
Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Overview of the ABC Transporters
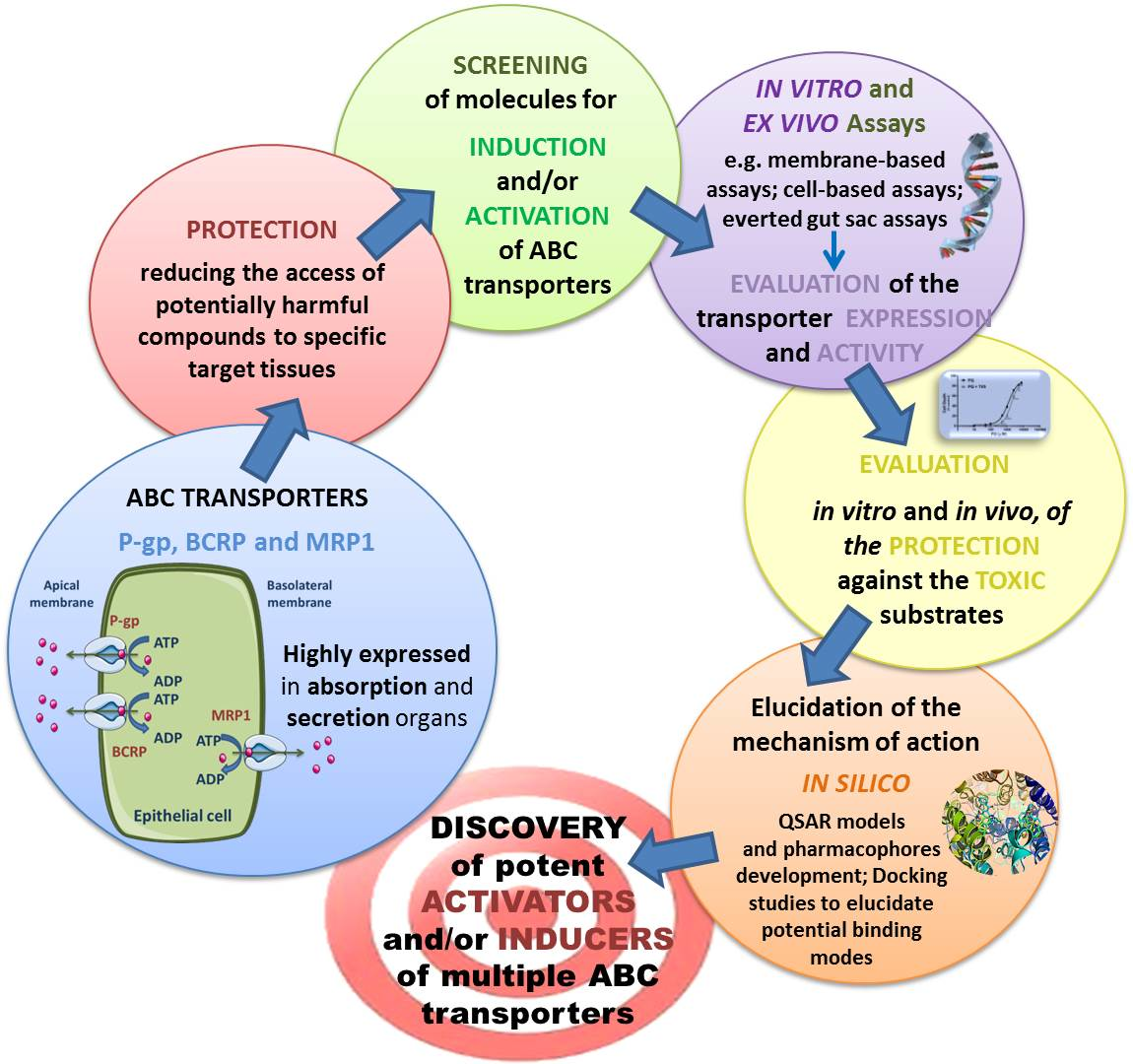
Overview of Modulators of the ABC Transporters: Activators and Inducers
3. Study Models for ABC Transporters
3.1. Cellular Models
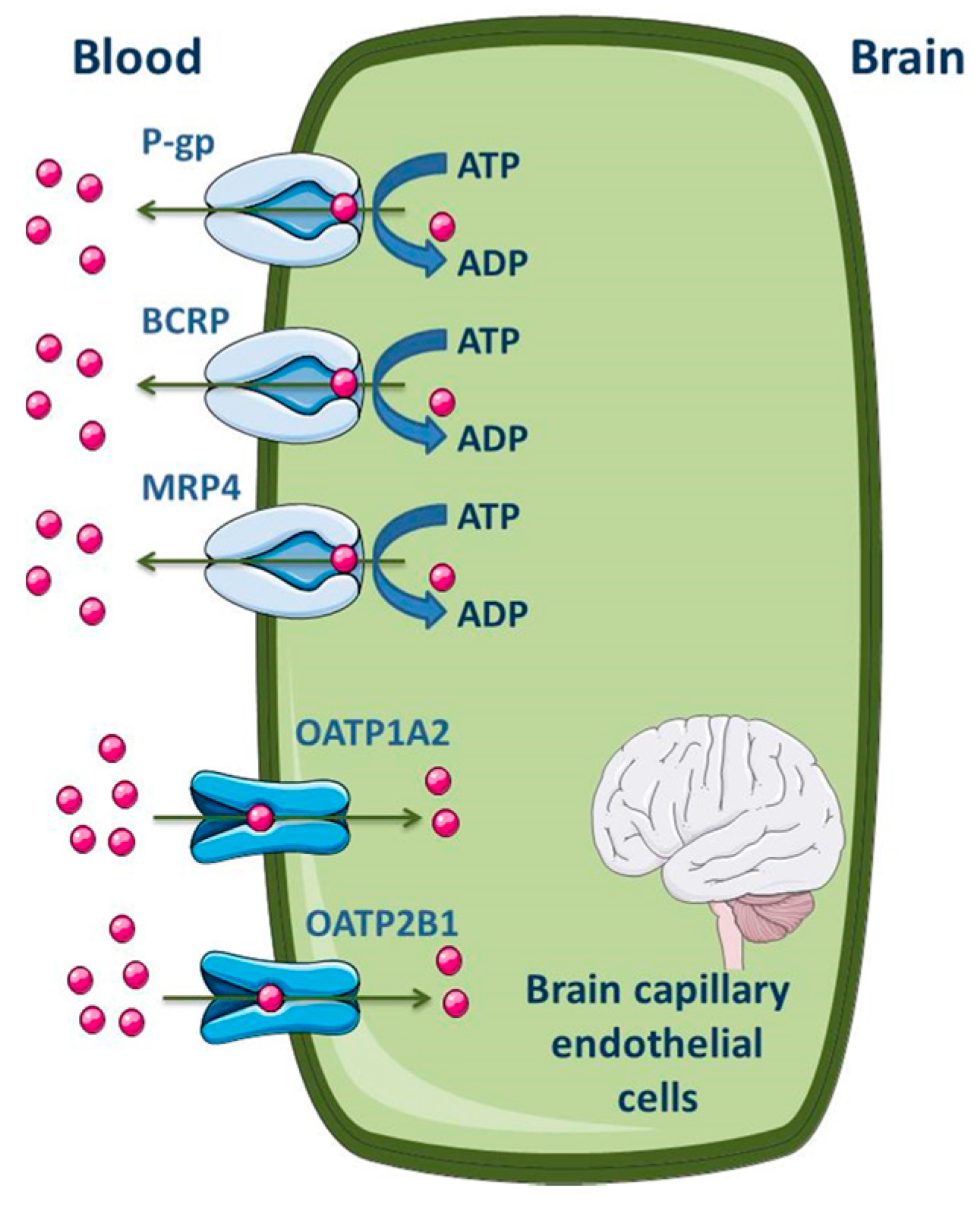
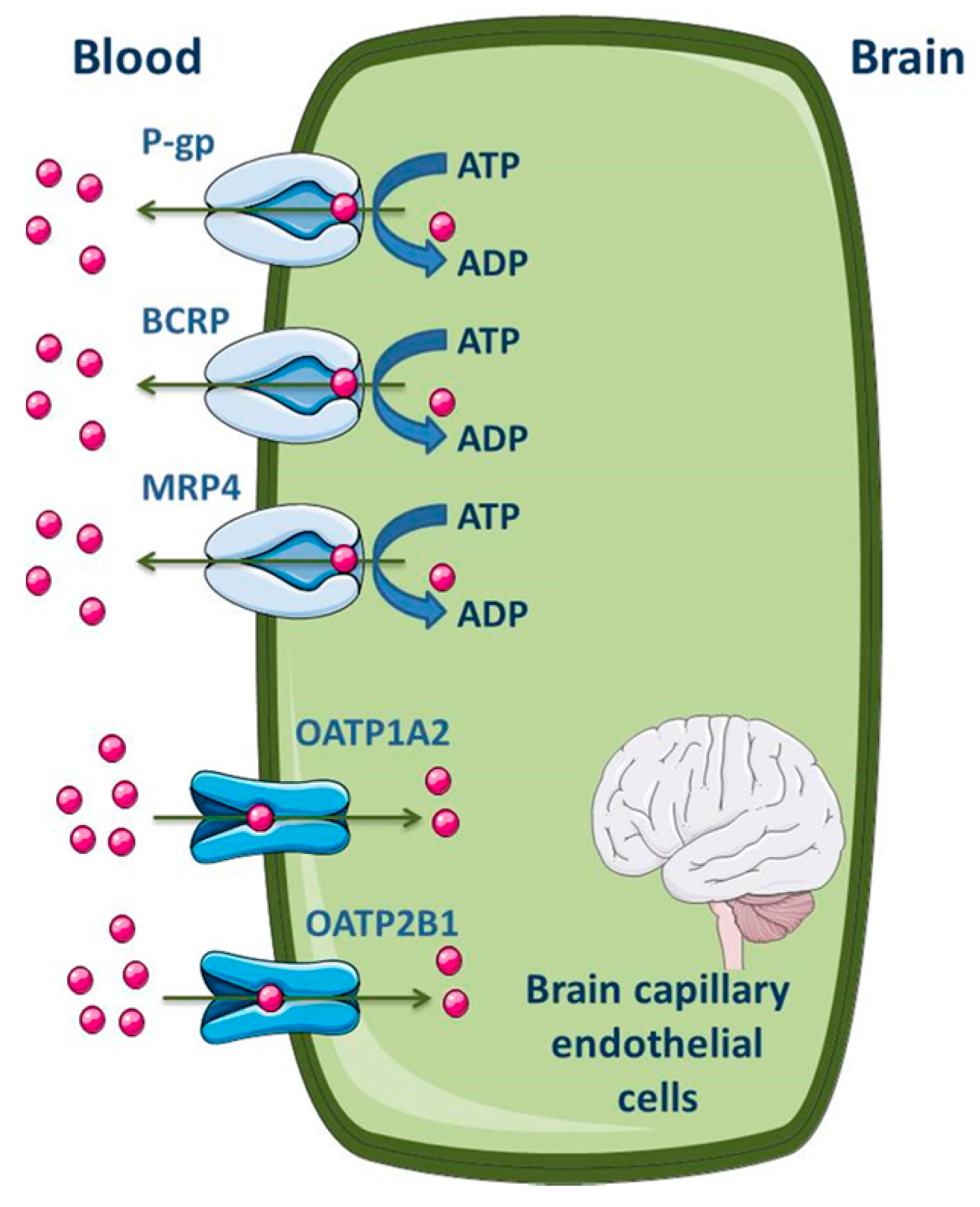
3.1.1. Blood-Brain Barrier
3.1.2. Cardiovascular System
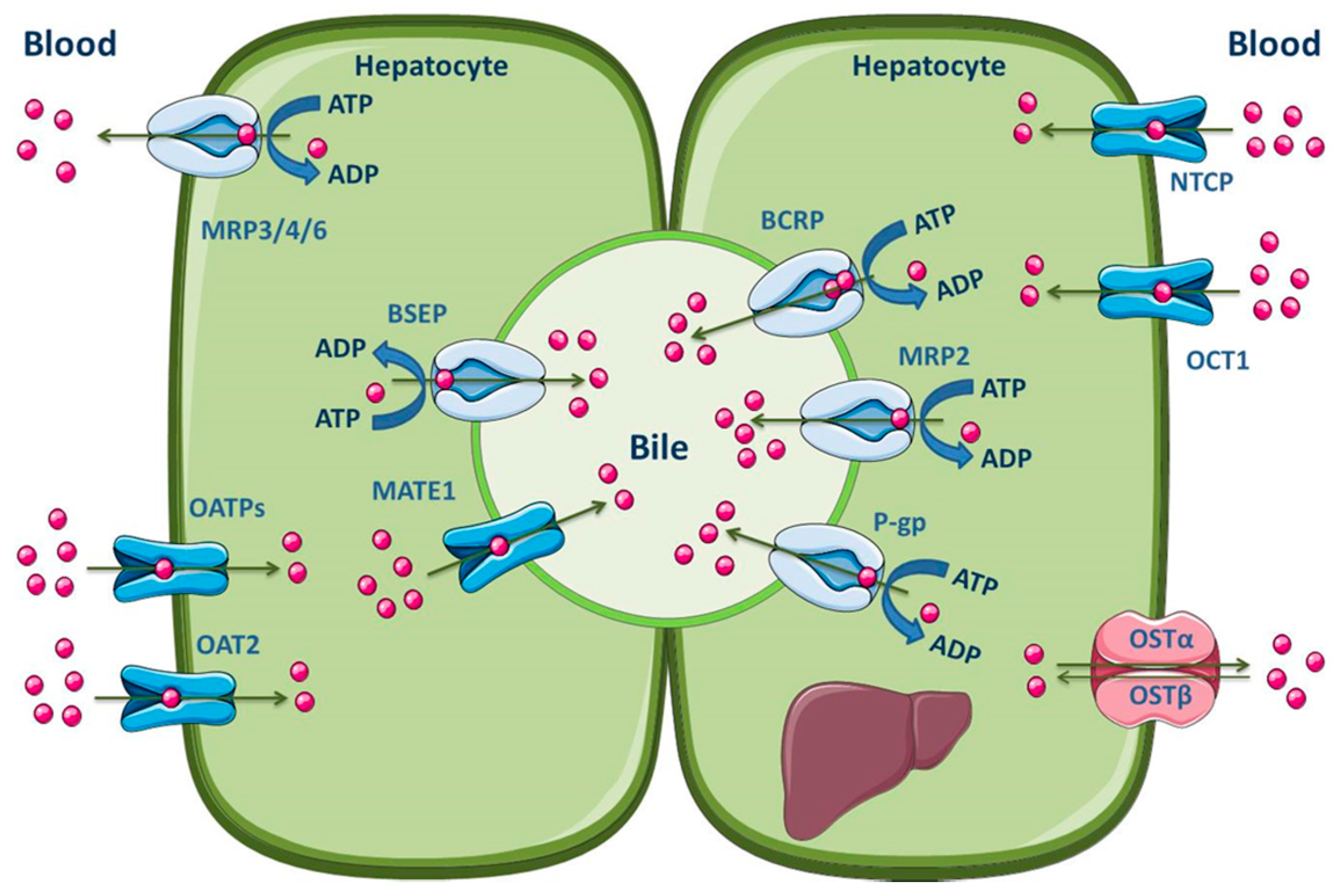
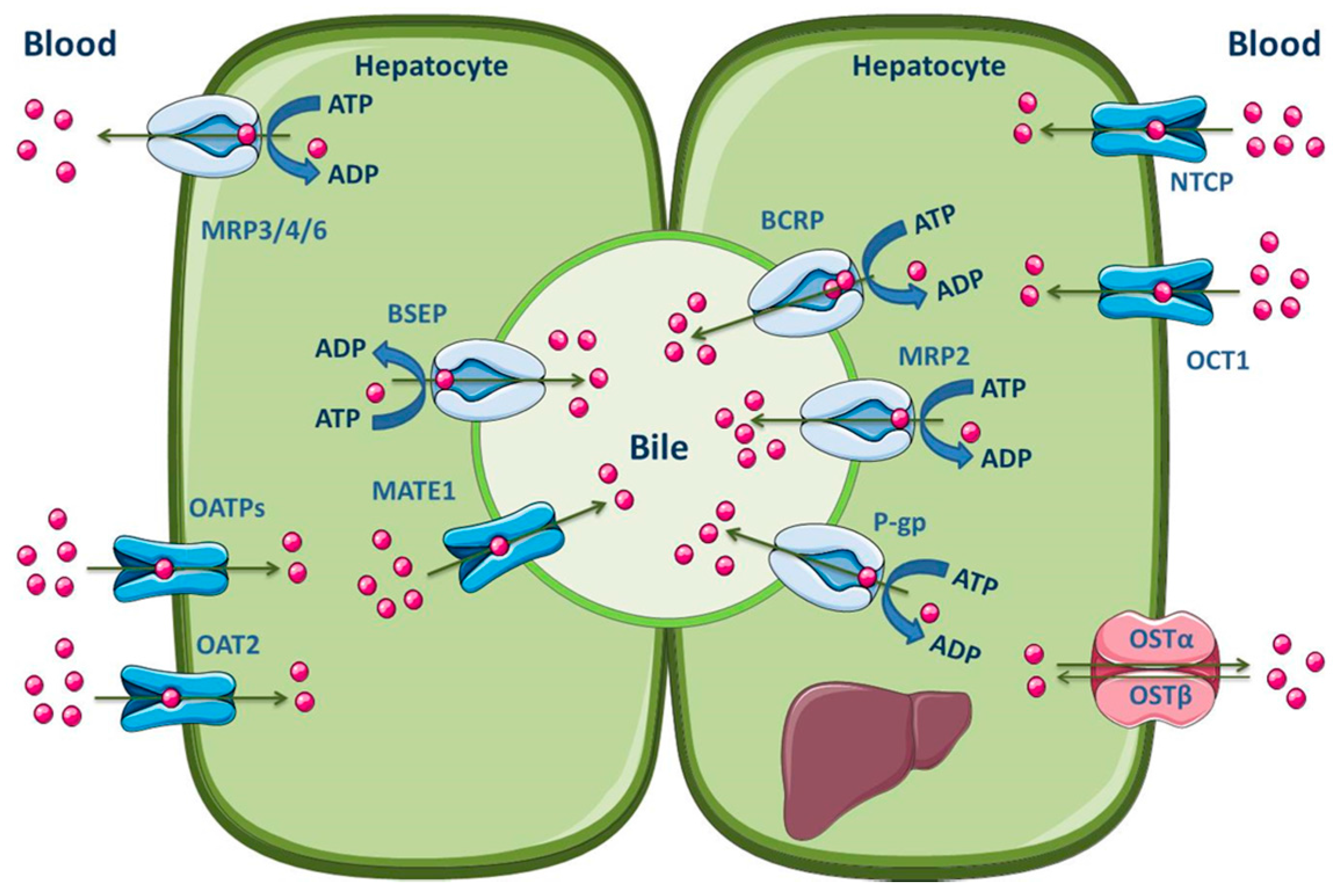
3.1.3. Liver
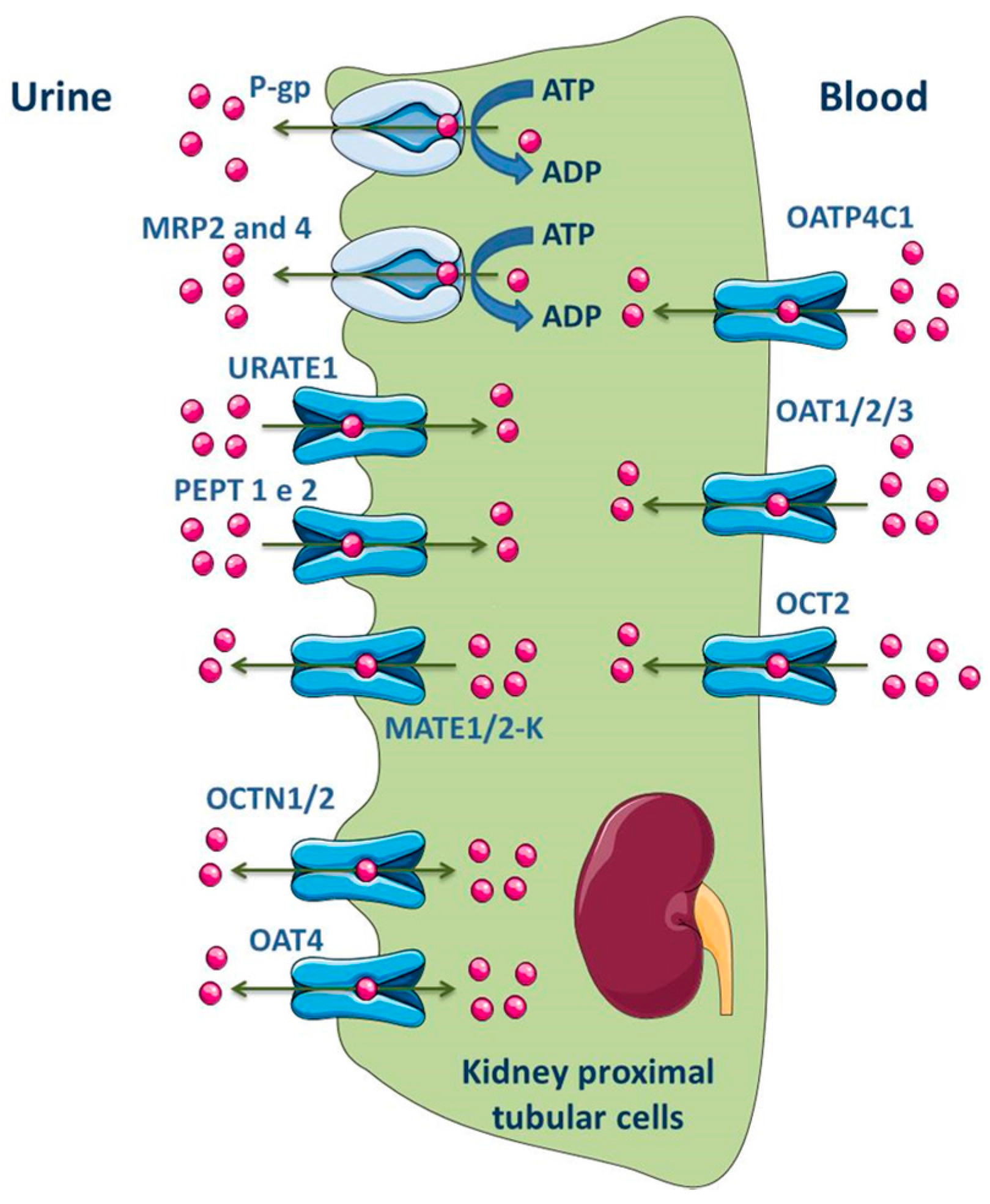
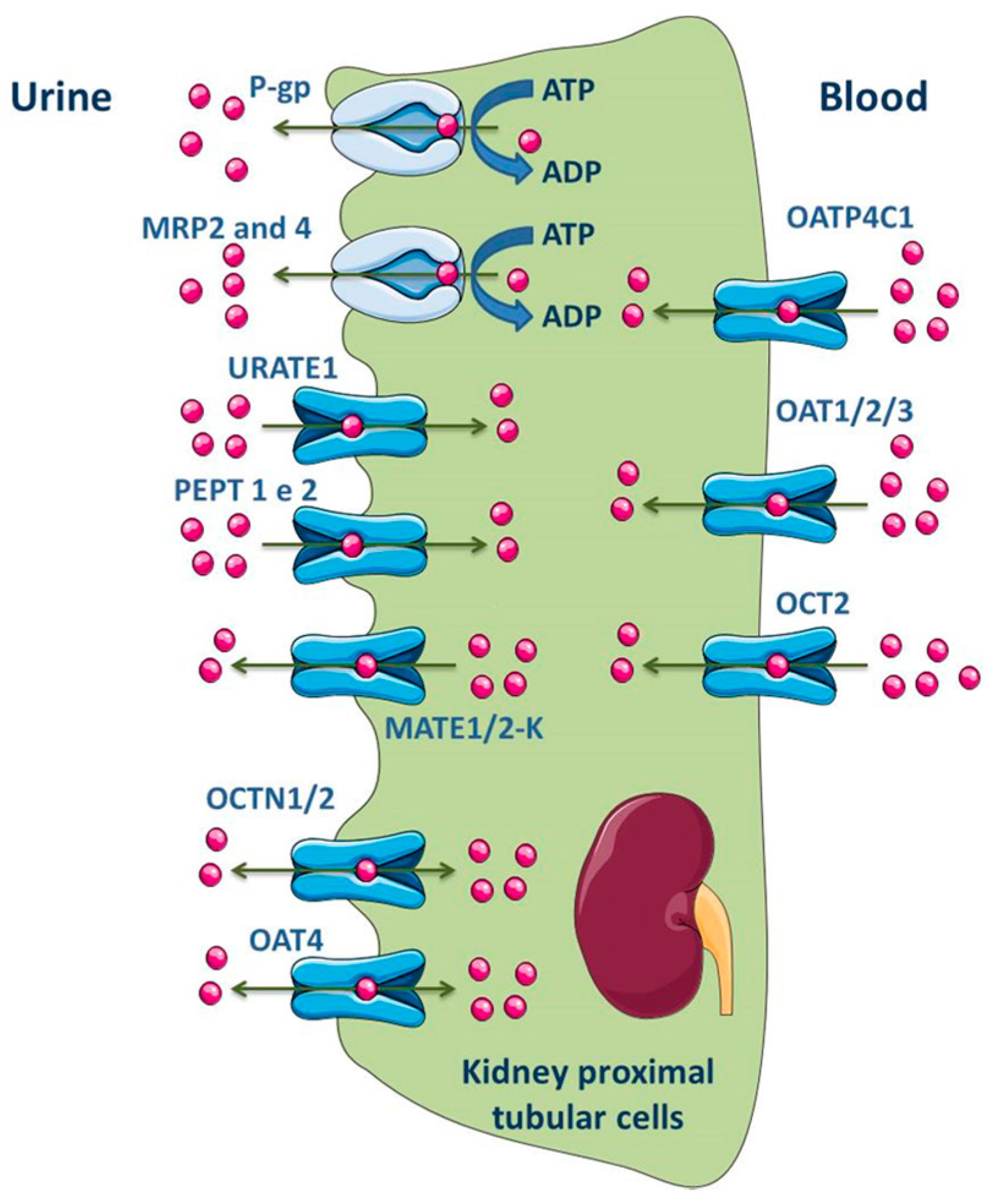
3.1.4. Kidney
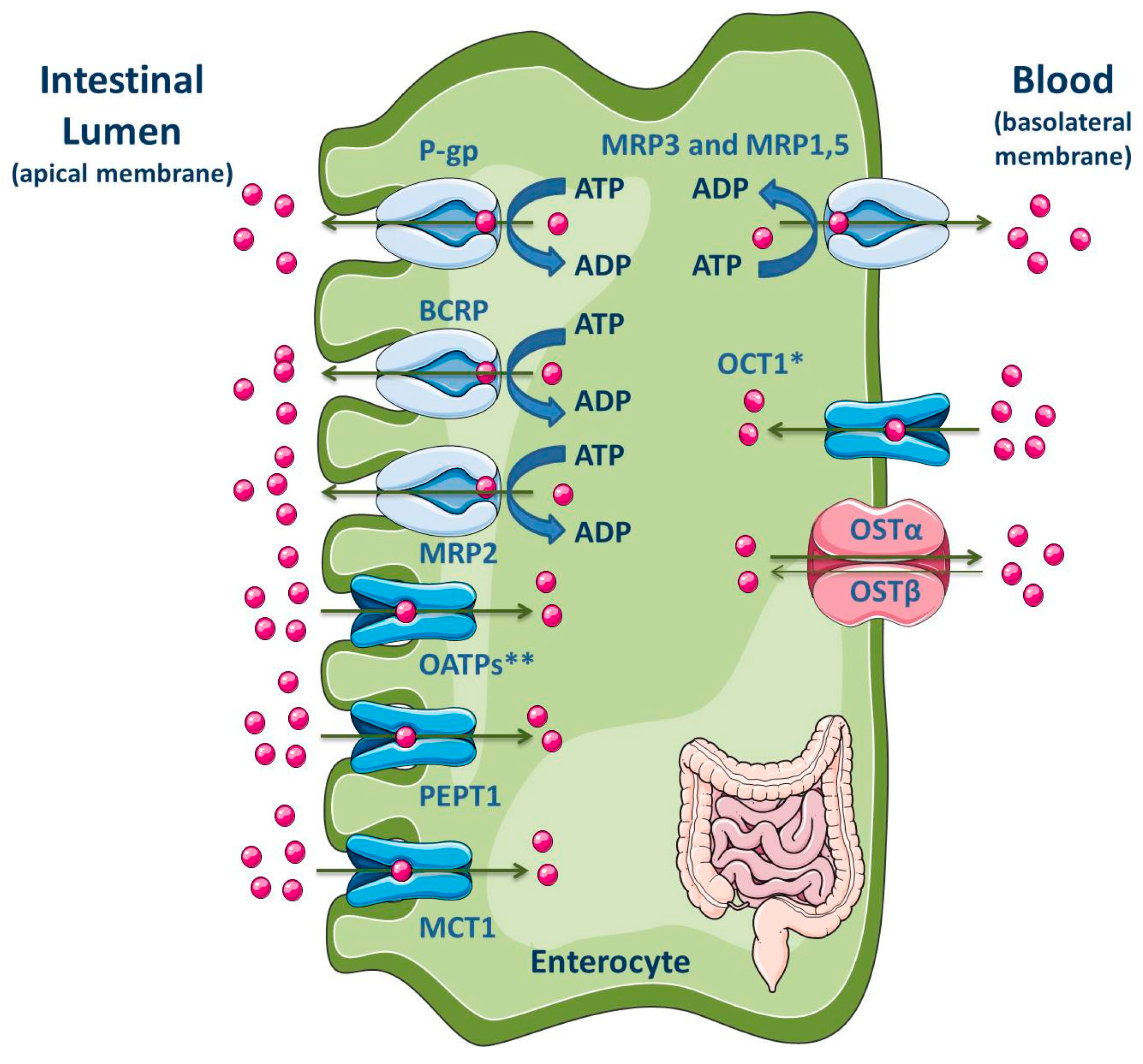
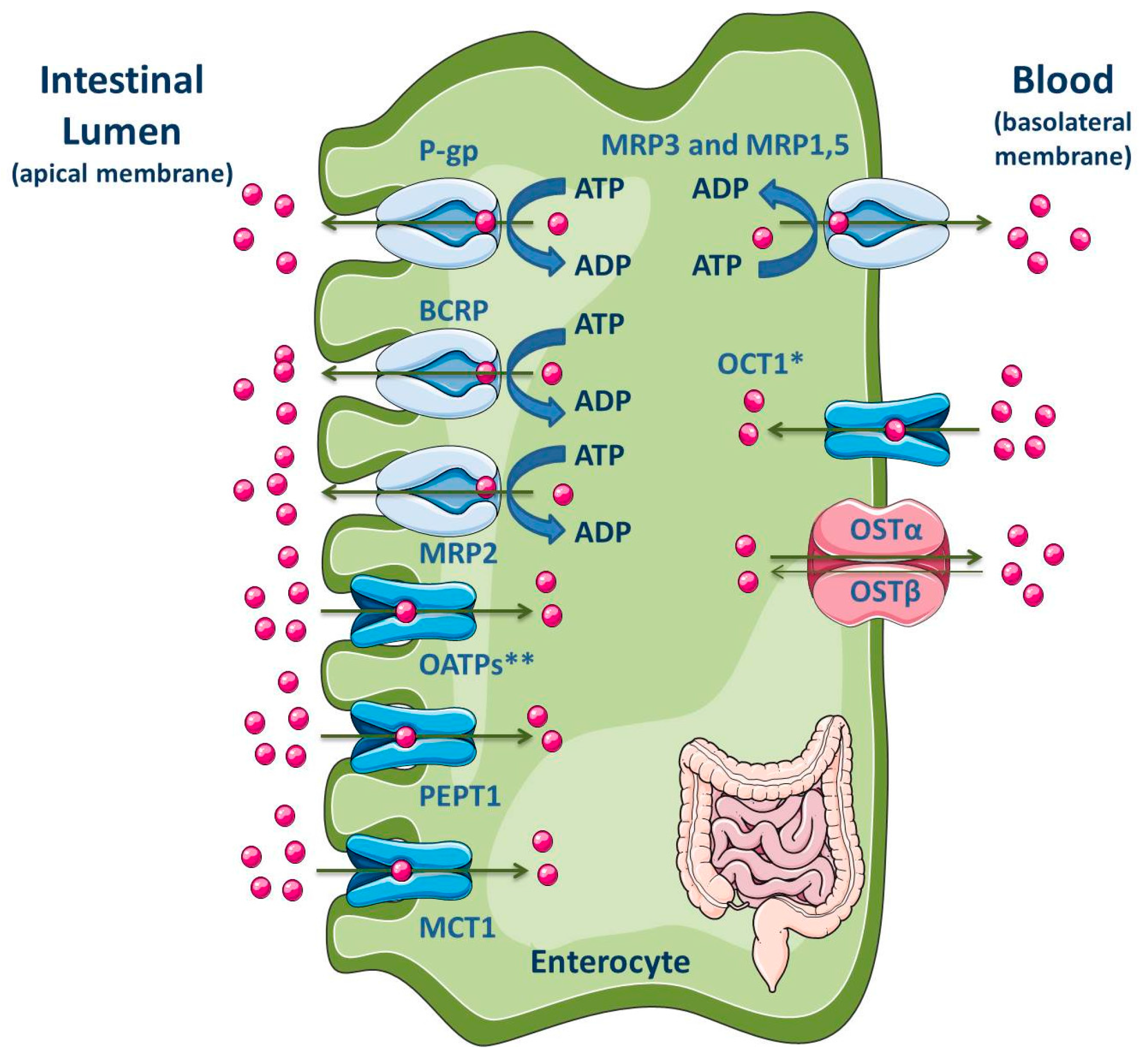
3.1.5. Intestine
3.2. In Vitro Assays
3.2.1. Membrane-Based Assays
ATPase Assays
Membrane Vesicular Transport Assays
Photoaffinity Labeling Assays
3.2.2. Cell-Based Assays
ABC Transporter Gene Expression
Real-Time Reverse Transcription-Polymerase Chain Reaction (Real-Time RT-PCR)
Flow Cytometry Assays
Accumulation and Efflux Assays
Western Blotting
Transport Assays Across Polarized Cell Monolayers
3.3. Ex Vivo Assays
3.4. In Silico Studies for ABC Transporters Inducers and Activators
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AB | amyloid-β peptide |
| ABC | adenosine triphosphate-binding cassette |
| AD | Alzheimer’s disease |
| ADMET | absorption, distribution, metabolism, elimination, transport |
| ADP | adenosine diphosphate |
| APP | amyloid precursor protein |
| ASBT | apical sodium–bile acid transporter |
| ATP | adenosine triphosphate |
| BBB | blood-brain barrier |
| BCRP | breast cancer resistance protein |
