1. Introduction
Increased oxidative stress and associated high levels of free radical generation have been described to occur during the pathogeneses of various diseases. Excessive reactive oxygen species (ROS) production plays a crucial role in liver diseases, including paracetamol-induced liver damage, alcoholic liver disease, nonalcoholic steatohepatitis, liver cirrhosis and fibrosis [
1,
2]. Chronic liver inflammation leads to oncogenic mutations in numerous cellular signaling cascades. Elevated levels of ROS, induced by hepatitis viruses, are considered as a key factor underlying the oncogenic effects of hepatitis B and C viruses [
3,
4]. Oxidative damage is the most likely causative process to alter and deplete mitochondria DNA, stimulate apoptotic pathways, and increase the tendency for hepatocellular necrosis [
5]. Therefore, antioxidant therapy alone or in combination with other pharmacological strategies has been proposed as a reasonable treatment of a variety of liver diseases.
Caffeic acid (CA) occurs widely in food plants and is thus widely consumed in the human diet. CA is absorbed in humans after oral intake and specific metabolites can be detected in the urine [
6]. CA exerts various biological activities, such as anti-oxidation, anti-inflammatory, and immune regulation effects [
7]. In addition, its derivatives also show unique pharmaceutical functions. For example, the phenethyl ester of caffeic acid (CAPE), an active component of honeybee propolis extract, possesses antimicrobial, antioxidant, anti-inflammatory, antitumoral, and neuroprotective properties [
8]. Due to their potential health benefits, CA and its derivatives have attracted considerable interest in the past few years. As a result, CA has been widely used as a template for the development of new chemical entities with a potential therapeutic interest in human diseases associated with oxidative stress [
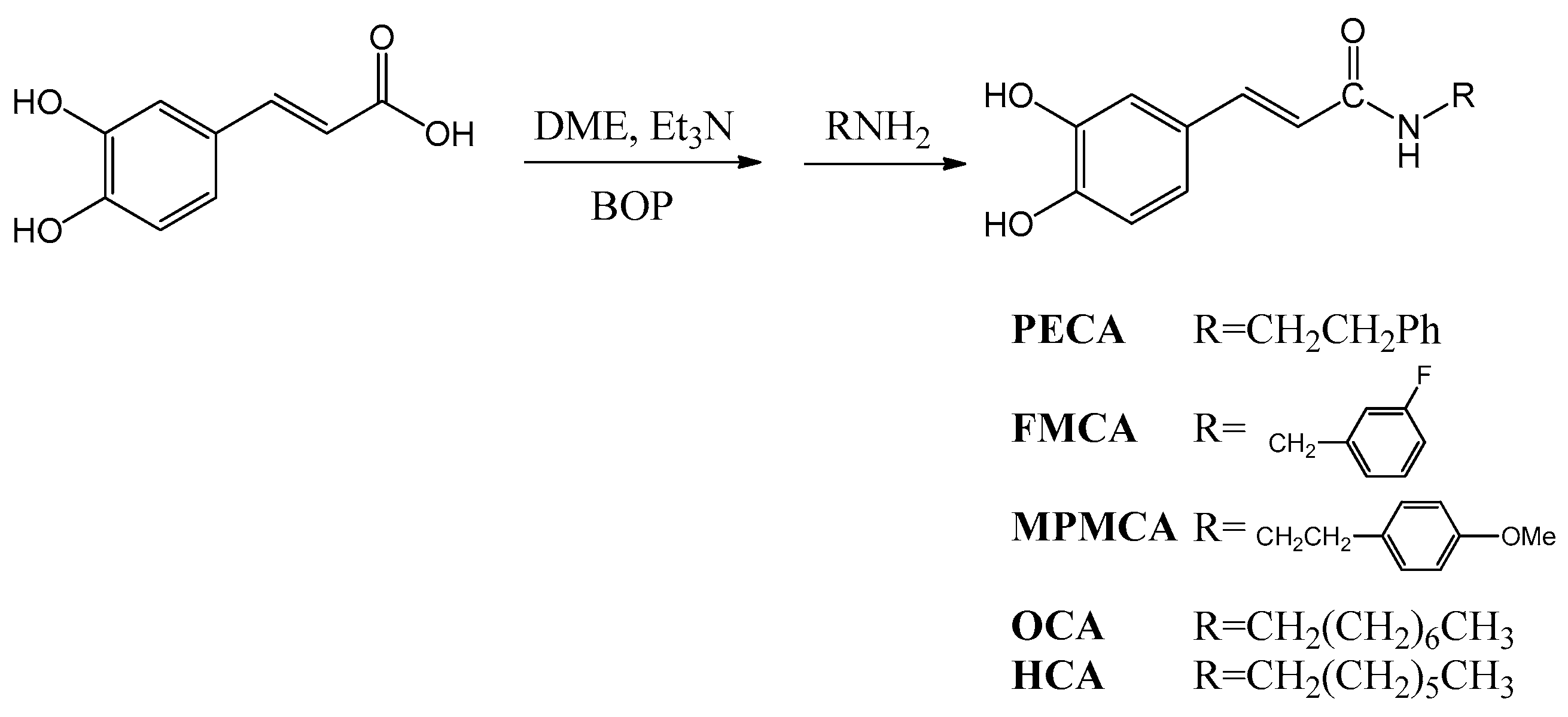
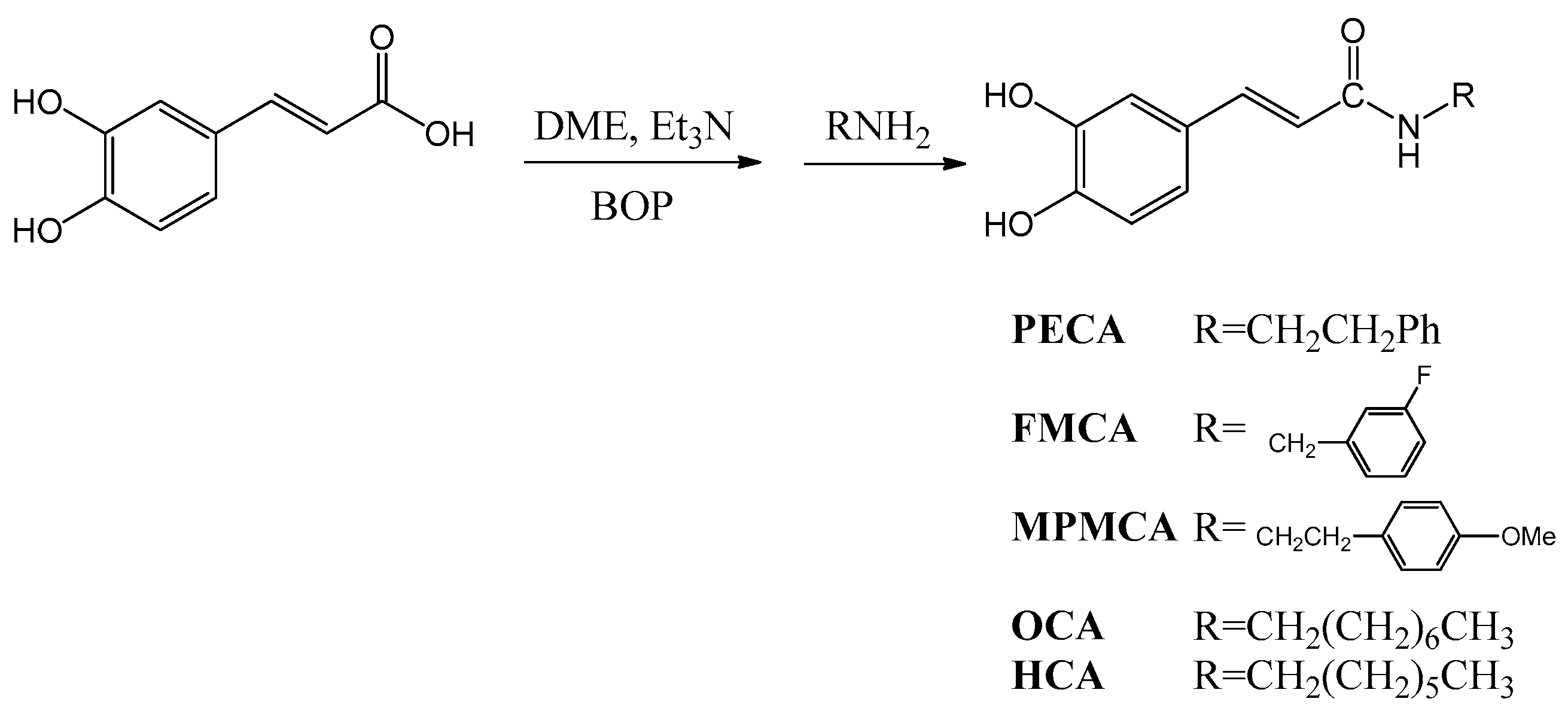
9]. As the synthesis of esters, amides and hybrids base on the CA scaffold could yield new or effective agents against oxidative stress, we prepared nine CA derivatives using CA as a substrate, including
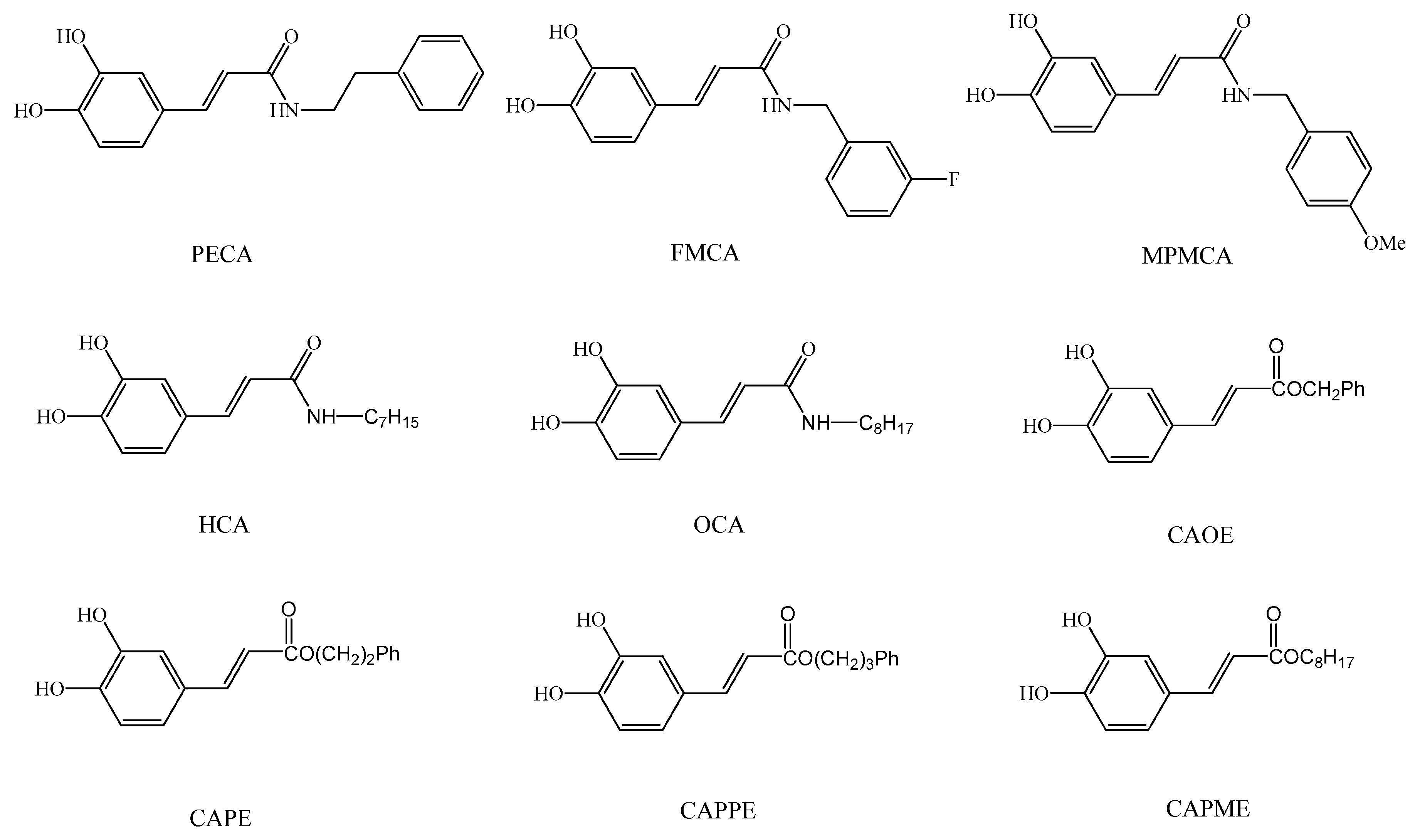
N-phenylethyl caffeamide (PECA),
N-(3-florophen)methyl caffeamide (FMCA),
N-(4-methoxyphen)methyl caffeamide (MPMCA),
N-heptyl caffeamide (HCA),
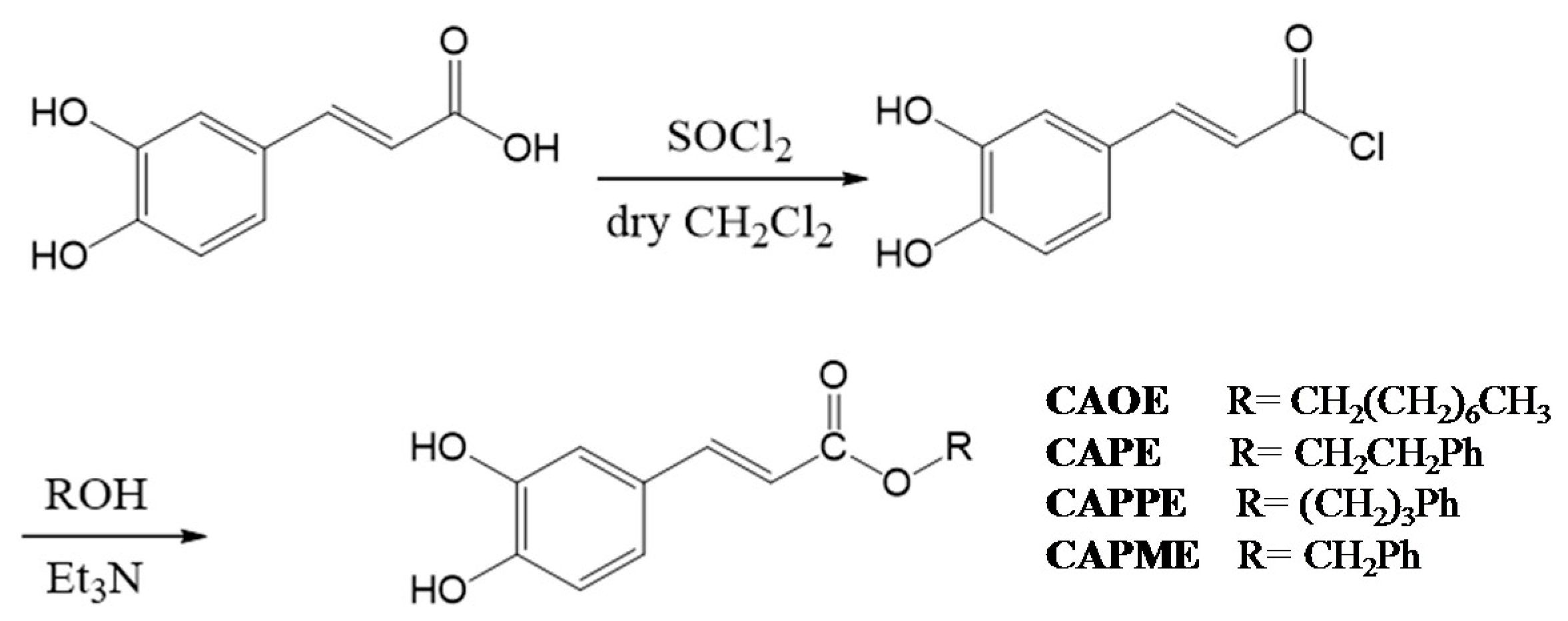
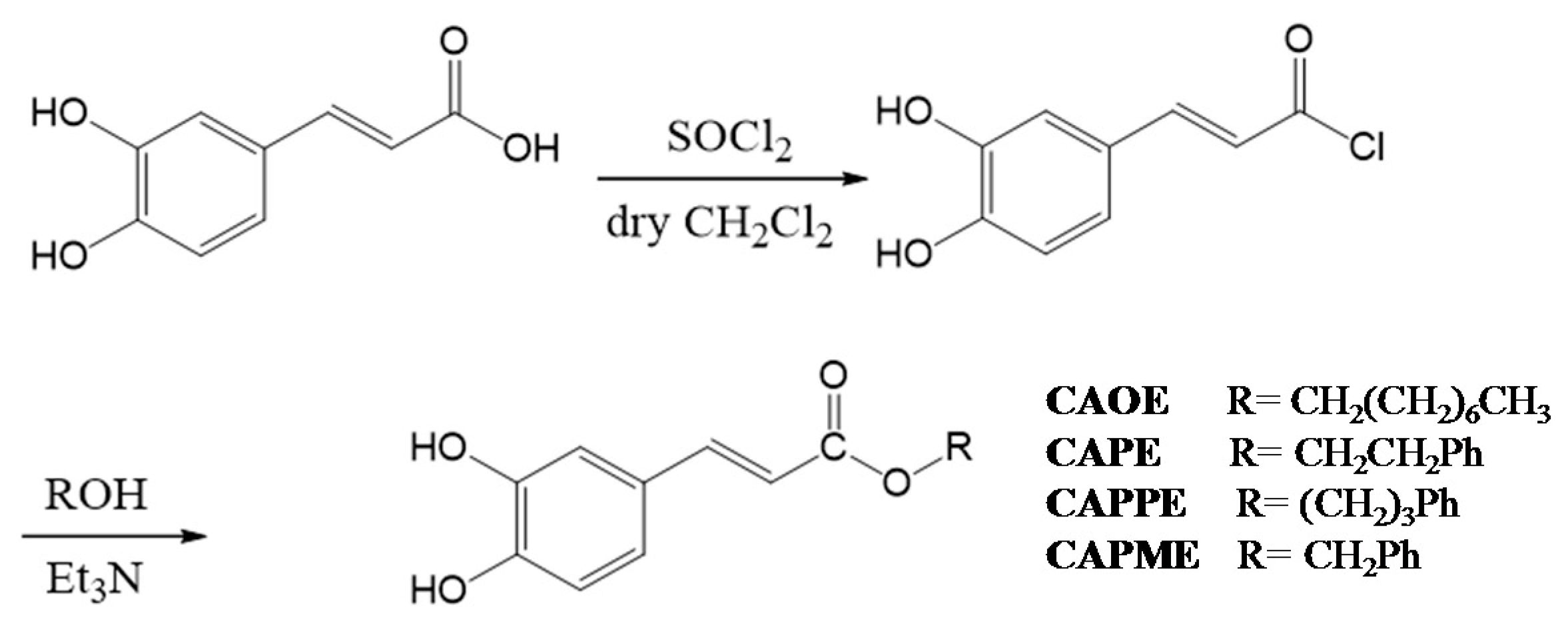
N-octyl caffeamide (OCA), octyl caffeate (CAOE), phenpropyl caffeate (CAPPE), phenethyl caffeate (CAPE; phenethyl ester of caffeic acid), and phenmethyl caffeate (CAPME) in this study (
Figure 1).
