
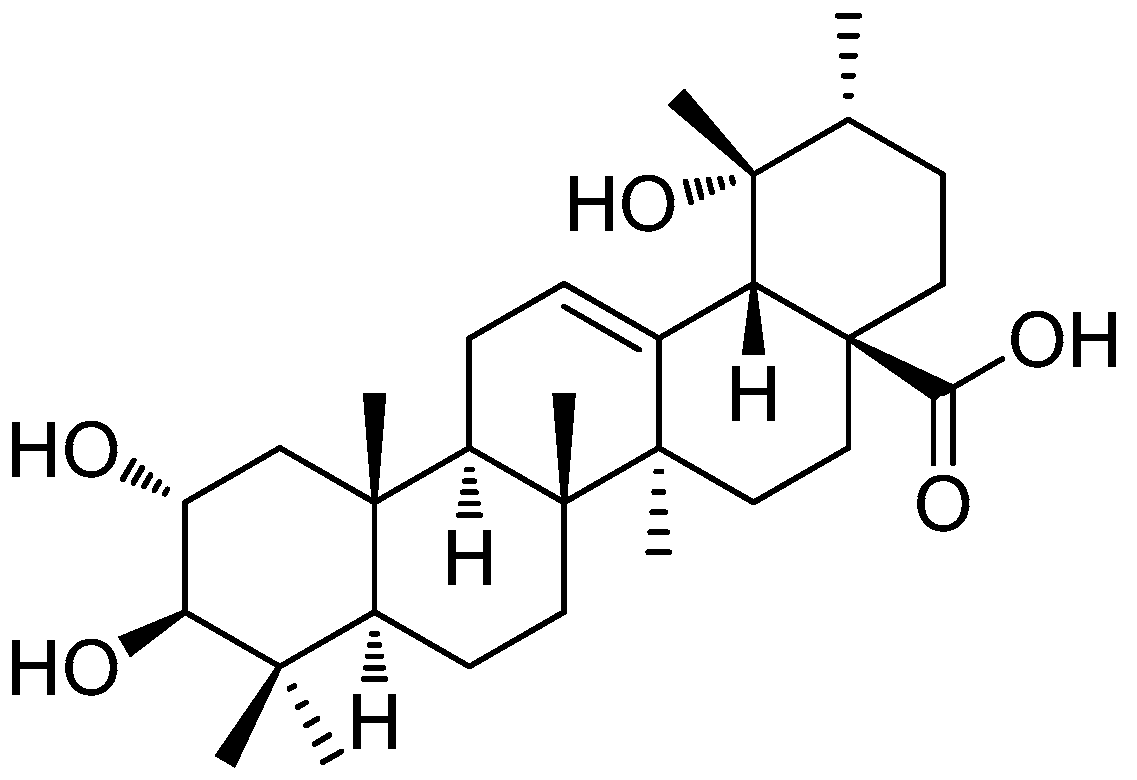
Protective Effects of Tormentic Acid, a Major Component of Suspension Cultures of Eriobotrya japonica Cells, on Acetaminophen-Induced Hepatotoxicity in Mice


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
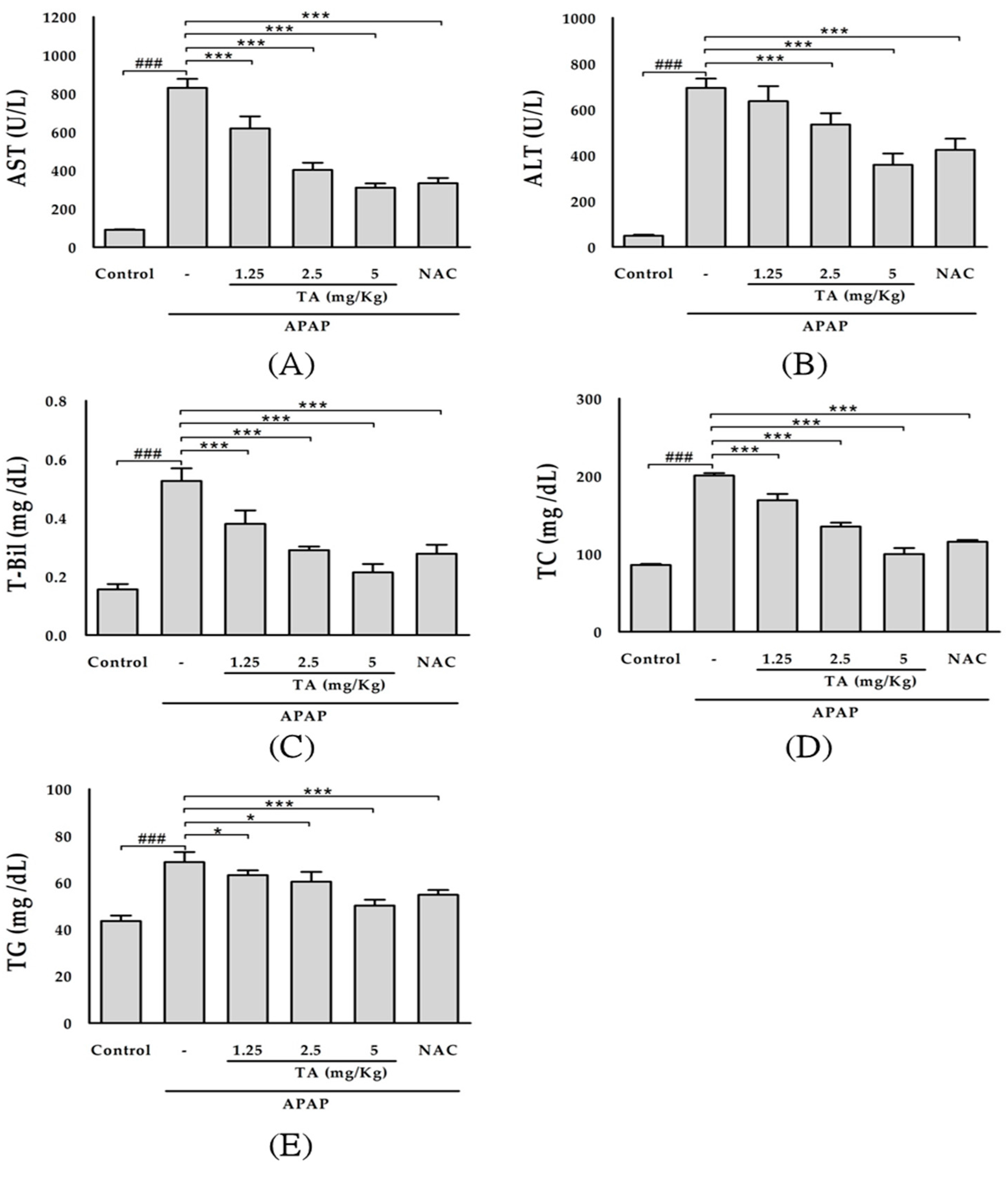
2.1. Effect of TA on Hepatotoxicity in APAP-Treated Mice
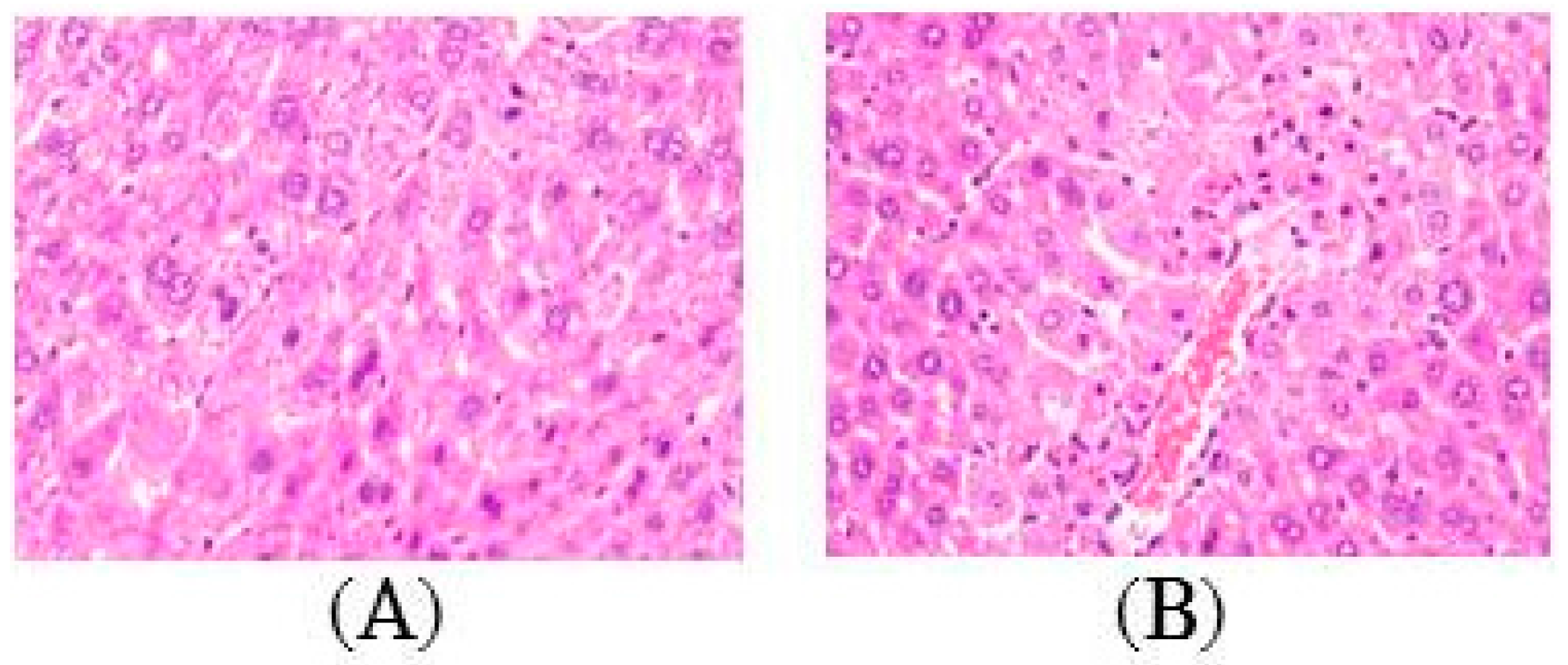
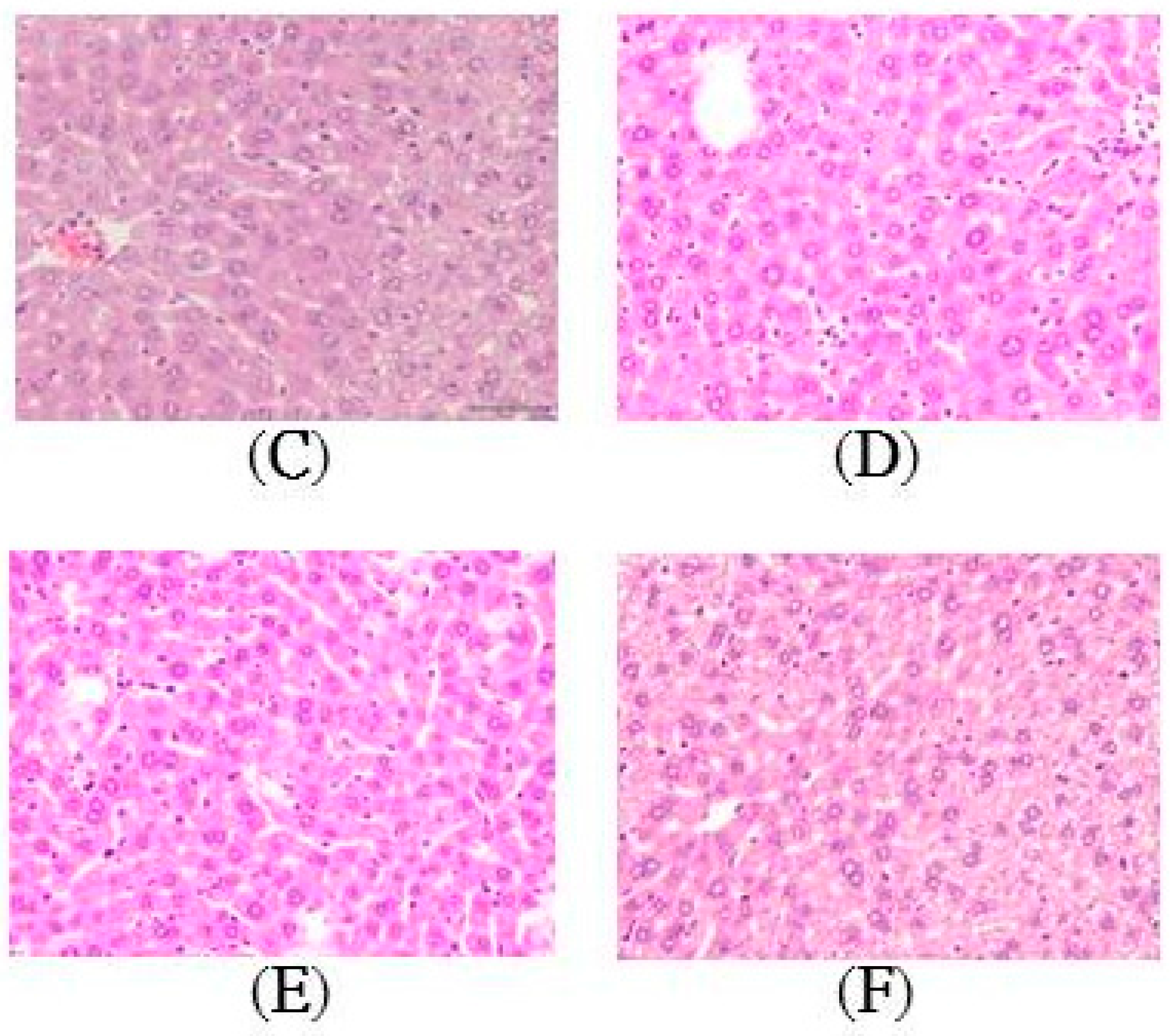
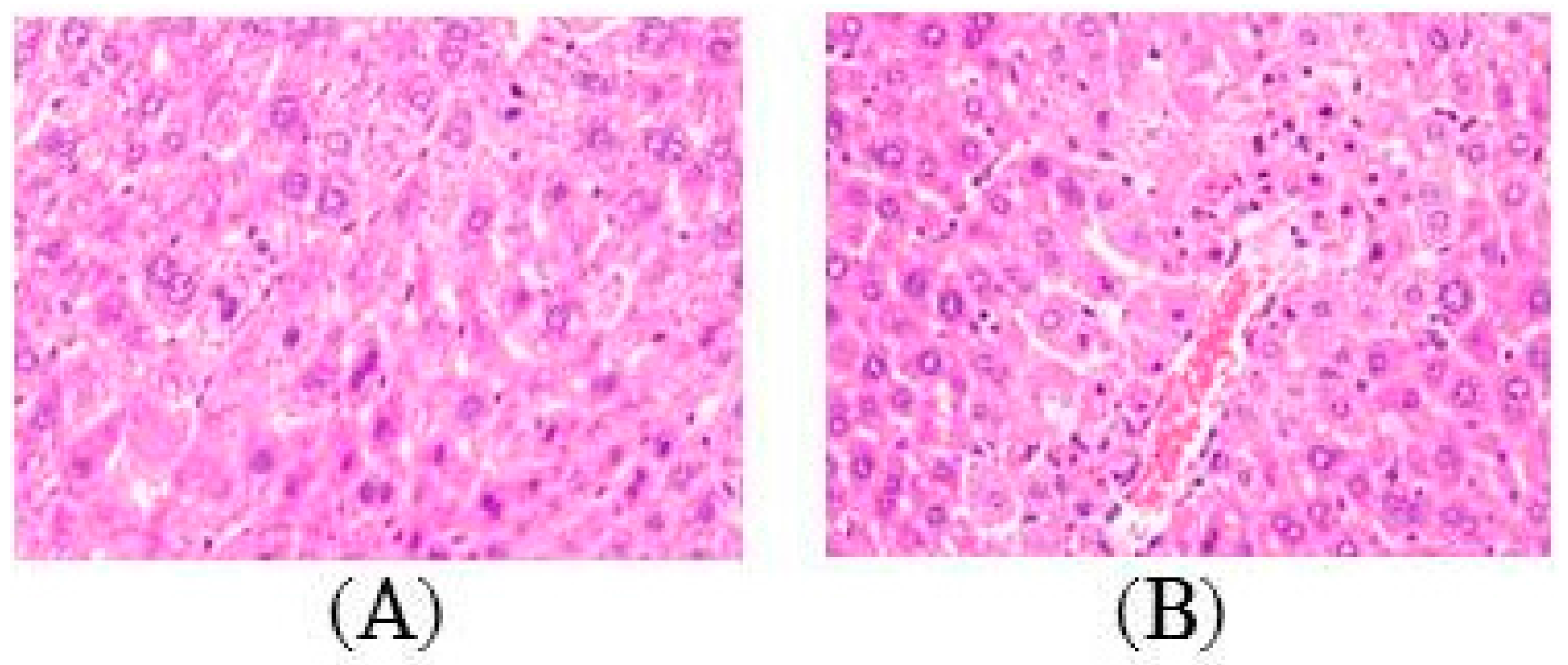
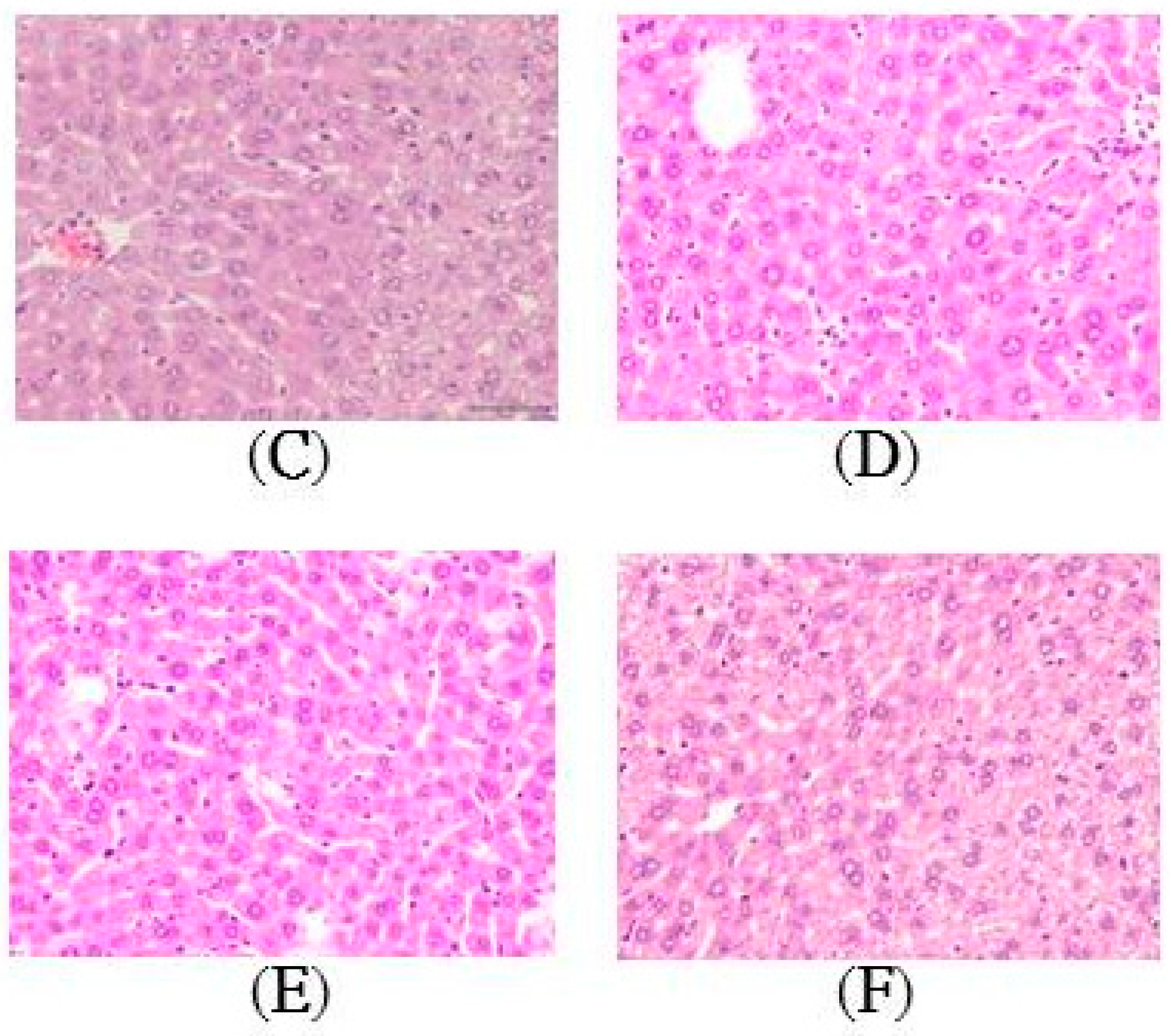
2.2. Histopathology of the Liver
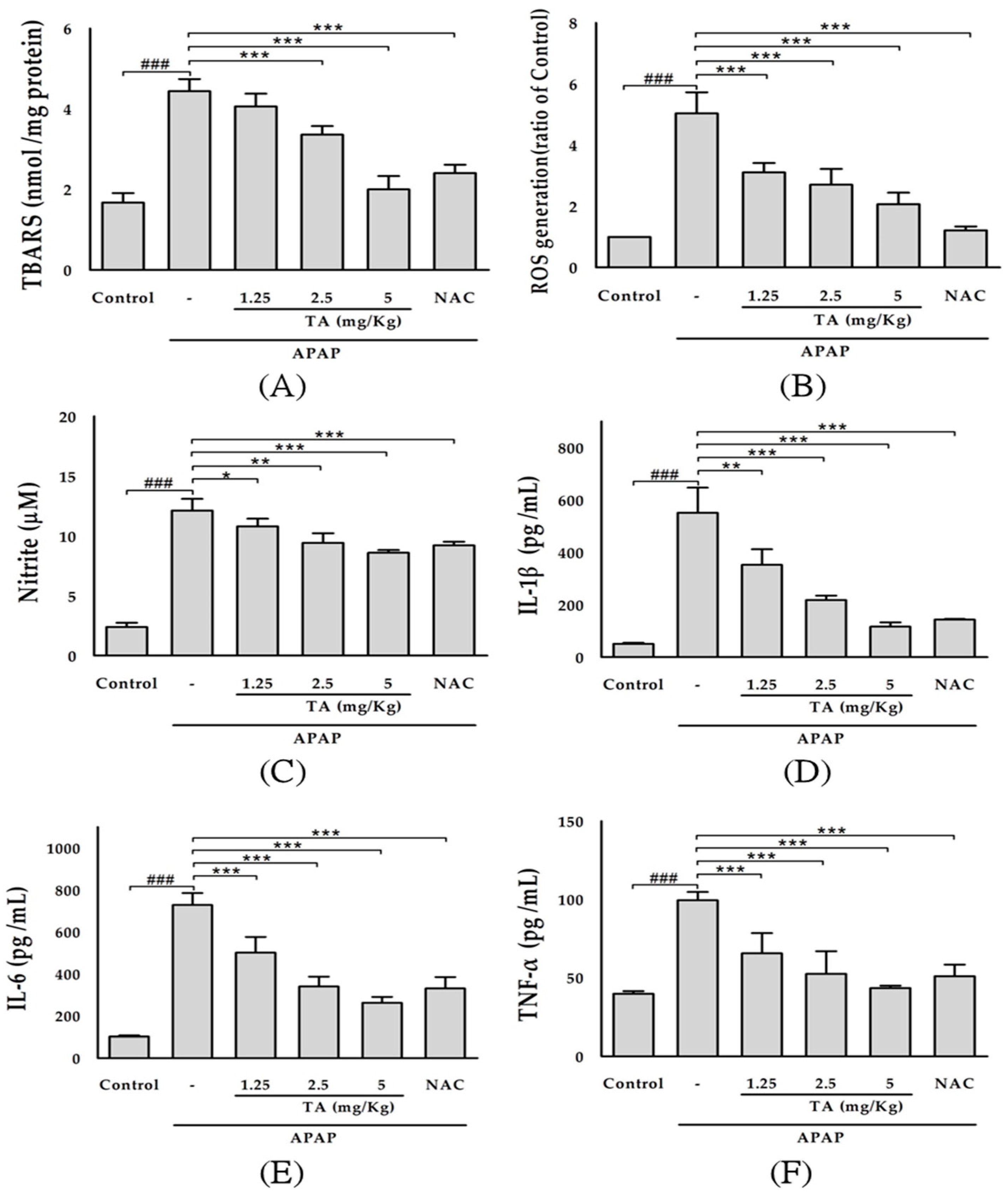
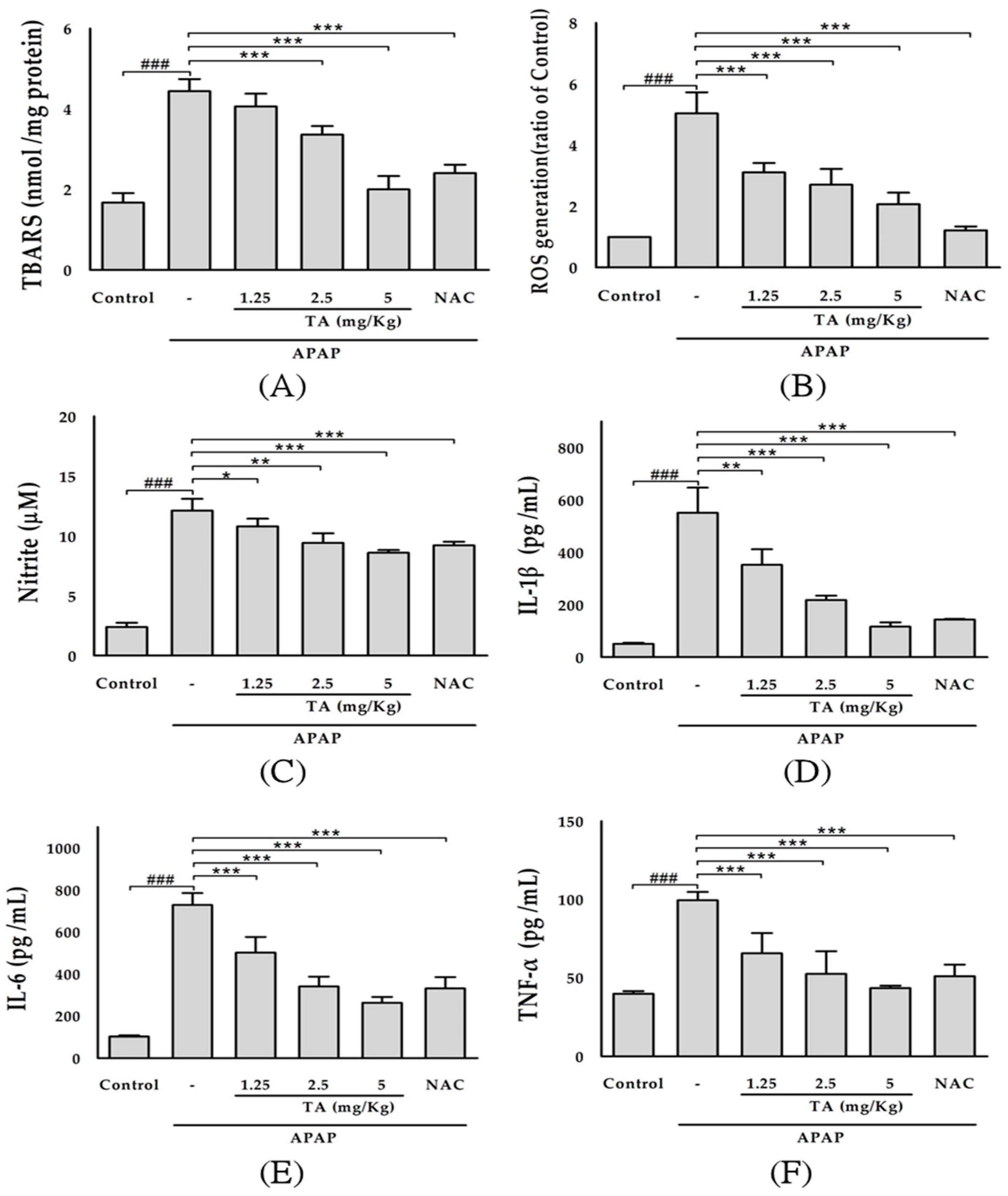
2.3. Effect of TA on Lipid Peroxidation in APAP-Treated Mouse Livers
2.4. Effect of TA on Serum ROS Levels in APAP-Treated Mice
2.5. Effect of TA on Serum Nitrite Levels in APAP-Treated Mice
2.6. Effects of TA on Serum IL-1β, IL-6, and TNF-α Levels
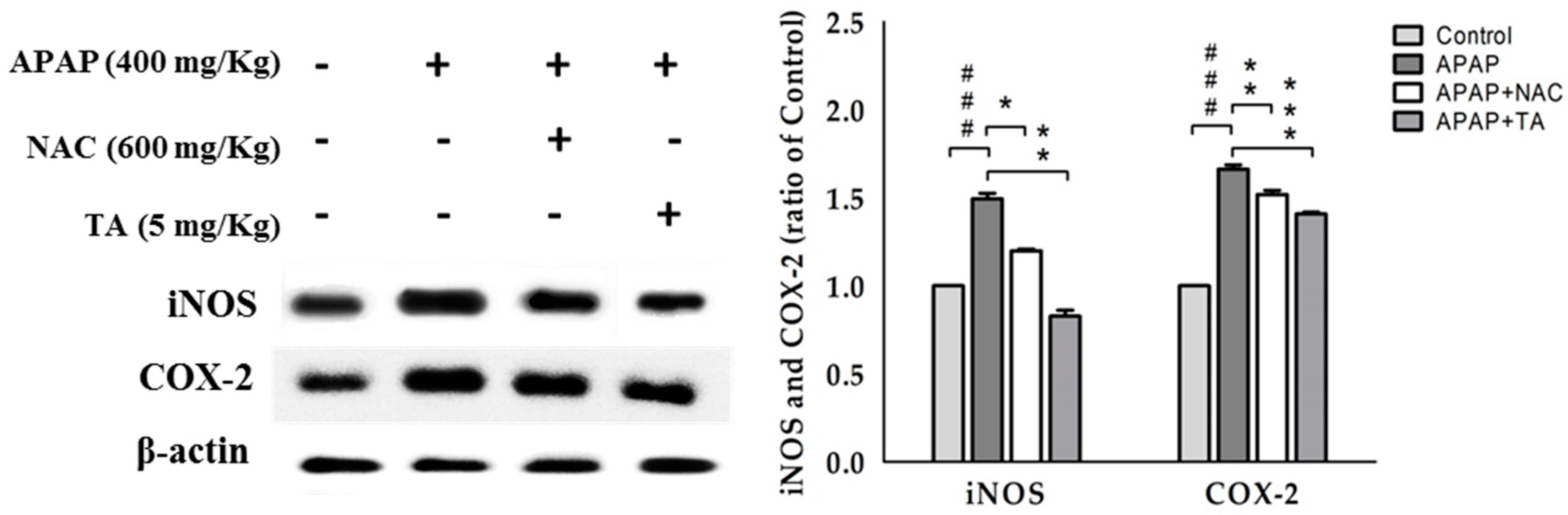
2.7. Effects of TA on iNOS and COX-2 Expression in APAP-Treated Mouse Liver Tissues
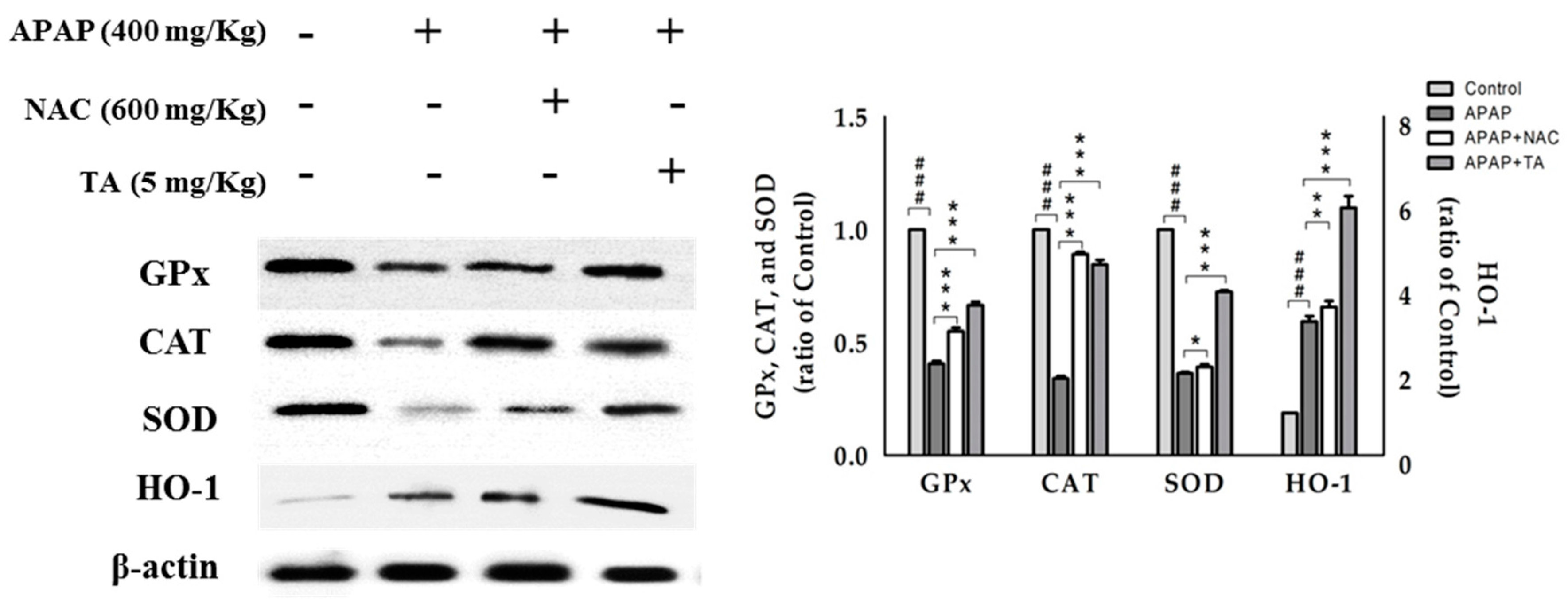
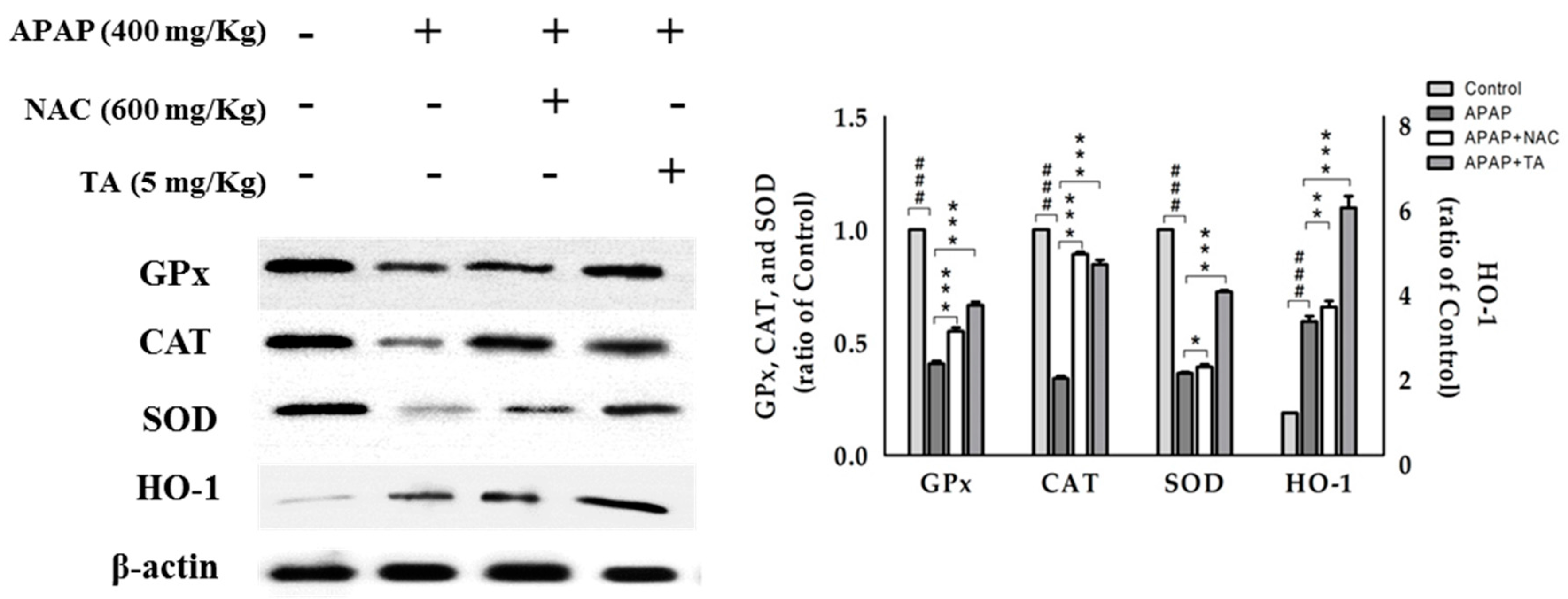
2.8. Effect of TA on the Expression of the Antioxidant Enzymes GPx, CAT, and SOD
2.9. Effects of TA on APAP-induced Expression of HO-1 Protein in the Mice Liver
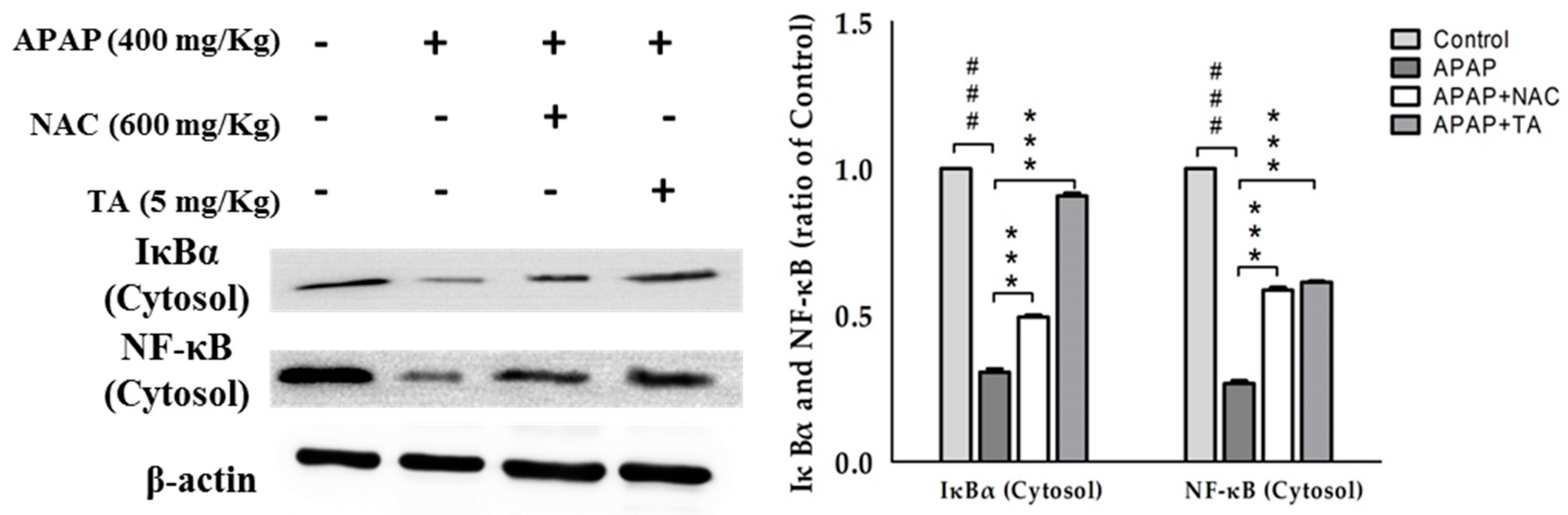
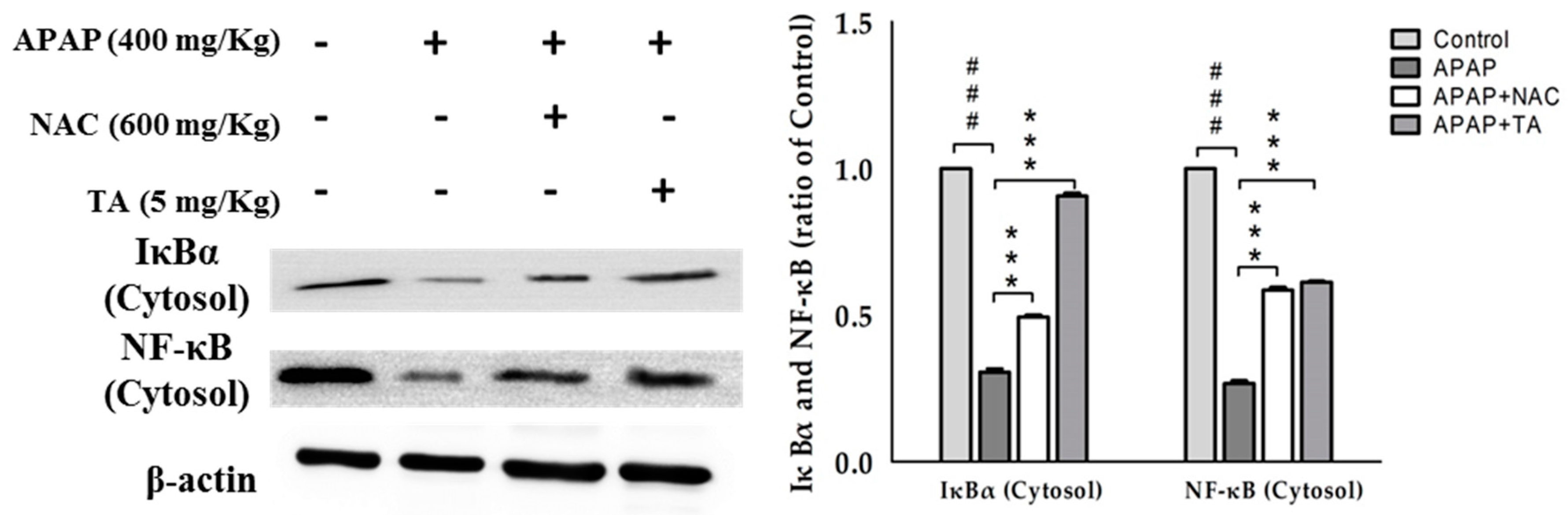
2.10. Effects of TA on NF-κB and IκBα in APAP-Induced Mice
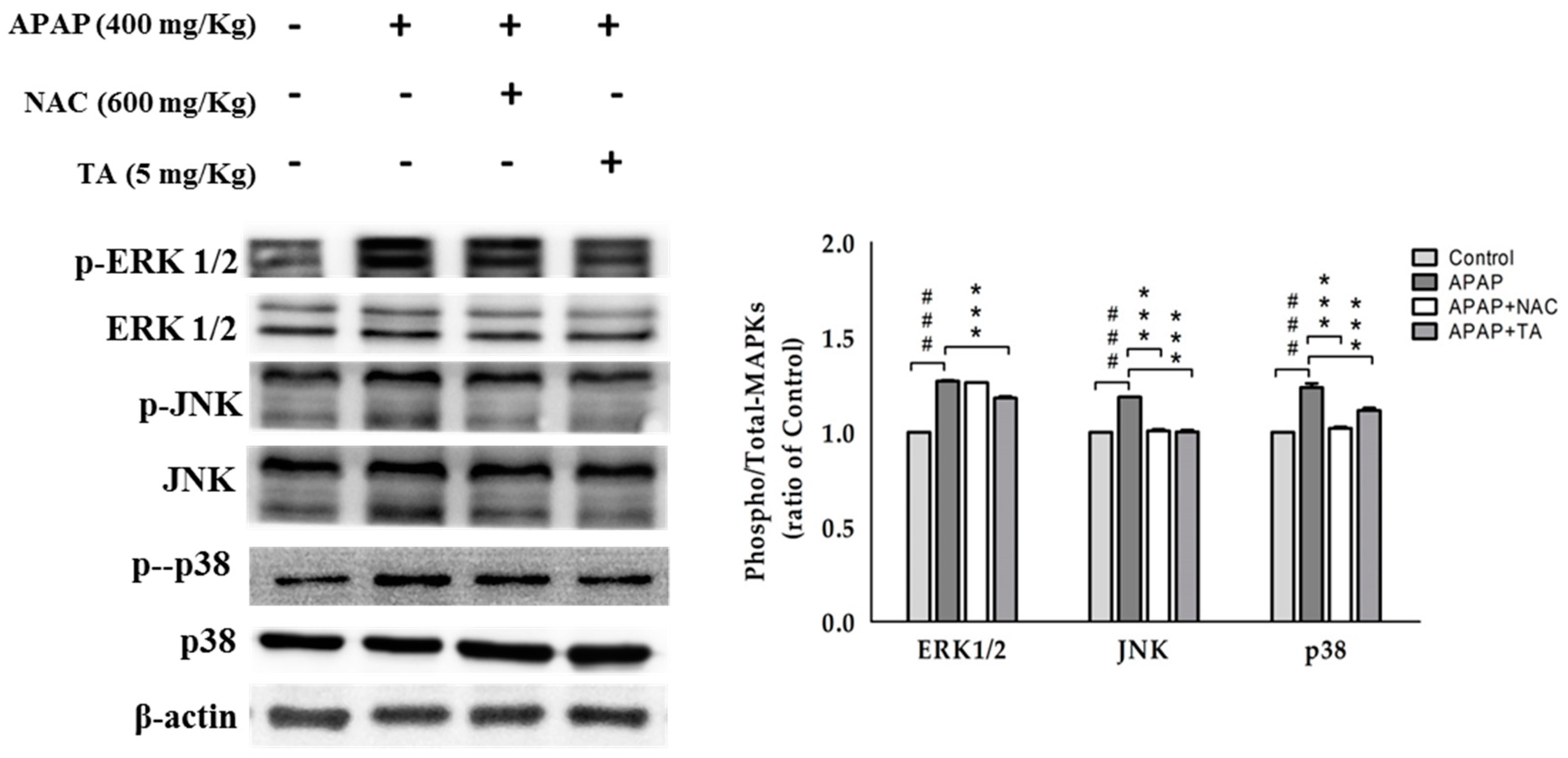
2.11. Effects of TA on the APAP-Induced Phosphorylation of MAPK
3. Discussion
4. Materials and Methods
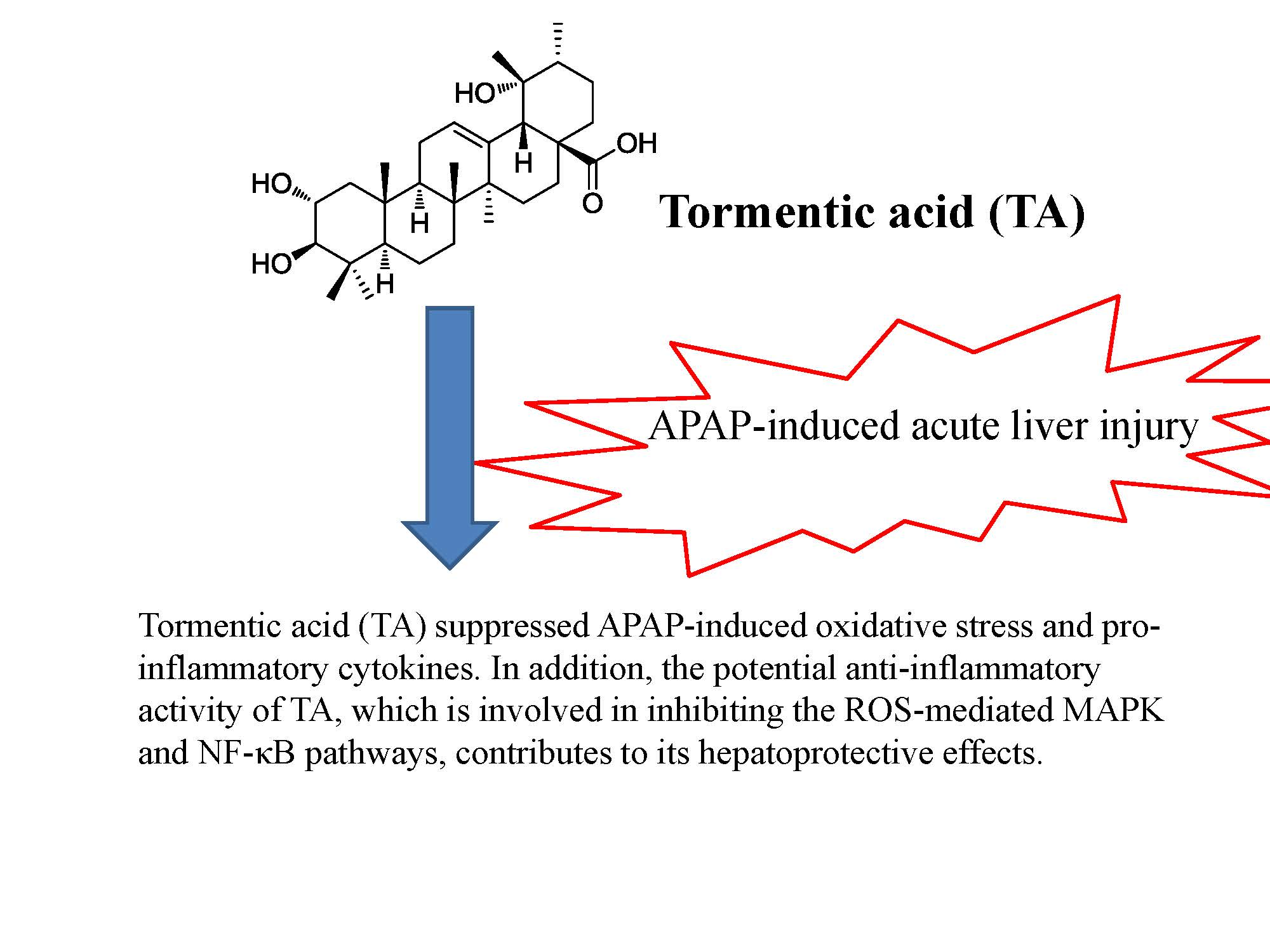
4.1. Isolation of TA, Callus Induction, and Suspension Cultures
4.2. Animals and Treatments
4.3. Assessment of Liver Functions
4.4. Histopathological Examination
4.5. Hepatic Lipid Peroxidation Assays
4.6. Measurement of the Levels of ROS in Serum
4.7. Measurement of NO/Nitrite Levels
4.8. Cytokine Assays