| BCSFB | blood-cerebrospinal fluid barrier |
| BSEP | bile salt export pump |
| Calcein-AM | calcein acetoxymethyl ester |
| CAR | constitutive androstane receptor |
| CD | cluster of differentiation |
| CNS | central nervous system |
| Ct | cycle threshold |
| CYP | cytochrome |
| DMSO | dimethyl sulfoxide |
| EC50 | half maximal effective concentration |
| EE | ethynylestradiol |
| ENT | equilibrative nucleoside transporter |
| FDA | food and drug administration |
| FRET | Förster resonance energy transfer |
| FXR | farnesoid X receptor |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GNT | genistein |
| h | hours |
| hAECs | human aortic endothelial cells |
| hCMEC/D3 | human brain endothelial capillary cells |
| HK-2 | human kidney-2 cells |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| hMVECs | human microvascular endothelial cells |
| HPLC | high performance liquid chromatography |
| HPT | intestinal peptide-associated transporter |
| HPV | human papilloma virus |
| hTERT IC50 | human telomerase reverse transcriptase half maximal inhibitory concentration |
| i.p. | intraperitoneally |
| Ki | inhibitory constant |
| Km | affinity constant |
| LC | liquid chromatography |
| LLC-PK1 | Lilly Laboratories cells - porcine kidney 1 |
| LRP1 | low-density lipoprotein receptor-related protein-1 |
| LXR | liver X receptor |
| MATE | multidrug and toxin extrusion |
| MCT | monocarboxylic acid transporter |
| MDCKII | Madin-Darby canine kidney strain II cell line |
| mRNA | messenger RNA |
| MRP | multidrug resistance-associated protein |
| MS | mass spectrometry |