tert-Butyl hydroperoxide (
t-BHP), a pro-oxidant, induces oxidative stress and cell injury that result from the intracellular production of ROS.
t-BHP has been used as a model compound to investigate the mechanisms of cell damage initiated by oxidative stress. In hepatocyte cultures and livers,
t-BHP can be metabolized to free radical intermediates, and these intermediates result in initiating lipid peroxidation, decreasing mitochondrial membrane potential, changes in mitochondrial membrane integrity, depletion of cellular stores of glutathione (GSH), damage to cell integrity and hepatic inflammation [
10,
11,
12].
HepG2 cells have been considered to be a good tool to study the in vitro xenobiotic metabolisms, and cytoprotective, genotoxic, and antigenotoxic effects of compounds, as they retain the activity of many phase I, phase II, and antioxidant enzymes [
13,
14,
15,
16]. Here, we evaluated the protective effects of the nine CA derivatives on
t-BHP-induced oxidative injury and mitochondrial dysfunction in HepG2 cells.
3. Discussion
Numerous studies have demonstrated the biological and pharmacological actions of CA derivatives, especially CAPE. CAPE is a potent antioxidant, which effectively scavenges ROS and protects the cell membrane against lipid peroxidation [
8]. Moreover, CAPE possesses hepato-protective activity against tetrachloride or cholestasis-induced liver injury [
18,
19] and
t-BHP-induced cytotoxicity in HepG2 cells and rat liver [
20]. In addition, CAOE exerts anti-virus activity against hepatitis C virus, which is a major causative agent of chronic liver disease including cirrhosis and hepatocellular carcinoma [
21]. Caffeamide derivatives are more stable compound than CAPE and could potentially extend the beneficial effects of CAPE [
22]. In particular, PECA prevents photo-damage and protects against the metabolic consequences of diabetes mellitus [
23,
24]. Several catechol ring-fluorinated derivatives of CA phenethylamide show cytoprotective activity against H
2O
2 induced oxidative stress in human umbilical vein endothelial cells [
25]. To the best of our knowledge, there is no study relating the biological and pharmacological activities of FMCA, MPMCA, HCA, OCA, and CAPME. Moreover, the information concerning the hepato-protective action of CA derivatives is still limited, with the exception of CAPE. Therefore, the possible cytoprotective effect of the nine CA derivatives against
t-BHP-induced cell death of hepatocytes was investigated in this study.
Silymarin displays antioxidant, anti-inflammatory, and immunomodulatory actions [
26] and has been widely used as a therapeutic agent for a variety of acute and chronic liver diseases. Silymarin reverses the GSH content and reduces lipid peroxides (malondialdehyde, MDA) in isolated rat hepatocytes and human erythrocytes that were altered by
t-BHP [
27,
28,
29]. Due to its proven hepatoprotective and antioxidant properties, we used silymarin as a positive control in this study. Death of liver cells is a characteristic feature of many liver diseases, such as cholestasis, hepatitis, or ischemia/reperfusion [
30]. In this study, all nine CA derivatives exerted similar effective cytoprotection as compared to silymarin (
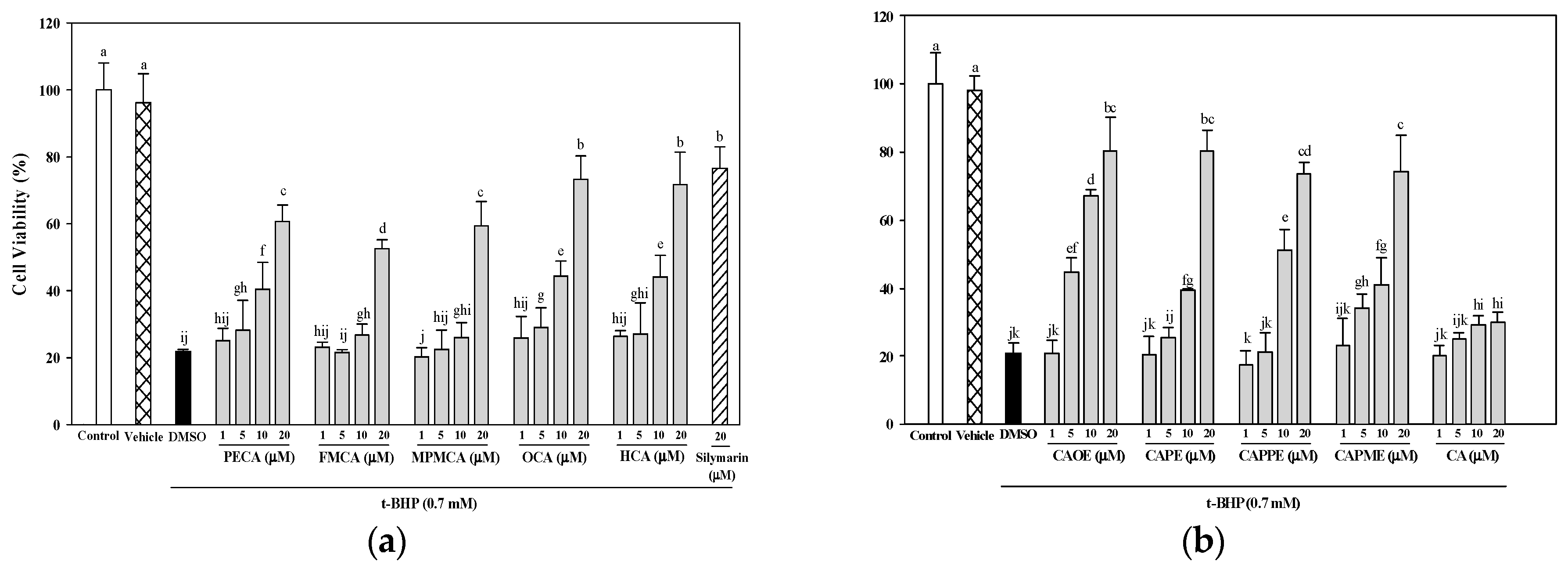
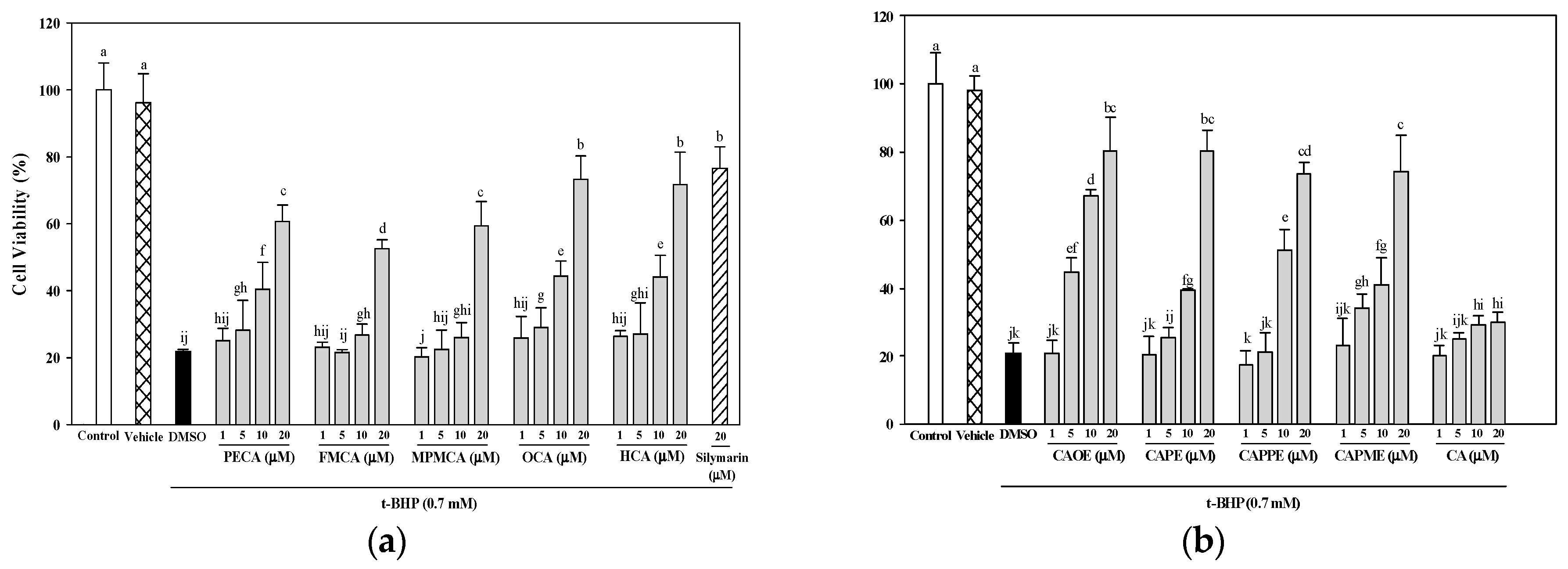
Figure 3). In addition, cells treated with the nine CA derivatives showed higher protective effect compared to cells treated with CA against oxidative cell injury (
Figure 3). Interestingly, CA-derived ester analogues are decomposed by esterases that lead to their low bioavailability in vivo [
31]. However, CAOE exhibited the most potent cytoprotective activity in this study. Concerning structure activity relationship, Wu et al. [
32] reported that the antioxidative activity of CAPE and its related compounds (CA, ferulic acid, and ethyl ferulate) depends on the hydrogen groups or catechol rings, the polarity, hydrophobicity (or partitioning properties between lipid and aqueous phases) and stability of the antioxidants. All CA derivatives used in this work have the same
ortho-dihydroxyl functionality in the catechol ring. In our present experimental condition, the crucial structural feature responsible for the better cytoprotection activities of CAOE, OCA and HCA is their hydrophobic side chains. For CAOE, OCA and HCA, the conversion of the acid group to the ester or amide group decrease the molecular polarity. The higher lipophilicity might facilitate the transport of these compounds through cell membrane and reach cell interior, which then these compounds could scavenge the ROS where they were produced.
