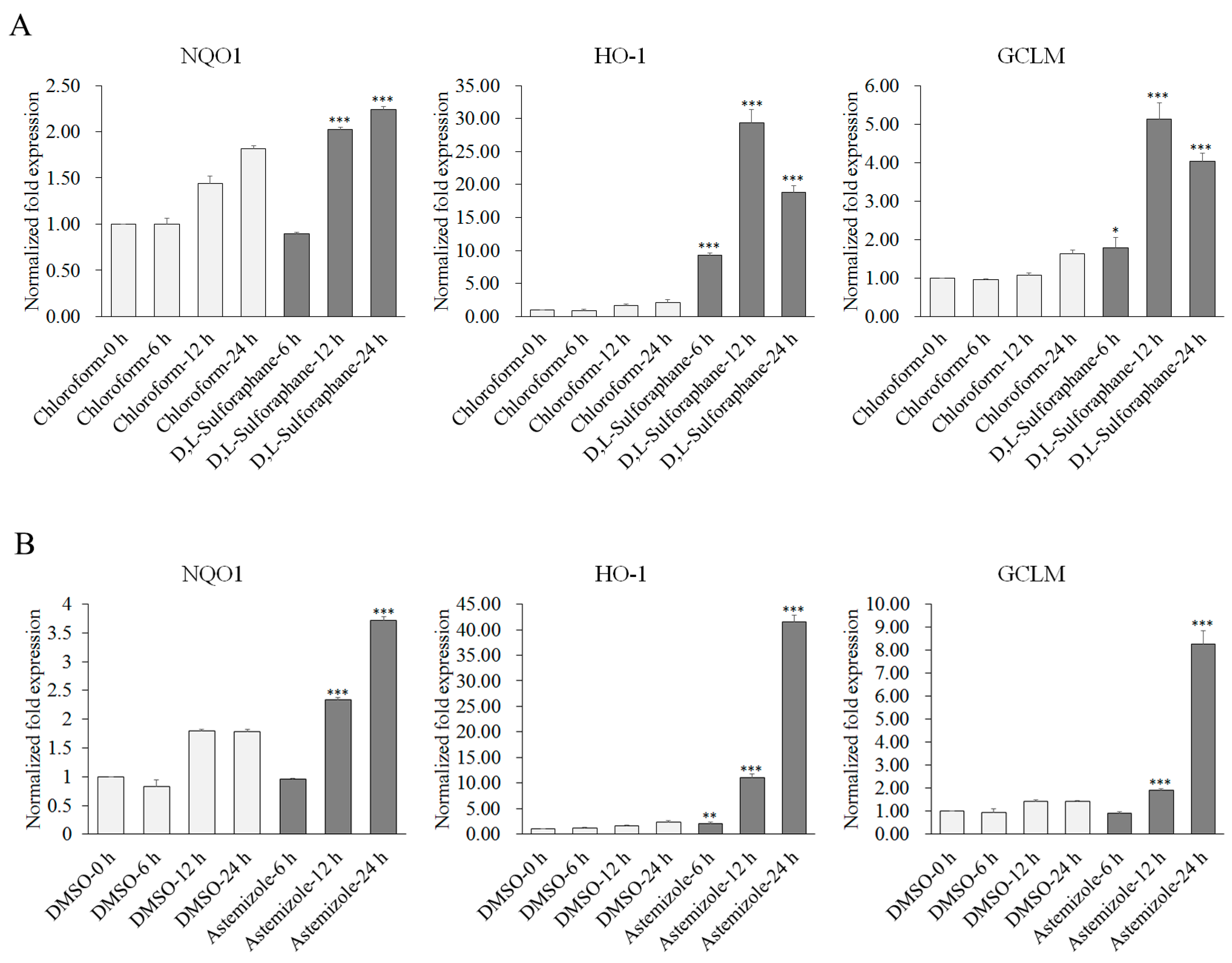
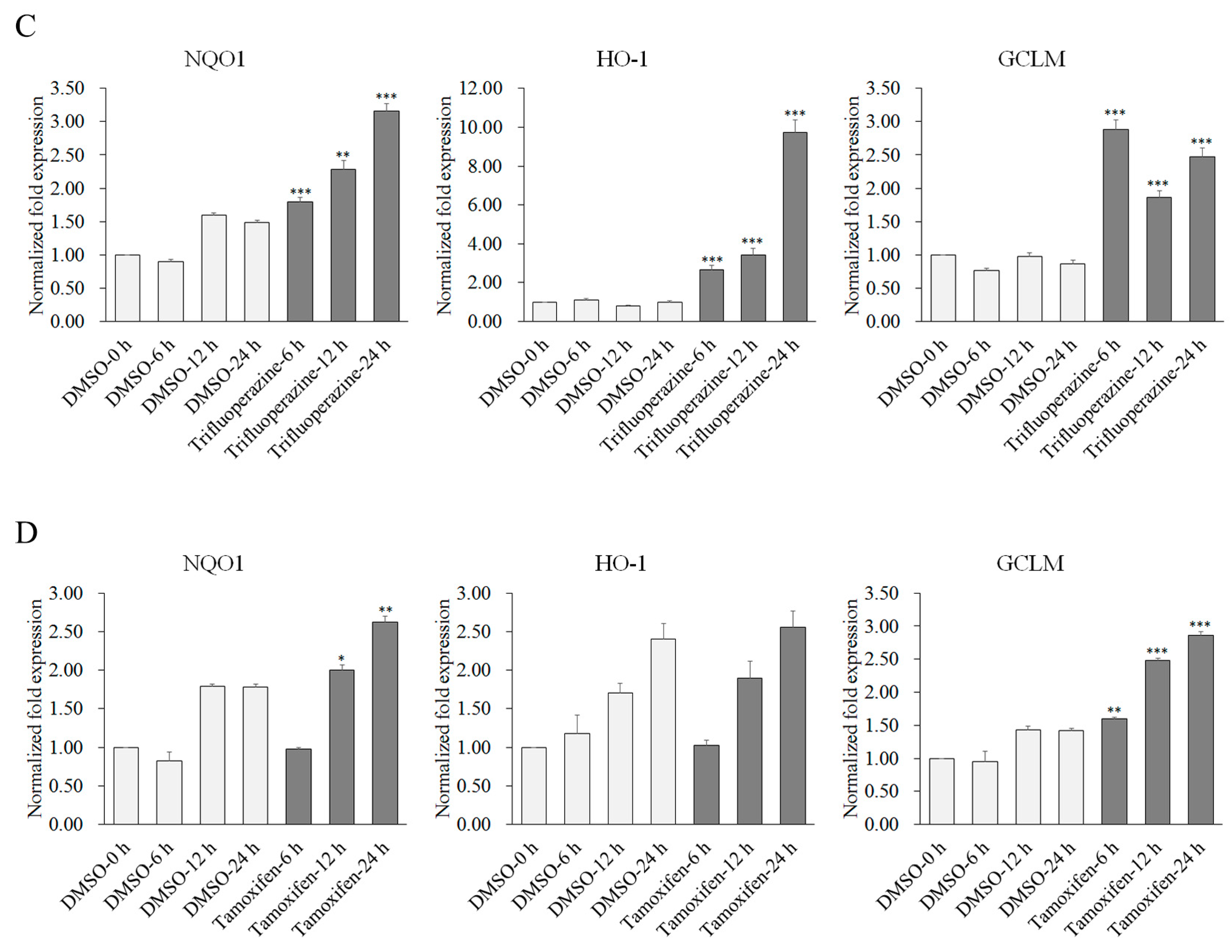
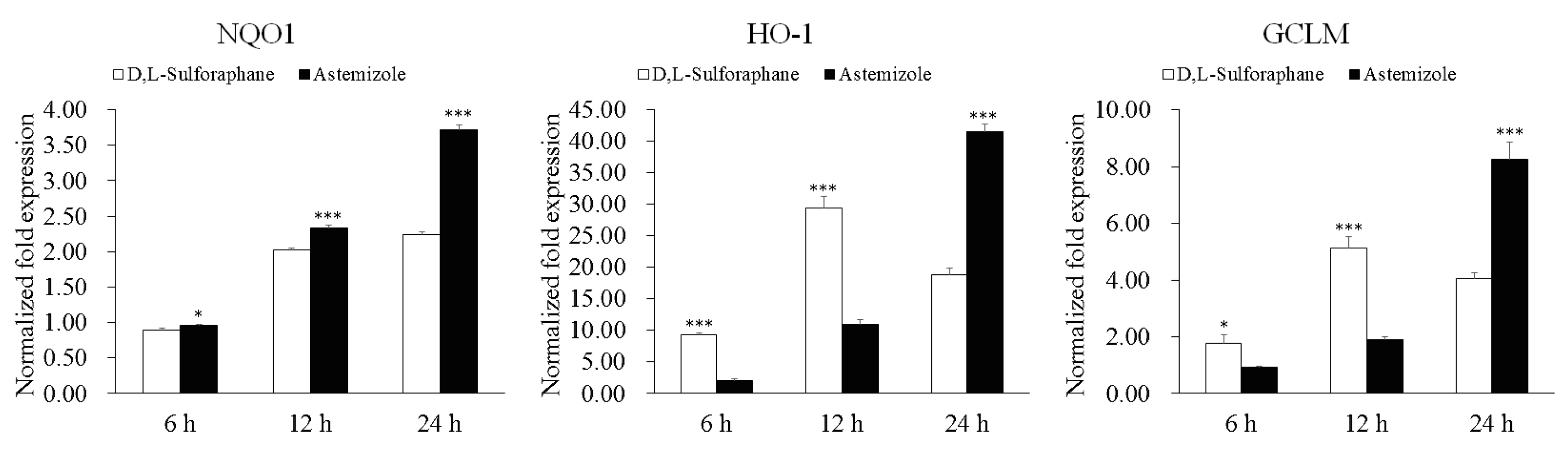
2.2. Potency of Candidate Compounds to Activate the Expression of Nrf2-Regulated Cytoprotective Genes
3. Materials and Methods
3.1. Data and Reagent Retrieval
3.2. Cell Culture Conditions
3.3. RNA Extraction and qRT-PCR Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| cMap | connectivity map |
| qRT-PCR | quantitative real time polymerase chain reaction |
| ARE | antioxidant response element |
| MEM | minimum essential medium |
| FBS | fetal bovine serum |
| DMSO | dimethyl sulfoxide |
| ROS | reactive oxygen species |
References
- Gacesa, R.; Dunlap, W.C.; Long, P.F. Bioinformatics analyses provide insight into distant homology of the Keap1-Nrf2 pathway. Free Radic. Biol. Med. 2015, 88, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Lee, J.S.; Wang, X.; Campbell, M.R.; Bell, D.A.; Slattery, M. Beyond antioxidant genes in the ancient Nrf2 regulatory network. Free Radic. Biol. Med. 2015, 88, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Speltz, T.E.; Moore, T.W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett. 2015, 25, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, P.; Zhao, Y.; Yang, C.; Clark, A.; Leung, T.C.; Chen, X.; Sang, S. Synthesis, evaluation, and metabolism of novel [6]-shogaol derivatives as potent Nrf2 activators. Free Radic. Biol. Med. 2016, 95, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox regulation by Keap1–Nrf2 system in toxicology and cancer. Curr. Opin. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1-Nrf2 protein-protein interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kim, I.-S.; More, S.V.; Kim, B.-W.; Choi, D.-K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014, 31, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Traustadóttir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer 2006, 55, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.A.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.; Torti, S. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Munday, R. Dithiolethiones for cancer chemoprevention: Where do we stand? Mol. Cancer Ther. 2008, 7, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Wille, E.; Calingasan, N.Y.; Tampellini, D.; Williams, C.; Gouras, G.K.; Liby, K.; Sporn, M.; Nathan, C.; Flint Beal, M.; et al. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009, 109, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [PubMed]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Charturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingson, R.; et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE 2009, 4, e5757. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Korner, S.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Kerns, J.K.; Callahan, J.F.; Moody, C.J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–7476. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochem. Mosc. 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An exceptionally potent inducer of cytoprotective enzymes: Elucidation of the structural features that determine inducer potency and reactivity with Keap1. J. Biol. Chem. 2010, 285, 33747–33755. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Dinkova-Kostova, A.T.; Stephenson, K.K.; Talalay, P. The ‘Prochaska’ microtiter plate bioassay for inducers of NQO1. Methods Enzymol. 2004, 382, 243–258. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Talalay, P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1). Methods Enzymol. 2004, 382, 423–448. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Zhang, Y.; Spencer, S.R.; Wilczak, C.A.; Talalay, P. The electrophile counterattack response: Protection against neoplasia and toxicity. Adv. Enzyme Regul. 1993, 33, 281–296. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Xu, L.L.; Lu, M.C.; Chen, Z.Y.; Yuan, Z.W.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; Sun, H.P.; You, Q.D. Structure-activity and structure-property relationship and exploratory in vivo evaluation of the Nanomolar Keap1-Nrf2 protein-protein interaction inhibitor. J. Med. Chem. 2015, 58, 6410–6421. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, A.; Yon, J.; Day, P.J.; Richardson, C.; Tickle, I.J.; Williams, P.A.; Callahan, J.F.; Carr, R.; Concha, N.; Kerns, J.K.; et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS ONE 2014, 9, e98896. [Google Scholar] [CrossRef] [PubMed]