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tien, Y.H.; Chen, B.H.; Wang Hsu, G.S.; Lin, W.T.; Huang, J.H.; Lu, Y.F. Hepatoprotective and anti-oxidant activities of Glossogyne tenuifolia against acetaminophen-induced hepatotoxicity in mice. Am. J. Chin. Med. 2014, 42, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.; Lee, C.H.; Wang, J.Y.; Lee, P.H.; Lai, H.S.; Hu, R.H. Nationwide longitudinal analysis of acute liver failure in taiwan. Medicine (Baltimore) 2014, 93, e35. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; He, L.; Chou, N.; Wan, Y.J. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol. Sci. 2006, 92, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Kakkar, P. Lupeol prevents acetaminophen-induced in vivo hepatotoxicity by altering the Bax/Bcl-2 and oxidative stress-mediated mitochondrial signaling cascade. Life Sci. 2012, 90, 561–570. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [PubMed]
- James, L.P.; Lamps, L.W.; McCullough, S.; Hinson, J.A. Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem. Biophys. Res. Commun. 2003, 309, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar]
- Zar, P.P.K.; Morishita, A.; Hashimoto, F.; Sakao, K.; Fujii, M.; Wada, K.; Hou, D.X. Anti-inflammatory effects and molecular mechanisms of loquat (Eriobotrya japonica) tea. J. Funct. Foods 2014, 6, 523–533. [Google Scholar] [CrossRef]
- Maher, K.; Yassine, B.A.; Sofiane, B. Anti-inflammatory and antioxidant properties of Eriobotrya japonica leaves extracts. Afr. Health Sci. 2015, 15, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, W.; Xu, C.; Li, X. Biological activities of extracts from loquat (Eriobotrya japonica Lindl.): A review. Int. J. Mol. Sci. 2016, 17, 1983. [Google Scholar] [CrossRef] [PubMed]
- Baljinder, S.; Seena, G.; Dharmendra, K.; Vikas, G.; Bansal, P. Pharmacological potential of Eriobotrya japonica—An overview. Int. Res. J. Pharm. 2010, 1, 95–99. [Google Scholar]