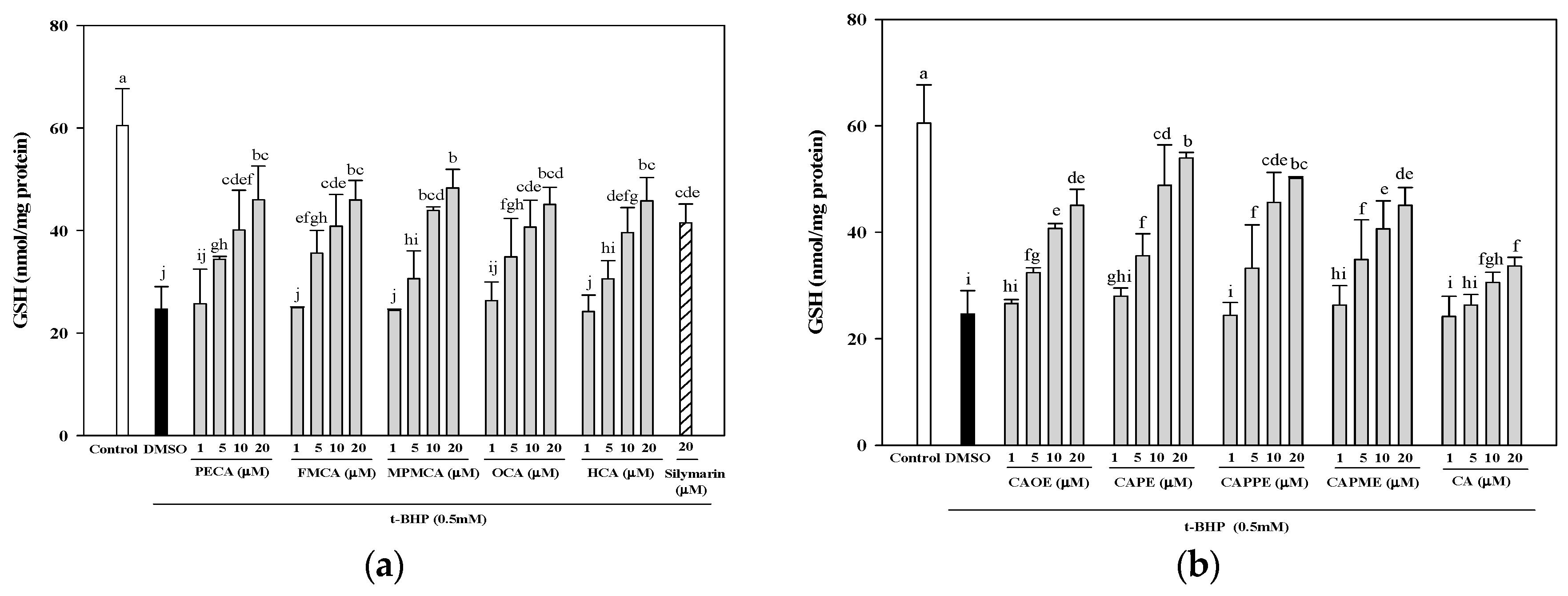
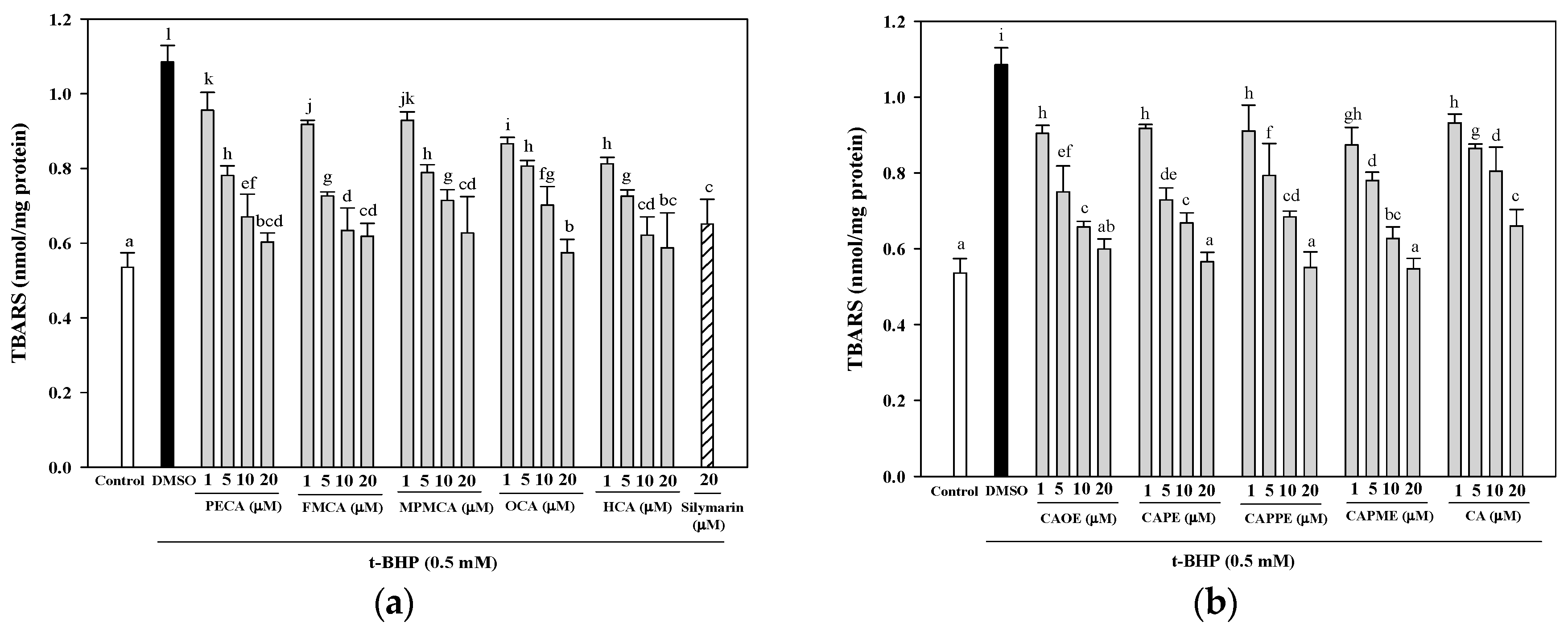
Next, investigation of whether cytoprotective action of CA derivatives is related to the antioxidant capacity was conducted. Oxidative stress is an imbalance between pro-oxidants and anti-oxidants. Overproduction of ROS or inadequate antioxidant defenses (e.g., low levels of vitamins, GSH), or both, can lead to liver injury. Intracellular MDA concentration is a direct result of membrane unsaturated fatty acid peroxidation and was used as a biomarker for
t-BHP-induced oxidation in HepG2 cells. All CA derivatives efficiently inhibited the TBARS formation (
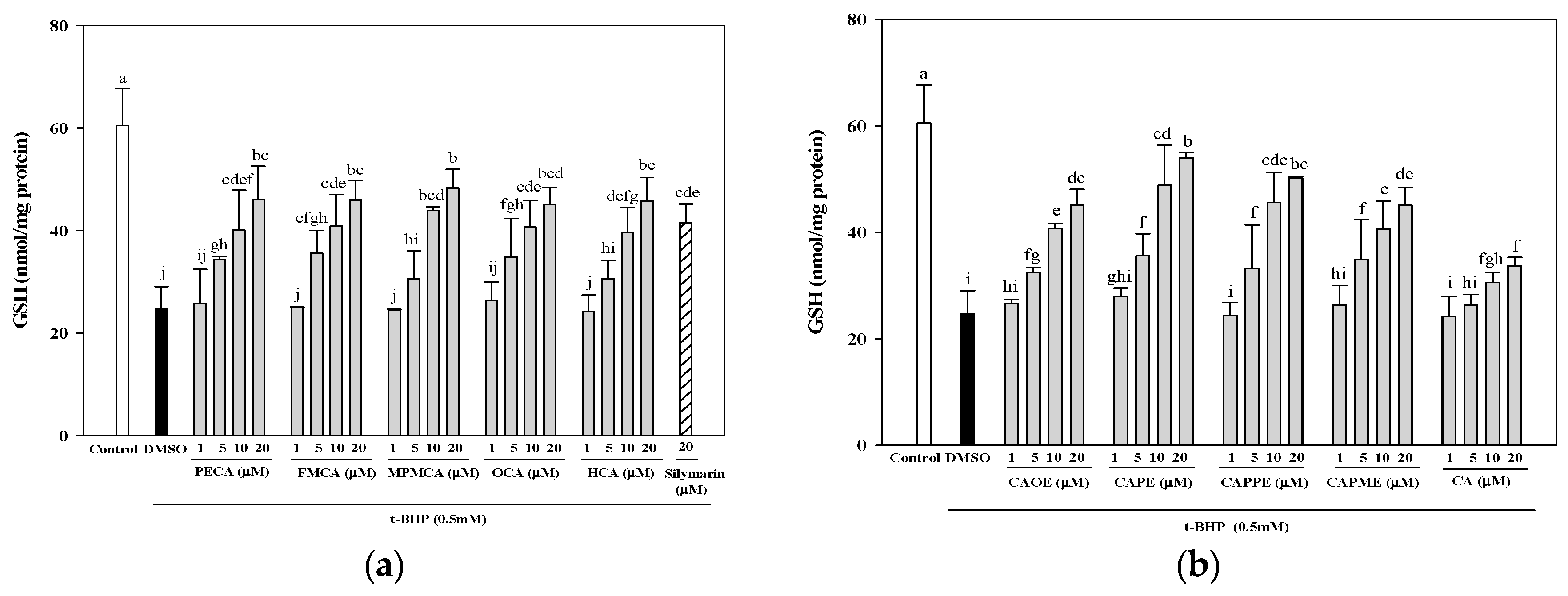
Figure 4). Moreover, GSH depletion is considered a potential biomarker of drug-induced hepatotoxicity. Because hepatic mitochondria lack catalase, GSH plays a critical role in protecting mitochondria against oxidative stress [
33]. Our data demonstrated that all CA derivatives prevented GSH depletion in
t-BHP-treated HepG2 cells (
Figure 5).