- Steel, R.; Cowan, J.; Payerne, E.; O’Connell, M.A.; Searcey, M. Anti-inflammatory effect of a cell-penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med. Chem. Lett. 2012, 3, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Jnoff, E.; Albrecht, C.; Barker, J.J.; Barker, O.; Beaumont, E.; Bromidge, S.; Brookfield, F.; Brooks, M.; Bubert, C.; Ceska, T.; et al. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. Chem. Med. Chem. 2014, 9, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-P.; Jiang, Z.-Y.; Zhang, M.-Y.; Lu, M.-C.; Yang, T.-T.; Pan, Y.; Huang, H.-Z.; Zhang, X.-J.; You, Q. Novel protein–protein interaction inhibitor of Nrf2–Keap1 discovered by structure-based virtual screening. Med. Chem. Commun. 2014, 5, 93–98. [Google Scholar] [CrossRef]
- Moehlenkamp, J.D.; Johnson, J.A. Activation of antioxidant/electrophile-responsive elements in IMR-32 human neuroblastoma cells. Arch. Biochem. Biophys. 1999, 363, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gidwani, V.; Sun, Z.; Zhang, D.D.; Wong, P.K. Development of a molecular assay for rapid screening of chemopreventive compounds targeting Nrf2. J. Lab. Autom. 2008, 13, 243–248. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Stevenson, J.C.R.; Horbach, G.J.; Schoonen, W.G.E.J. The development of RAD51C, cystatin A, p53 and Nrf2 luciferase-reporter assays in metabolically competent HepG2 cells for the assessment of mechanism-based genotoxicity and of oxidative stress in the early research phase of drug development. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 696, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Radical medicine: Treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Li, B.; Zhu, Q.; Wang, Y.X.; Zhang, H.Y. Identification of transcription factors for drug-associated gene modules and biomedical implications. Bioinformatics 2014, 30, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Miura, T.; Phuc, B.H.; Shinkai, Y.; Kumagai, Y. Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol. Appl. Pharmacol. 2011, 255, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Connectivity Map 02. Available online: https://portals.broadinstitute.org/cmap/ (accessed on 15 July 2016).
- Zhang, Y.; Talalay, P.; Chot, C.-G.; Posnert, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Oh, J.H.; Park, H.J.; Kim, D.G.; Lee, J.H.; Kim, C.Y.; Kwon, M.S.; Yoon, S. Simultaneous gene expression signature of heart and peripheral blood mononuclear cells in astemizole-treated rats. Arch. Toxicol. 2010, 84, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.M.; Brogden, R.N.; Heel, R.C. Astemizole. Drugs 1984, 28, 38–61. [Google Scholar] [CrossRef] [PubMed]