- Banno, N.; Akihisa, T.; Tokuda, H.; Yasukawa, K.; Taguchi, Y.; Akazawa, H.; Ukiya, M.; Kimura, Y.; Suzuki, T.; Nishino, H. Anti-inflammatory and antitumor-promoting effects of the triterpene acids from the leaves of Eriobotrya japonica. Biol. Pharm. Bull. 2005, 28, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.F.; Wang, T.Y.; Zhao, B.; Lv, X.W.; Jin, Y.; Peng, L.; Yu, S.C.; Li, J. Anti-inflammatory effect of triterpenoic acids of Eriobotrya japonica (Thunb.) Lindl. Leaf on rat model of chronic bronchitis. Am. J. Chin. Med. 2009, 37, 309. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, J.; Wang, R.; Wu, Q.; Li, Y.H.; Yu, S.C.; Cheng, W.M.; Wang, Y.Y. Effect of triterpene acids of Eriobotrya japonica (Thunb.) Lindl. leaf on inflammatory cytokine and mediator induction from alveolar macrophages of chronic bronchitic rats. Inflamm. Res. 2007, 56, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, J.; Cao, Q.; Yu, S.C.; Lv, X.W.; Jin, Y.; Zhang, L.; Zou, Y.H.; Ge, J.F. Anti-oxidative effect of triterpene acids of Eriobotrya japonica (Thunb.) Lindl. leaf in chronic bronchitis rats. Life Sci. 2006, 78, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Oleanolic acid and ursolic acid: Research perspectives. J. Ethnopharmacol. 2005, 100, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Imayoshi, Y.; Kobayashi, E.; Takamatsu, Y.; Ito, H.; Hatano, T.; Sakagami, H.; Tokuda, H.; Nishino, H.; Sugita, D.; et al. Production of bioactive triterpenes by Eriobotrya japonica calli. Phytochemistry 2002, 59, 315–323. [Google Scholar] [CrossRef]
- Shih, C.C.; Ciou, J.L.; Lin, C.H.; Wu, J.B.; Ho, H.Y. Cell suspension culture of Eriobotrya japonica regulates the diabetic and hyperlipidemic signs of high-fat-fed mice. Molecules 2013, 18, 2726–2753. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Liang, K.Y.; Lin, W.C.; Kitanaka, S.; Wu, J.B. Regulation and improvement of triterpene formation in plant cultured cells of Eriobotrya japonica Lindl. J. Biosci. Bioeng. 