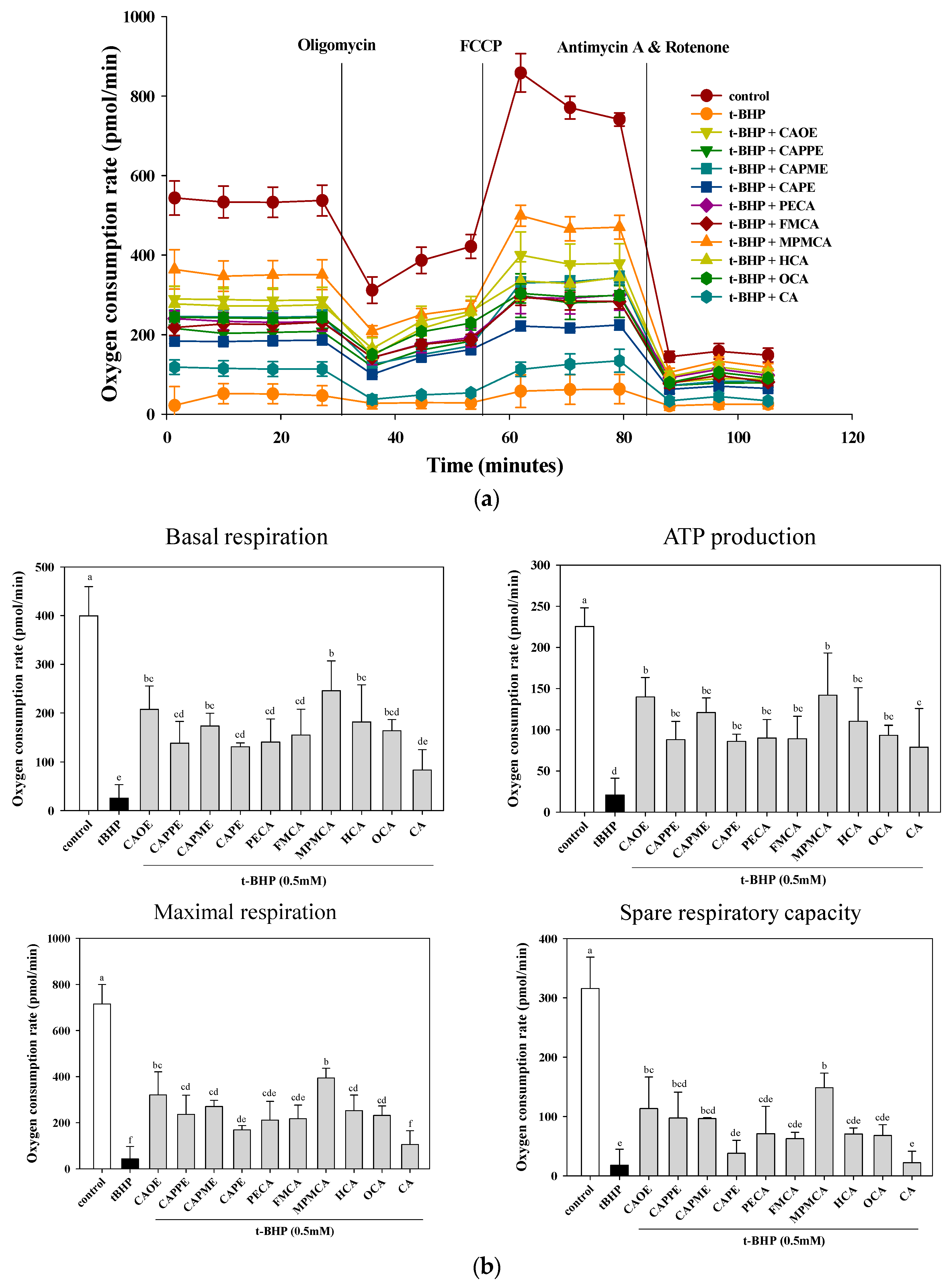
Hepatocytes are normally rich in mitochondria and each hepatocyte contains about 800 mitochondria occupying about 18% of the entire liver cell volume. Mitochondria play an important role in hepatocyte metabolism and ATP production. The mitochondrial dysfunction contributes to oxidative stress. Oxidative stress is believed to play an important role in the development of steatosis and liver lesion [
34]. We further examined whether CA derivatives preserve mitochondrial respiration and energy production capacity in the presence of
t-BHP. Mitochondrial metabolic function was determined by measuring mitochondrial oxygen consumption rate (OCR) using an XF24 analyzer. After
t-BHP treatment, OCR of HepG2 cells rapidly declined, confirming mitochondrial dysfunction (
Figure 6) and consistent with previous studies in myotubes [
35]. Pre-treatment with CA derivatives preserved mitochondrial OCR in
t-BHP-treated cells (
Figure 6). All CA derivatives herein prevent mitochondrial dysfunction and energy deficits caused by oxidant exposure. Mitochondrial dysfunction is a common mechanism in the etiology of organ injuries and diseases caused by metabolic insufficiency. Our results highlight the potential of CA derivatives to treat disorders characterized by mitochondrial impairment.