| NBD | nucleotide-binding domain |
| NF-κB | nuclear factor-κB |
| NF-Y | nuclear factor Y |
| Nrf2 | nuclear factor erythroid-derived 2-related factor |
| NTCP | sodium-dependent taurocholate co-transporting polypeptide |
| OAT | organic anion transporter |
| OATP | organic anion-transporting polypeptide |
| OCT | organic cation transporter |
| OST | organic solute transporter |
| Papp | apparent permeability coefficient |
| PCR | polymerase chain reaction |
| PD | Parkinson’s disease |
| PDB | protein data bank |
| PEPT | peptide transporter |
| P-gp | P-glycoprotein |
| Pi | inorganic phosphate |
| PPAR | peroxisome proliferator-activated receptors |
| PQ | paraquat |
| PXR | pregnane X receptor |
| QSAR | quantitative structure-activity relationship |
| RAGE | receptor for advanced glycation end products |
| RedRif | reduced derivative |
| RT-PCR | reverse transcription-polymerase chain reaction |
| SLC | solute carrier |
| SV40 | simian vacuolating virus 40 |
| TEER | transendothelial electrical resistance |
| TMD | transmembrane domain |
| TMH | transmembrane-spanning α-helices |
| TXs | thioxanthones |
| URAT | urate transporter |
| Vi | inorganic vanadate |
| Vmax | maximal velocity |
| Xs | dihydroxylated xanthones |
| YB-1 | Y-box binding protein-1 |
References
- Xia, C.Q.; Milton, M.N.; Gan, L.S. Evaluation of drug-transporter interactions using in vitro and in vivo models. Curr. Drug Metab. 2007, 8, 341–363. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Hesselson, S.E.; Matsson, P.; Shima, J.E.; Fukushima, H.; Yee, S.W.; Kobayashi, Y.; Gow, J.M.; Ha, C.; Ma, B.; Poon, A.; et al. Genetic variation in the proximal promoter of ABC and SLC superfamilies: Liver and kidney specific expression and promoter activity predict variation. PLoS ONE 2009, 4, e6942. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Huls, M.; Russel, F.G.; Masereeuw, R. The role of ATP binding cassette transporters in tissue defense and organ regeneration. J. Pharmacol. Exp. Ther. 2009, 328, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Cheepala, S.; Hulot, J.S.; Morgan, J.A.; Sassi, Y.; Zhang, W.; Naren, A.P.; Schuetz, J.D. Cyclic nucleotide compartmentalization: Contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-glycoprotein transport system and cardiovascular drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Estudante, M.; Morais, J.G.; Soveral, G.; Benet, L.Z. Intestinal drug transporters: An overview. Adv. Drug Deliv. Rev. 2013, 65, 1340–1356. [Google Scholar] [CrossRef] [PubMed]
- Doring, B.; Petzinger, E. Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Khuri, N.; Giacomini, K.M.; Sali, A. Molecular modeling and ligand docking for solute carrier (SLC) transporters. Curr. Top. Med. Chem. 2013, 13, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A call for systematic research on solute carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Couture, L.; Nash, J.A.; Turgeon, J. The ATP-binding cassette transporters and their implication in drug disposition: A special look at the heart. Pharmacol. Rev. 2006, 58, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J.; Remiao, F.; Duarte, J.A.; Ferreira, R.; Sanchez