2010, 110, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Bonesi, M.; Passalacqua, N.G.; Saab, A.; Menichini, F.; Tundis, R. Antiproliferative activities on renal, prostate and melanoma cancer cell lines of Sarcopoterium spinosum aerial parts and its major constituent tormentic acid. Anticancer Agents Med. Chem. 2013, 13, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Su, M.H.; Yao, D.H.; Zeng, B.Y.; Chang, Q.; Wang, W.; Xu, J. Anti-hepatocellular carcinoma activity of tormentic acid derived from suspension cells of Eriobotrya japonica (Thunb.) Lindl. Plant. Cell. Tissue Organ. Cult. 2017, 2017, 1–7. [Google Scholar] [CrossRef]
- Rocha Gda, G.; Simoes, M.; Lucio, K.A.; Oliveira, R.R.; Coelho Kaplan, M.A.; Gattass, C.R. Natural triterpenoids from Cecropia lyratiloba are cytotoxic to both sensitive and multidrug resistant leukemia cell lines. Bioorg. Med. Chem. 2007, 15, 7355–7360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, F.; Hu, J.; Liang, S.; Zhang, Y.; Du, G.; Zhang, C.; Cheng, Y. Antibacterial lignans and triterpenoids from Rostellularia procumbens. Planta Med. 2007, 73, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.T.; Huang, S.S.; Lin, S.S.; Amagaya, S.; Ho, H.Y.; Hou, W.C.; Shie, P.H.; Wu, J.B.; Huang, G.J. Anti-inflammatory activities of tormentic acid from suspension cells of Eriobotrya Japonica ex vivo and in vivo. Food Chem. 2011, 127, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Banno, N.; Akihisa, T.; Tokuda, H.; Yasukawa, K.; Higashihara, H.; Ukiya, M.; Watanabe, K.; Kimura, Y.; Hasegawa, J.; Nishino, H. Triterpene acids from the leaves of Perilla frutescens and their anti-inflammatory and antitumor-promoting effects. Biosci. Biotechnol. Biochem. 2004, 68, 85–90. [Google Scholar] [CrossRef] [PubMed]
- An, H.J.; Kim, I.T.; Park, H.J.; Kim, H.M.; Choi, J.H.; Lee, K.T. Tormentic acid, a triterpenoid saponin, isolated from Rosa rugosa, inhibited LPS-induced iNOS, COX-2, and TNF-alpha expression through inactivation of the nuclear factor-kappab pathway in RAW 264.7 macrophages. Int. Immunopharmacol. 