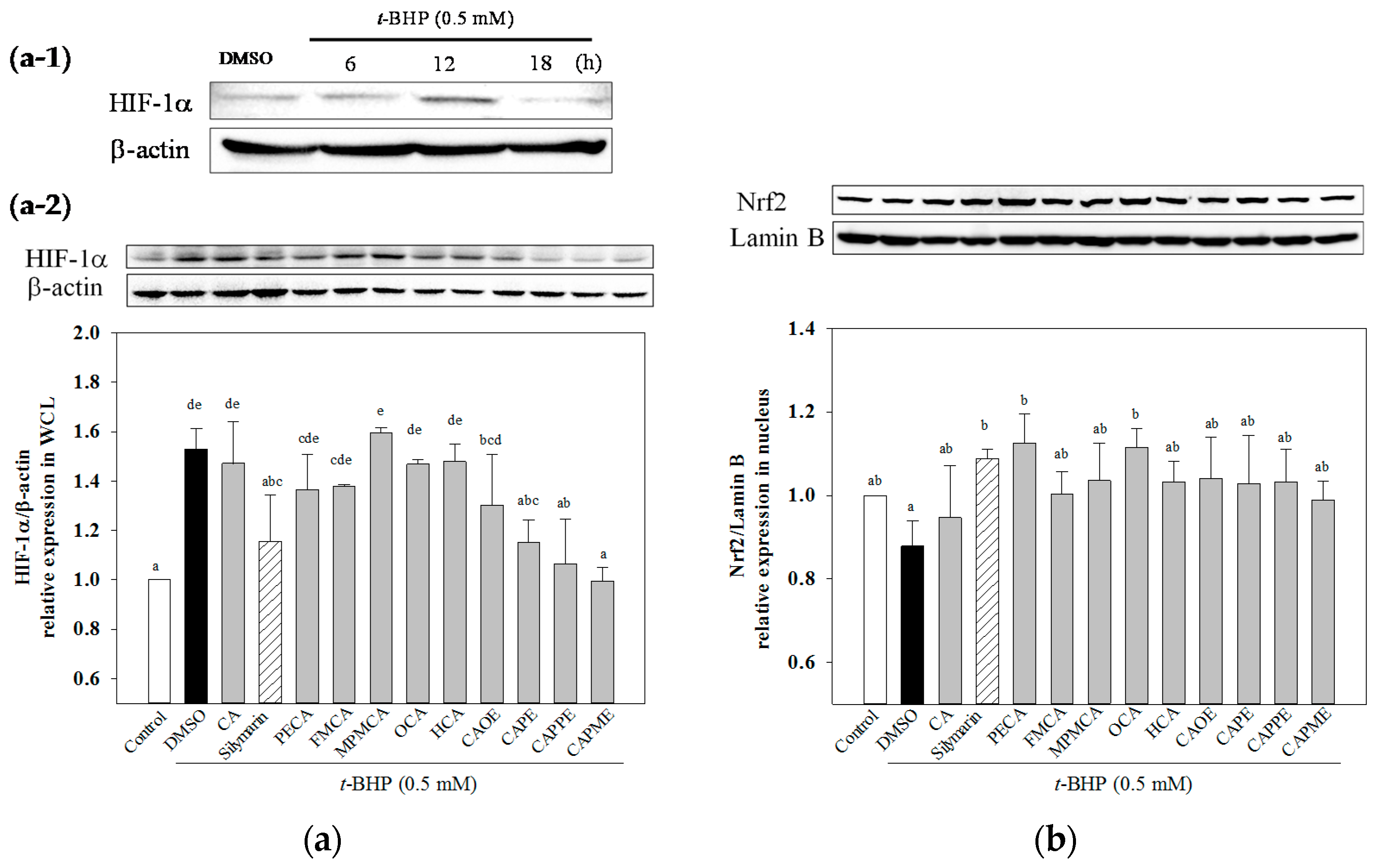
HIF-1α is a heterodimeric transcription factor that plays a key role in the signaling pathways transmitting information regarding cellular oxygen levels. There is evidence to suggest that ROS regulate HIF-1α stabilization under normoxia. Addition of exogenous H
2O
2 stabilizes HIF-1α protein under normoxia [
36]. After 12 h treatment of
t-BHP, an exogenous inducer of oxidative stress, increased HIF-1α protein level (
Figure 7a-1). While, CA derivatives except OCA and HCA significantly decreased
t-BHP-induced HIF-1α protein level under normoxia (
Figure 7a-2). However, the mechanisms explaining how HIF-1α could mediate
t-BHP-induced damage remains unclear. Nrf2 signal plays a crucial role against oxidative stress and cytotoxicity. The enhanced activation of Nrf2 exerts hepatoprotective activity in different oxidative stress models, such as acetaminophen or CCl
4-induced liver injury [
5,
37]. Activation of Nrf2 protects mitochondria from oxidative stress [
5]. CAPE has been shown to protect liver cells against CCl
4-induced oxidative stress and inhibit activation of hepatic stellate cells through up-regulated Nfr2 expression [
38]. In this study, we demonstrated that all CA derivatives increased nuclear Nrf2 accumulation (
Figure 7b). Taken together, these findings suggested that Nrf2 signaling pathways could play a key role in mediating the hepatoprotective effects of CA derivatives.
In conclusion, our data suggested that CA derivatives have protective effects against oxidant-induced hepatotoxicity, at least in part, by attenuation of lipoperoxidation, preservation of glutathione, inhibition of mitochondrial dysfunction, and regulation of the master regulator of antioxidant response Nrf2. However, further studies are still required to validate the detailed molecular mechanisms involved in the protective effect of CA derivatives.
4. Materials and Methods
4.1. General Information
All chemicals, including caffeic acid, N,N-dimethylformamide, trimethylamine, BOP, CH2Cl2, EtOAc, HCl, thionyl chloride, SOCl2, NaHCO3 (aq.), and MgSO4, were of analytical-grade purity. They were purchased from the branch of Merck in Taipei, Taiwan, and the branch of Sigma Aldrich in Taichung, Taiwan. UV spectra were recorded on a Shimadzu UV-1601PC spectrophotometer. Infrared (IR) spectra were measured on a Perkin-Elmer-983G FT-IR spectrophotometer. 1H- and 13C-NMR and 2D NMR spectra were obtained on a Bruker DRX-500 FT-NMR spectrometer with tetramethylsilane (TMS) as the internal standard. EI-MS were measured with a Jeol-JMSHX300 mass spectrometer. Silica gel (230–400 mesh; Merck & Co., Inc.) was used for column chromatography (CC). Pre-coated silica gel (60 F-254; Merck & Co., Inc.) plates were used for TLC. The spots on TLC were detected by spraying with 5% H2SO4 and then heating at 100 °C.
4.2. Synthesis of Caffeamide Derivatives
Compounds were obtained by the following method: caffeic acid (100 mg, 0.56 mmol) was dissolved in the mixture of
N,
N-dimethylformamide (DMF, 1 mL) and trimethylamine (80 μL), and then the appropriate amine derivative (80 μL, assumes that they all have the same density and molecular weight, 1.2 eq.) was added to the reaction solution cooled in an ice bath (0 °C), and then BOP (295 mg, 1.2 eq.) in CH
2Cl
2 (5 mL) was added to the reaction mixture, followed by stirring for 30 min (
Scheme 1). The reaction mixture was kept at room temperature for 2 h. The solvent was then evaporated under vacuum, and the crude mixture was partitioned between EtOAc and H
2O. The organic layer was washed with 3 N aqueous HCl and then with 10% aqueous NaHCO
3 solution. The product was purified by chromatography on silica gel. The yields of
PECA,
FMCA,
MPMCA,
OCA, and
HCA were 70%~80%.
N-Phenylethyl caffeamide (PECA): Pale white needle-like crystals; m.p.: 148–149 °C; IR νmax (cm−1): 3288, 1642, 1591, 1523, 1361, 1279, 1036, 975, 849. 1H-NMR (CD3COCD3, 500 MHz): δ 2.84 (2H, t, J = 6.8 Hz), 3.53 (2H, q, J = 6.8 Hz), 6.43 (1H, d, J = 15.2 Hz), 6.83 (1H, d, J = 8.1 Hz), 6.92 (1H, dd, J = 8.1, 1.8 Hz), 7.07 (1H, d, J = 1.8 Hz), 7.15–7.30 (5H, m), 7.35 (-NH, br. s), 7.43 (1H, d, J = 15.2 Hz), 8.20 (-OH, s), 8.42 (-OH, s). EI-MS m/z (%): 283 (M+, 17), 178 (22), 163 (100); UV (MeOH) λmax (logε): 322 (4.42), 296 (4.36), 245 (4.30), 216 (4.61)nm.
N-(3-Florophen)methyl caffeamide (FMCA): White solid; m.p.: 186–188 °C; IR νmax (cm−1): 3436, 1652, 1619, 1520, 1440, 1358, 1115, 1015, 976, 852, 818. 1H-NMR (CD3OD, 400 MHz): δ 4.47 (2H, s), 6.40 (1H, d, J = 15.6 Hz), 6.75 (1H, d, J = 8.0Hz), 6.90 (1H, dd, J = 8.0, 2.0 Hz), 6.98 (1H, m), 7.01 (1H, d, J = 2.0 Hz), 7.03 (1H, m), 7.12 (1H, m), 7.33 (1H, m), 7.43 (1H, d, J = 15.6 Hz). EI-MS m/z (%): 287 (M+, 100), 247 (35), 163 (95), 124 (90), 109 (50); UV(MeOH) λmax (logε): 324 (4.37), 296 (4.30), 245 (4.28), 251(4.54) nm.
N-(4-Methoxyphen)methyl caffeamide (MPMCA): Solid; m.p.: 170–171 °C; IR νmax (cm−1): 3283, 1653, 1613, 1520, 1447, 1374, 1116, 1009, 850. 1H-NMR (CD3COCD3, 500 MHz): 3.75 (3H, s), 4.44 (2H, d, J = 6.2 Hz), 6.49 (1H, d, J = 15.8 Hz), 6.81–6.94 (4H, m), 7.07 (1H, d, J = 1.6 Hz), 7.25 (2H, d, J = 8.8 Hz), 7.45 (1H, d, J = 15.8 Hz), 7.59 (1H, br. s, -NH), 8.17 (1H, s, -OH), 8.38 (1H, s, -OH). EI-MS m/z (%): 299 (M+, 7), 163 (100); UV (MeOH) λmax (logε): 321(4.16), 295 (4.13), 284 (4.13), 245 (4.16) nm.
N-Octyl caffeamide (OCA): White solid; m.p.: 111–112 °C; IR νmax (cm−1): 3286, 1642, 1588, 1520, 1363, 1277, 1112, 975, 811. 1H-NMR (CD3COCD3, 400 MHz): δ 0.84 (3H, t, J = 6.6 Hz), 1.24 (10H, m), 1.52 (2H, quin, J = 6.6 Hz), 3.30 (2H, q, J = 6.6 Hz), 6.47, 7.42 (each 1H, d, J = 15.6 Hz), 6.82 (1H, d, J = 8.2 Hz), 6.90 (1H, dd, J = 8.2, 1.8 Hz), 7.09 (1H, d, J = 1.8 Hz). EI-MS m/z (%): 291 (M+, 18), 220 (8), 193 (11), 178 (31), 163 (100), 145 (8), 135 (13), 128 (22), 117 (11), 98 (8), 89 (19), 84 (12); UV (MeOH) λmax (logε): 322 (4.32), 294 (4.26), 238 (3.92), 219 (4.37) nm.