Navarro, A.; Bastos, M.L.; Carvalho, F. P-glycoprotein induction: An antidotal pathway for paraquat-induced lung toxicity. Free Radic. Biol. Med. 2006, 41, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Dinis-Oliveira, R.; Cordeiro-da-Silva, A.; Lima, S.C.; Carvalho, F.; Bastos Mde, L.; Remiao, F. In vitro study of P-glycoprotein induction as an antidotal pathway to prevent cytotoxicity in Caco-2 cells. Arch. Toxicol. 2011, 85, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; de Pinho, P.G.; Dinis-Oliveira, R.J.; Carvalho, F.; Silva, I.; Correia-de-Sa, P.; Bastos Mde, L.; Remiao, F. Doxorubicin decreases paraquat accumulation and toxicity in Caco-2 cells. Toxicol. Lett. 2013, 217, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Silva, R.; Palmeira, A.; Sousa, E.; Ferreira, L.M.; Branco, P.S.; Carvalho, F.; Bastos Mde, L.; Remiao, F. Development of novel rifampicin-derived P-glycoprotein activators/inducers. Synthesis, in silico analysis and application in the RBE4 cell model, using paraquat as substrate. PLoS ONE 2013, 8, e74425. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; Bastos Mde, L.; Remiao, F. Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Palmeira, A.; Carmo, H.; Barbosa, D.J.; Gameiro, M.; Gomes, A.; Paiva, A.M.; Sousa, E.; Pinto, M.; Bastos Mde, L.; et al. P-glycoprotein induction in Caco-2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity. Arch. Toxicol. 2015, 89, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Pick, A.; Klinkhammer, W.; Wiese, M. Specific inhibitors of the breast cancer resistance protein (BCRP). ChemMedChem 2010, 5, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.; Fernandes, M.X. Structure and ligand-based design of P-glycoprotein inhibitors: A historical perspective. Curr. Pharm. Des. 2012, 18, 4197–4214. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Chen, Z.S.; Ambudkar, S.V. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist. Updates 2012, 15, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Pokharel, D.; Bebawy, M. A novel mechanism governing the transcriptional regulation of ABC transporters in MDR cancer cells. Drug Deliv. Transl. Res. 2017, 7, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Complete characterization of the human ABC gene family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Higgins, C.F.; Linton, K.J. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004, 11, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J. Structure and function of ABC transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J.; Higgins, C.F. Structure and function of ABC transporters: The ATP switch provides flexible control. Pflugers Archiv Eur. J. Physiol. 2007, 453, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Szakacs, G.; Homolya, L.; Orban, T.I.; Telbisz, A.; Jani, M.; Sarkadi, B. Ins and outs of the ABCG2 multidrug transporter: An update on in vitro functional assays. Adv. Drug Deliv. Rev. 2009, 61, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; van Veen, H.W. Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta 2009, 1794, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Zutz, A.; Hoffmann, J.; Hellmich, U.A.; Glaubitz, C.; Ludwig, B.; Brutschy, B.; Tampe, R. Asymmetric ATP hydrolysis cycle of the heterodimeric multidrug ABC transport complex TmrAB from Thermus thermophilus. J. Biol. Chem. 2011, 286, 7104–7115. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New uses for old drugs: Pharmacophore-based screening for the discovery of P-glycoprotein inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T; Zhang, Q.; Urbatsch, I.L. Structure of P-Glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Ramaen, O.; Leulliot, N.; Sizun, C.; Ulryck, N.; Pamlard, O.; Lallemand, J.-Y.; van Tilbeugh, H.; Jacquet, E. Structure of the Human Multidrug Resistance Protein 1 Nucleotide Binding Domain 1 bound to Mg2+/ATP Reveals a Non-productive Catalytic Site. J. Mol. Biol. 2006, 359, 940–949. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Fox, K.; Lam, P.; Ling, V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 1999, 259, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Berridge, G.; Higgins, C.F.; Mistry, P.; Charlton, P.; Callaghan, R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 2000, 58, 624–632. [Google Scholar] [PubMed]
- Daoud, R.; Julien, M.; Gros, P.; Georges, E. Major photoaffinity drug binding sites in multidrug resistance protein 1 (MRP1) are within transmembrane domains 10–11 and 16–17. J. Biol. Chem. 2001, 276, 12324–12330. [Google Scholar] [CrossRef] [PubMed]
- Daoud, R.; Kast, C.; Gros, P.; Georges, E. Rhodamine 123 binds to multiple sites in the multidrug resistance protein (MRP1). Biochemistry 2000, 39, 15344–15352. [Google Scholar] [CrossRef] [PubMed]