2011, 11, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chang, Z.; Wang, Q. Ursane triterpenoids inhibit atherosclerosis and xanthoma in LDL receptor knockout mice. Cardiovasc. Drugs Ther. 2006, 20, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Fogo, A.S.; Antonioli, E.; Calixto, J.B.; Campos, A.H. Tormentic acid reduces vascular smooth muscle cell proliferation and survival. Eur. J. Pharmacol. 2009, 615, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, S.; Huang, R.; Tan, S.; Liang, S.; Wu, X.; Zhuo, L.; Huang, Q. Protective effect of tormentic acid from Potentilla chinensis against lipopolysaccharide/d-galactosamine induced fulminant hepatic failure in mice. Int. Immunopharmacol. 2014, 19, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.B.; Kuo, Y.H.; Lin, C.H.; Ho, H.Y.; Shih, C.C. Tormentic acid, a major component of suspension cells of Eriobotrya japonica, suppresses high-fat diet-induced diabetes and hyperlipidemia by glucose transporter 4 and AMP-activated protein kinase phosphorylation. J. Agric. Food Chem. 2014, 62, 10717–10726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, F.; Wang, K.; Liu, G.; Yang, M.; Luan, Y.; Zhao, Z. Protective effect of allyl methyl disulfide on acetaminophen-induced hepatotoxicity in mice. Chem. Biol. Interact. 2016, 249, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Suangkaew, N.; Morinaga, O.; Kariyazono, H.; Oiso, S.; Shoyama, Y. Eriobotryae folium extract suppresses LPS-induced iNOS and COX-2 expression by inhibition of NF-kappaB and MAPK activation in murine macrophages. Am. J. Chin. Med. 2010, 38, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Lauterburg, B.H.; Corcoran, G.B.; Mitchell, J.R. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in Vivo. J. Clin. Investig. 1983, 71, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.Y.; Huang, J.; Wang, H.Y.; Du, X.W.; Cheng, S.M.; Han, Y.; Wang, L.F.; Li, G.Y.; Wang, J.H. Protective effect of Zhuyeqing liquor, a Chinese traditional health liquor, on acute alcohol-induced liver injury in mice. J. Inflamm. (Lond.) 2013, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Islam, M.A.; Tanvir, E.M.; Ahmed, R.; Das, S.; Rumpa, N.E.; Hossen, M.S.; Parvez, M.; Gan, S.H.; Khalil, M.I. Satkara (Citrus macroptera) fruit protects against acetaminophen-induced hepatorenal toxicity in rats. Evid. Based Complement. Altern. Med. 2016, 2016, 9470954. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Krausz, K.W.; Shah, Y.M.; Idle, J.R.; Gonzalez, F.J. Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem. Res. Toxicol. 2009, 22, 699–707. [Google Scholar] [CrossRef] [PubMed]
- El-Shafey, M.M.; Abd-Allah, G.M.; Mohamadin, A.M.; Harisa, G.I.; Mariee, A.D. Quercetin protects against acetaminophen-induced hepatorenal toxicity by reducing reactive oxygen and nitrogen species. Pathophysiology 2015, 22, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Coito, A.J. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr. Opin. Organ. Transplant. 2011, 16, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Michael Brown, J.; Ball, J.G.; Wright, M.S.; Van Meter, S.; Valentovic, M.A. Novel protective mechanisms for S-adenosyl-L-methionine against acetaminophen hepatotoxicity: Improvement of key antioxidant enzymatic function. Toxicol. Lett. 2012, 212, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, K.M.; Park, J.; Kwak, J.H.; Kim, Y.S.; Lee, S.M. Geniposidic acid protects against d-galactosamine and lipopolysaccharide-induced hepatic failure in mice. J. Ethnopharmacol. 2013, 146, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.R.; Heck, D.E.; Yang, C.S.; Thomas, P.E.; Zhang, X.J.; DeGeorge, G.L.; Laskin, J.D.; Laskin, D.L. Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology 1998, 27, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Zentella, M.L.; Hernandez-Munoz, R. Is liver enzyme release really associated with cell necrosis induced by oxidant stress? Oxid. Med. Cell. Longev. 2016, 2016, 3529149. [Google Scholar] [CrossRef] [PubMed]