N-Heptyl caffeamide (HCA): White solid; m.p.: 126–127 °C; IR νmax (cm−1): 3347, 1642, 1588, 1545, 1510, 1363, 1266, 1112, 975, 809. 1H-NMR (CD3COCD3, 400 MHz): δ 0.82 (3H, t, J = 6.6 Hz), 1.20 (8H, m), 1.52 (2H, quin, J = 6.6 Hz), 3.32 (2H, q, J = 6.6 Hz), 6.51, 7.48 (each 1H, d, J = 15.6 Hz), 6.83 (1H, d, J = 8.1 Hz), 6.92 (1H, dd, J = 8.1, 1.5 Hz), 7.11 (1H, d, J = 1.5 Hz). EI-MS m/z (%): 277 (M+, 42), 192 (10), 178 (32), 163 (100), 145 (12), 135 (20), 114 (14), 98 (8); UV (MeOH) λmax (logε): 322 (4.23), 295 (4.17), 2.34 (4.16), 217 (4.30) nm.
4.3. Synthesis of Caffeate Derivatives
Caffeate derivatives were obtained as follows: Caffeic acid (200 mg) and thionyl chloride (4 mL) dissolved in dry dichloromethane (10 mL), were heated under reflux for 4 h. The solvent and excess SOCl
2 was removed under vacuum, and then ROH (1.2 equiv.) in triethylamine (0.08 mL) was added dropwise under dry conditions. The reaction mixture was stirred for 24 h at ambient temperature, and then evaporated under vacuum. The residue was partitioned successively between EtOAc and H
2O, and then the EtOAc layer was washed with 3N aqueous HCl then 10% NaHCO
3 (aq.), dried over MgSO
4 and concentrated under vacuum. The product was purified by column chromatography on silica gel. The final products (65%~75% yield) were recrystallized from acetone to obtain pure crystals. (
Scheme 2).
Octyl caffeate (CAOE): White solid; m.p.: 98–100 °C; IR νmax (cm−1): 3488, 3340, 1675, 1630, 1275, 1184, 972, 812; 1H-NMR (CD3COCD3): δ 0.85 (3H, t, J = 6.7 Hz), 1.26 (10H, m), 1.67 (2H, quin, J = 6.7 Hz), 4.16 (2H, t, J = 6.7 Hz), 6.23, 7.54 (each 1H, d, J = 15.9 Hz), 6.84 (1H, d, J = 8.2 Hz), 6.96 (1H, dd, J = 8.2, 2.0 Hz), 7.06 (1H, d, J = 2.0 Hz), 8.26 (2H, brs, -OH). EI-MS m/z (%): 292 (M+, 27), 180 (100), 163 (47), 145 (8), 136 (18), 134 (12), 89 (13).
Phenethyl caffeate (CAPE): Pale white needle-like crystals; m.p.: 126–128 °C, IR (KBr) νmax 3400, 1709, 1605, 1519, 1352, 1282, 1192 cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 2.95 (2H, t, J = 6.9 Hz, -OCH2CH2Ph), 4.32 (2H, t, J = 6.9 Hz, -OCH2CH2Ph), 6.23, 7.58 (each 1H, d, J = 15.9 Hz), 6.76 (1H, d, J = 8.1 Hz), 6.99 (1H, d, J = 8.2, 2.0 Hz), 7.03 (1H, d, J = 2.0 Hz), 7.22 (1H, m, H-4’), 7.18–7.30 (5H, m); EI-MS m/z (%): 284 (M+), 179, 161, 135 (100%), 117.
Phenpropyl caffeate (CAPPE): Yellow solid; m.p.: 116–118 °C; IR (KBr) νmax (cm−1): 3482, 3327, 1671, 1629, 1597, 1179, 973, 809, 696; 1H-NMR (CDCl3): δ 2.01 (2H, quin, J = 6.8 Hz, -OCH2CH2CH2Ph), 2.72, 4.20 (each 2H, t, J = 6.8 Hz), 6.25, 7.55 (each 1H, d, J = 15.9 Hz), 6.85 (1H, d, J = 8.2 Hz), 6.98 (1H, dd, J = 8.2, 1.8 Hz), 7.08 (1H, d, J = 1.8 Hz), 7.10–7.30 (5H, m); EI-MS m/z (%): 298 (M+, 18), 180 (100), 163 (19), 135 (8), 118 (30), 117 (30), 91 (24).
Phenmethyl caffeate (CAPME): Light yellowish powder; m.p.: 150–152 °C, IR νmax (cm−1): 3464, 3327, 1689, 1600, 1277, 1185. 1H-NMR (DMSO-d6, 400 MHz): δH 5.19 (1H, s), 6.32, 7.52 (each 1H, d, J = 15.9 Hz), 6.75 (1H, d, J = 8.1 Hz), 7.02 (1H, dd, J = 2.0, 8.1 Hz), 7.05 (1H, d, J = 2.0 Hz), 7.34–7.41 (5H, m), 9.20, 9.59 (each 1H, s, -OH). EI-MS m/z (%): 270 (9%, M+), 208 (19), 163 (50), 136 (33), 91 (100), 89 (32), 77 (19), 65 (20), 51 (18).
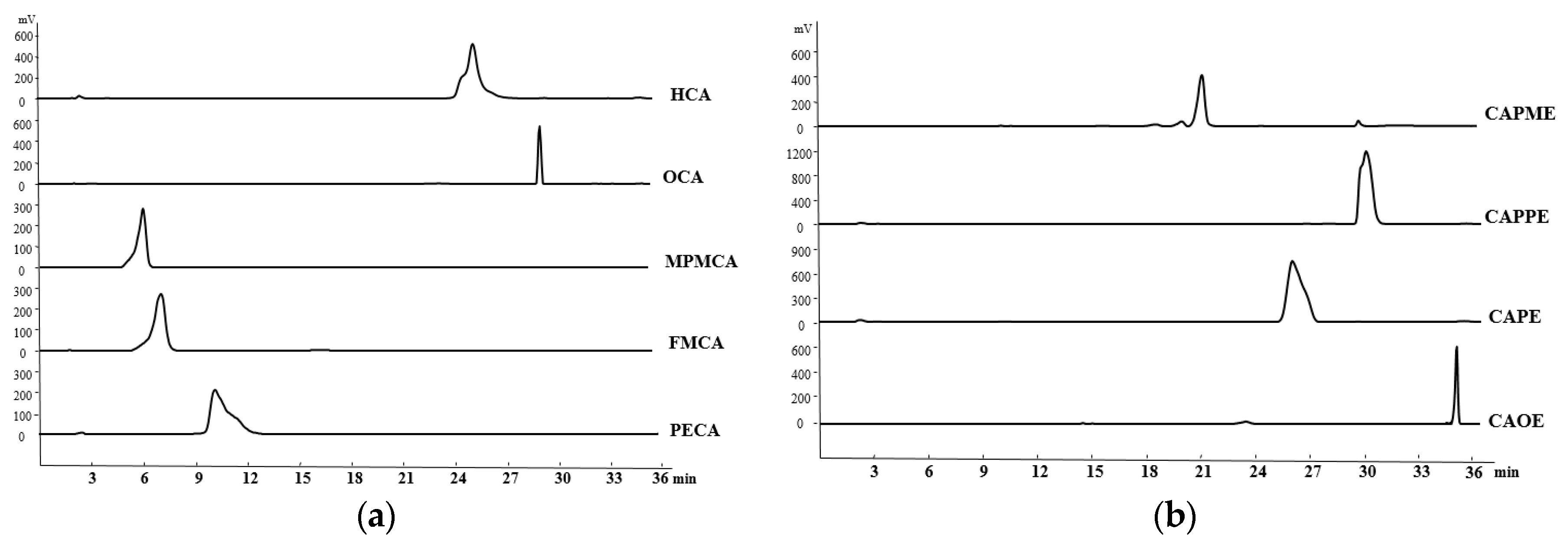
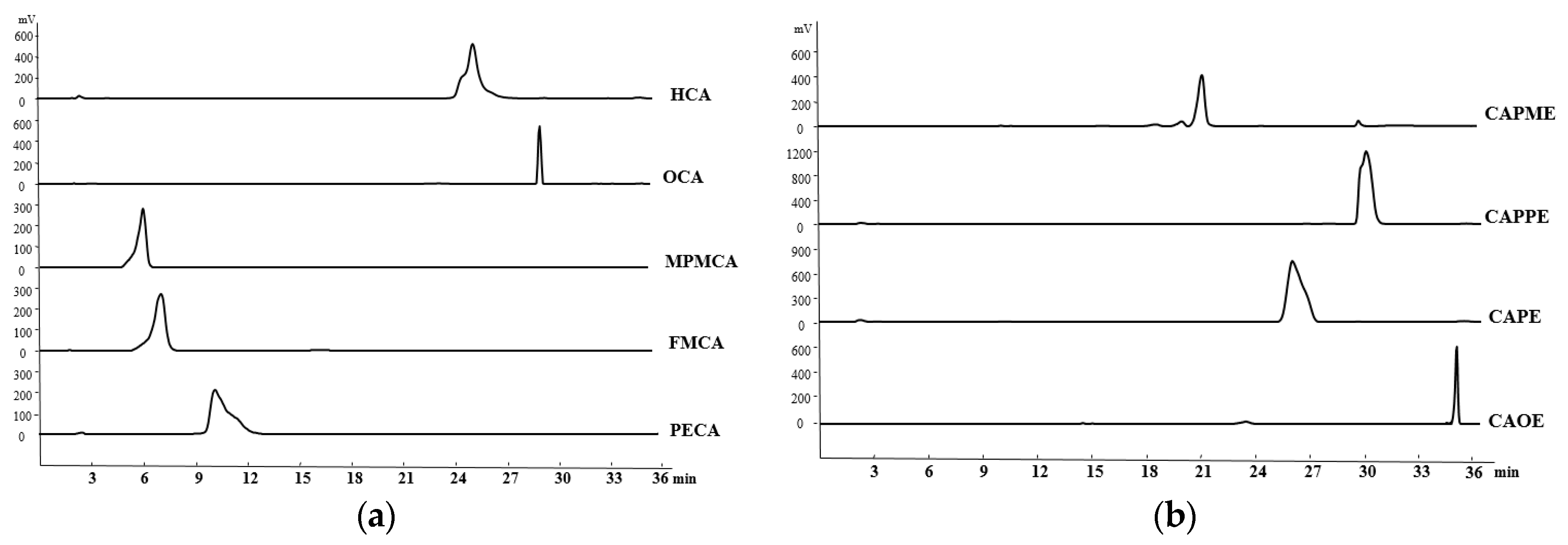
4.4. HPLC Determination of the CA Derivatives
The purity of CA derivatives was measured by HPLC on a system (Ecom, Prague, Czech Republic) equipped with gradient pumps (Ecom LCP 4100), a UV detector (Ecom LCD 2084) and a LiChrospher
® 100 RP-18E (5 µm) HPLC column (125 mm × 4 mm i.d., Merck Millipore (Darmstadt, Germany). The mobile phase consisting of a mixture of solvent A (water/methanol, 98:2), and solvent B (methanol/acetic acid, 98:2) was run in the following gradient mode: 0–7 min, from 50% A to 40% A with a flow rate of 0.5 mL/min; 7–12 min, from 40% A to 30% A with a flow rate of 0.3 mL/min; and 12–28 min, from 30% A to 20% A with a flow rate of 0.2 mL/min. The UV detector was at 280 nm. Chromatographic processing was done using the Peak-ABC Chromatography Data Handling System. The purities of PECA, FMCA, MPMCA, OCA, HCA, CAOE, CAPE, CAPPE, and CAPME were 96.4%, 97.1%, 99.7%, 95.2%, 95.6%, 98.6%, 96.7%, 96.4%, and 98.3%, respectively (
Figure 8).