- Özben, T. Pathophysiology of Cerebral Ischemia: Mechanisms involved in neuronal damage. In Free Radicals, Oxidative Stress, and Antioxidants: Pathological and Physiological Significance, 1st ed.; Özben, T., Ed.; Plenum Press: New York, NY, USA, 1998; Volume 296, pp. 163–188. [Google Scholar]
- Ramirez, A.; Vazquez-Sanchez, A.Y.; Camacho, J.; Carrion-Robalino, N. Ion channels and oxidative stress as a potential link for the diagnosis or treatment of liver diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3928714. [Google Scholar] [CrossRef] [PubMed]
- De Guadalupe Chávez-López, M.; Pérez-Carreón, J.I.; Zuñiga-García, V.; Díaz-Chávez, J.; Herrera, L.A.; Caro-Sánchez, C.H.; Acuña-Macías, I.; Gariglio, P.; Hernández-Gallegos, E.; Chiliquinga, A.J.; et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 2015, 36, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar]
- Sun, B.; Karin, M. Inflammation and liver tumorigenesis. Front. Med. 2013, 7, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Tardy, M.; Dold, M.; Rr, E.; Leucht, S. Trifluoperazine versus low-potency first-generation antipsychotic drugs for schizophrenia. Cochrane Database Syst. Rev. 2014, 7, CD009396. [Google Scholar]
- Khan, S.Z.; Longland, C.L.; Michelangeli, F. The effects of phenothiazines and other calmodulin antagonists on the sarcoplasmic and endoplasmic reticulum Ca2+ pumps. Biochem. Pharmacol. 2000, 60, 1797–1806. [Google Scholar] [CrossRef]
- Pan, G.; Zhou, T.; Radding, W.; Saag, M.S.; Mountz, J.D.; McDonald, J.M. Calmodulin antagonists inhibit apoptosis of CD4+ T-cells from patients with AIDS. Immunopharmacology 1998, 40, 91–103. [Google Scholar] [CrossRef]
- Castilho, R.F.; Carvalho-Alves, P.C.; Vercesi, A.E.; Ferreira, S.T. Oxidative damage to sarcoplasmic reticulum Ca2+-pump induced by Fe2+/H2O2/ascorbate is not mediated by lipid peroxidation or thiol oxidation and leads to protein fragmentation. Mol. Cell. Biochem. 1996, 159, 105–114. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Chen, Y.; Li, B.; Qin, G.; Tian, S. Ca2+-CaM regulating viability of Candida guilliermondii under oxidative stress by acting on detergent resistant membrane proteins. J. Proteom. 