- Carayol, N.; Chen, J.; Yang, F.; Jin, T.; Jin, L.; States, D.; Wang, C.Y. A dominant function of IKK/NF-kappaB signaling in global lipopolysaccharide-induced gene expression. J. Biol. Chem. 2006, 281, 31142–31151. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Huang, S.S.; Lee, M.M.; Deng, J.S.; Huang, G.J. Anti-inflammatory activities of cardamonin from Alpinia katsumadai through heme oxygenase-1 induction and inhibition of NF-kappaB and MAPK signaling pathway in the carrageenan-induced paw edema. Int. Immunopharmacol. 2015, 25, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shen, Z.; Yu, H.; Lu, G.; Yu, Y.; Liu, X.; Zheng, P. Carnosic acid protects against acetaminophen-induced hepatotoxicity by potentiating Nrf2-mediated antioxidant capacity in mice. Korean J. Physiol. Pharmacol. 2016, 20, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yiang, G.T.; Yu, Y.L.; Lin, K.T.; Chen, J.N.; Chang, W.J.; Wei, C.W. Acetaminophen induces JNK/p38 signaling and activates the caspase-9–3-dependent cell death pathway in human mesenchymal stem cells. Int. J. Mol. Med. 2015, 36, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Bourdi, M.; Korrapati, M.C.; Chakraborty, M.; Yee, S.B.; Pohl, L.R. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochem. Biophys. Res. Commun. 2008, 374, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Conde de la Rosa, L.; Schoemaker, M.H.; Vrenken, T.E.; Buist-Homan, M.; Havinga, R.; Jansen, P.L.; Moshage, H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J. Hepatol. 2006, 44, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Williams, C.D.; McGill, M.R.; Jaeschke, H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2014, 281, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.R.; Kim, Y.H.; Hwang, J.H.; Gang, G.T.; Kim, K.S.; Lee, I.K.; Yun, B.S.; Lee, C.H. Davallialactone protects against acetaminophen overdose-induced liver injuries in mice. Food Chem. Toxicol. 2013, 58, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Ho, H.Y.; Lin, W.C.; Kitanaka, S.; Chang, C.T.; Wu, J.B. Analysis of bioactive triterpenes in Eriobotrya japonica Lindl. by high-performance liquid chromatography. J. Food Drug Anal. 2008, 16, 4. [Google Scholar]