4.5. Determination of the Viability of HepG2 Cells
The human HepG2 cell line (BCRC 60025) was obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). Cells were maintained in DMEM (with 5.5 mM glucose) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified atmosphere with 5% CO2. HepG2 cells (5 × 104 cells/well) were cultured in 96-well culture plates and 24 h later treated with various concentrations of tested samples for the indicated times. Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. MTT (Sigma-Aldrich, St. Louis, MO, USA; 100 µL, 0.5 mg/mL) was added to each well and incubated at 37 °C for 3 h. The reaction was terminated by replacing the MTT-containing medium with 500 µL of dimethyl sulfoxide (Sigma-Aldrich), and the formazan salts were dissolved by gentle shaking for approximately 5 min at room temperature. The optical density (OD) of each well was measured at 595 nm using a microplate reader. Each assay was completed in triplicate wells, and each experiment was repeated three times.
4.6. Cytoprotective Effects of CA Derivatives
HepG2 cells (5 × 104 cells/well) were seeded in 96-well plates. After 24 h, the culture medium was replaced containing various concentrations of caffeic acid and its derivatives (1, 5, 10, or 20 μM) or 20 μM silymarin (Sigma-Aldrich; as a positive control). Commercially available caffeic acid (Sigma-Aldrich) was used as a reference. After 24 h, the culture medium containing the samples was discarded, and the cells were treated for 20 h with 0.7 mM t-BHP (Sigma-Aldrich) to induce oxidative stress. Cell viability, as an indication of the cytoprotective effects of each of the samples, was then evaluated using the MTT assay.
4.7. Measurement of Lipid Peroxidation in HepG2 Cells
HepG2 cells were seeded in a 6-well plate at a density of 7.5 × 105 cells/well. After 24 h, the culture medium was replaced with FBS-free medium containing different concentrations of CA and its derivatives (1, 5, 10, or 20 μM). After 24 h, the cells were treated for 24 h with 0.5 mM t-BHP to induce oxidative stress. After treatment, the cells were harvested and lysed by sonication. The lysates were centrifuged at 10,000× g for 10 min at 4 °C and used for the lipid peroxidation assay using TBARS assay. The absorbance was measured at 535 nm calibrated using solutions of authentic malondialdehyde (Sigma-Aldrich). The protein concentration was determined using the Bradford dye-binding method (Bio-Rad, Hercules, CA, USA). The results were expressed as nmol/mg protein.
4.8. Determination of Total GSH Level
HepG2 cells were seeded in 6-well plates at a density of 7.5 × 105 cells/well. After 24 h, the culture medium was replaced with FBS-free medium containing various concentrations of CA and its derivatives (1, 5, 10, and 20 μM). After 24 h, the cells were treated for 24 h with 0.5 mM t-BHP to induce oxidative stress. After treatment, the cells were harvested and lysed using a sonicator. The lysates were centrifuged at 10,000× g for 10 min, and used for the determinations of protein and GSH. The GSH levels in HepG2 cells were determined by a commercially available GSH assay kit (Cayman Chemical, Ann Arbor, MI, USA). The results were expressed as nmol GSH/mg of protein.
4.9. Measurement of Mitochondrial Oxygen Consumption Rate
HepG2 cells were cultured at 1.5 × 104 cells/well in XF24 cell culture plates (Seahorse Bioscience, Billerica, MA, USA). After 42 h, cells were treated with 20 μM of CA and its derivatives. After 6 h incubation, the media was replaced with 250 μL DMEM in the presence of 0.5 mM t-BHP for 2 h. Next, the media was exchanged with 675 μL XF Assay Medium-modified DMEM (Seahorse Bioscience) incubated at 37 °C without CO2 for 1 h. Mitochondrial oxygen consumption rate (OCR) was measured using an XF24 analyzer and software (Seahorse Bioscience). The cellular metabolic capacity and mitochondrial coupling were evaluated by injecting the following metabolic probes using the liquid injection ports on the XF instrument sensor plate: oligomycin, which inhibits the mitochondrial F1F0-ATPase, FCCP (carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone), which is a protonophore that depolarizes the inner mitochondrial membrane, and the mitochondrial electron transport chain (ETC) inhibitors of complex I (rotenone) and complex III (antimycin A). The final concentrations of mitochondrial inhibitors were at 1 μM oligomycin, 1 µM FCCP, and 0.5 μM antimycin/rotenone. Basal respiration is the baseline oxygen consumption reading before compounds are injected. Maximal respiration represents the maximum OCR measurement value after FCCP injection. Spare respiratory capacity is calculated by noting the OCR response to FCCP.
4.10. Immunoblotting Analysis
HepG2 cells were grown in 100 mm culture dishes (2 × 106 cells/dish) for 24 h, and then incubated with silymarin, CA, and CA derivatives (20 μM). After 24 h incubation, the cells were treated with 0.5 mM t-BHP to induce oxidative stress for 12 h. Cells were then harvested and washed with PBS. Whole cell lysates were prepared in a lysis buffer (Cell Signaling, Beverly, MA, USA) containing 10 mM phenylmethylsulfonyl fluoride (PMSF). The cell lysates were sonicated and cleared by centrifugation at 4 °C, 14,000× g for 10 min. Whole cell lysates were examined for HIF-1α expression. The nuclear proteins from harvested cells were prepared using the nuclear fractionation kit (Imgenex; San Diego, CA, USA) and were examined for Nrf2 expression. Protein concentrations were determined by DC protein assay (Bio Rad). Aliquots of the cell lysates (each containing 20 μg of protein) or nuclear protein (10 μg) were boiled for 5 min and electrophoresed on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel. Following SDS-polyacrylamide gel electrophoresis, proteins were transferred to PVDF membranes. Membranes were blocked by incubation in gelatin-NET buffer at room temperature, and then incubated with 1:1000 dilution of primary antibodies of Nrf2 (GeneTex, Irvine, CA, USA), HIF-1α (GeneTex), lamin B (GeneTex) and β-actin (Sigma-Aldrich), followed by horseradish peroxidase-conjugated secondary antibody according to the manufacturer’s instructions. The immunoreactive proteins were detected using the enhanced ECL chemiluminescence Western Blotting Detection System (ChemiDoc XRS, Bio-Rad). Signal strengths were quantified using densitometric program (Image Lab, Bio-Rad).
4.11. Statistical Analysis
All data are presented as means ± SD. Statistical analyses were performed using the SPSS 19.0 statistical package (Chicago, IL, USA). The data were evaluated for statistical significance with the one-way ANOVA and Duncan multiple comparison test. A p value of <0.05 was considered statistically significant. The concentration of tested samples required to inhibit 50% of the activity under the assay conditions was determined from dose-response curves and defined as the IC50 value.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}