2014, 109, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Souza dos Santos, P.; Saraiva, D.F.; Ferraz da Costa, D.C.; Scofano, H.M.; de Carvalho-Alves, P.C. Trifluoperazine protects brain plasma membrane Ca2+-ATPase from oxidative damaging. Exp. Brain Res. 2007, 177, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Zenebe, W.J.; Parihar, A.; Larson, S.K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007, 67, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Bekele, R.T.; Venkatraman, G.; Liu, R.-Z.; Tang, X.; Mi, S.; Benesch, M.G.K.; MacKey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.W.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
| Type-I | 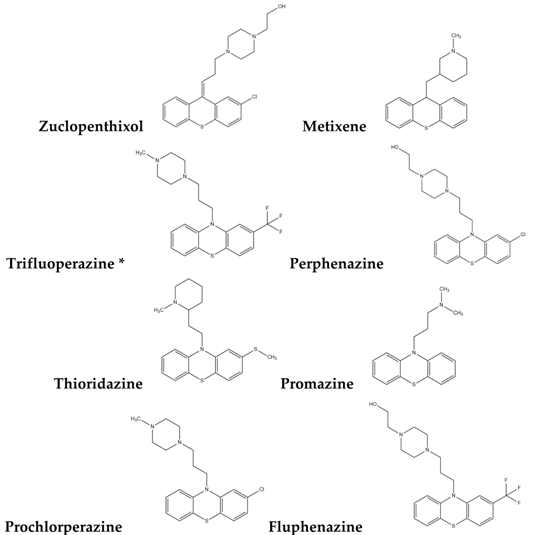 |
| Type-II |  |
| Type-III |  |
| Type-IV |  |
| Type-V |  |
| Type-VI |  |
| Type-VII |  |
| Drug | Solvent | Concentration | Sample |
|---|---|---|---|
| Terfenadine | Chloroform | 8 μM [49] | 6 h/12 h/24 h |
| Trimipramine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Quinidine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Hexetidine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Dosulepin | Chloroform | 12 μM [49] | 6 h/12 h/24 h |
| Diphenylpyraline | Chloroform | 12 μM [49] | 6 h/12 h/24 h |
| d,l-Sulforaphane | Chloroform | 15 μM [50] | 6 h/12 h/24 h |
| Tamoxifen | DMSO | 1 μM [49] | 6 h/12 h/24 h |
| Astemizole | DMSO | 8 μM [49] | 6 h/12 h/24 h |
| Trifluoperazine | DMSO | 10 μM [49] | 6 h/12 h/24 h |
| Chloroform (control) | 0 h/6 h/12 h/24 h | ||
| DMSO (control) | 0 h/6 h/12 h/24 h |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.-Y.; Chu, X.-Y.; Jiang, L.-H.; Liu, M.-Y.; Mei, Z.-L.; Zhang, H.-Y. Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules 2017, 22, 883. https://doi.org/10.3390/molecules22060883
Zhang Q-Y, Chu X-Y, Jiang L-H, Liu M-Y, Mei Z-L, Zhang H-Y. Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules. 2017; 22(6):883. https://doi.org/10.3390/molecules22060883
Chicago/Turabian StyleZhang, Qing-Ye, Xin-Yi Chu, Ling-Han Jiang, Meng-Yuan Liu, Zhi-Ling Mei, and Hong-Yu Zhang. 2017. "Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs" Molecules 22, no. 6: 883. https://doi.org/10.3390/molecules22060883
APA StyleZhang, Q.-Y., Chu, X.-Y., Jiang, L.-H., Liu, M.-Y., Mei, Z.-L., & Zhang, H.-Y. (2017). Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules, 22(6), 883. https://doi.org/10.3390/molecules22060883