- Huang, G.J.; Deng, J.S.; Huang, S.S.; Lee, C.Y.; Hou, W.C.; Wang, S.Y.; Sung, P.J.; Kuo, Y.H. Hepatoprotective effects of eburicoic acid and dehydroeburicoic acid from Antrodia camphorata in a mouse model of acute hepatic injury. Food Chem. 2013, 141, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.J.; Deng, J.S.; Huang, S.S.; Shao, Y.Y.; Chen, C.C.; Kuo, Y.H. Protective effect of antrosterol from Antrodia camphorata submerged whole broth against carbon tetrachloride-induced acute liver injury in mice. Food Chem. 2012, 132, 709–716. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound tormentic acid are available from the authors. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.-P.; Huang, S.-S.; Matsuda, Y.; Saito, H.; Uramaru, N.; Ho, H.-Y.; Wu, J.-B.; Huang, G.-J. Protective Effects of Tormentic Acid, a Major Component of Suspension Cultures of Eriobotrya japonica Cells, on Acetaminophen-Induced Hepatotoxicity in Mice. Molecules 2017, 22, 830. https://doi.org/10.3390/molecules22050830
Jiang W-P, Huang S-S, Matsuda Y, Saito H, Uramaru N, Ho H-Y, Wu J-B, Huang G-J. Protective Effects of Tormentic Acid, a Major Component of Suspension Cultures of Eriobotrya japonica Cells, on Acetaminophen-Induced Hepatotoxicity in Mice. Molecules. 2017; 22(5):830. https://doi.org/10.3390/molecules22050830
Chicago/Turabian StyleJiang, Wen-Ping, Shyh-Shyun Huang, Yoshikazu Matsuda, Hiroshi Saito, Naoto Uramaru, Hui-Ya Ho, Jin-Bin Wu, and Guan-Jhong Huang. 2017. "Protective Effects of Tormentic Acid, a Major Component of Suspension Cultures of Eriobotrya japonica Cells, on Acetaminophen-Induced Hepatotoxicity in Mice" Molecules 22, no. 5: 830. https://doi.org/10.3390/molecules22050830
APA StyleJiang, W.-P., Huang, S.-S., Matsuda, Y., Saito, H., Uramaru, N., Ho, H.-Y., Wu, J.-B., & Huang, G.-J. (2017). Protective Effects of Tormentic Acid, a Major Component of Suspension Cultures of Eriobotrya japonica Cells, on Acetaminophen-Induced Hepatotoxicity in Mice. Molecules, 22(5), 830. https://doi.org/10.3390/molecules22050830