- Hazai, E.; Bikadi, Z. Homology modeling of breast cancer resistance protein (ABCG2). J. Struct. Biol. 2008, 162, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Gout, T. Role of ATP binding and hydrolysis in the gating of the cystic fibrosis transmembrane conductance regulator. Ann. Thorac. Med. 2012, 7, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Albermann, N.; Schmitz-Winnenthal, F.H.; Z’Graggen, K.; Volk, C.; Hoffmann, M.M.; Haefeli, W.E.; Weiss, J. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 2005, 70, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Sato, H.; Sugiyama, Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Sugiyama, Y. Transporter biology in drug approval: Regulatory aspects. Mol. Asp. Med. 2013, 34, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Yang, X.; Yang, Z.; Gao, S.; Yin, T.; Liu, W.; Wang, F.; Hu, M.; Liu, Z. The role of efflux transporters on the transport of highly toxic aconitine, mesaconitine, hypaconitine, and their hydrolysates, as determined in cultured Caco-2 and transfected MDCKII cells. Toxicol. Lett. 2013, 216, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Role of multidrug resistance associated proteins in drug development. Drug Discov. Ther. 2008, 2, 305–332. [Google Scholar] [PubMed]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, S.; Sinicrope, F.A.; Kuo, M.T. Induction of multidrug resistance proteins MRP1 and MRP3 and gamma-glutamylcysteine synthetase gene expression by nonsteroidal anti-inflammatory drugs in human colon cancer cells. Biochem. Biophys. Res. Commun. 2002, 290, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.M.; Li, C.G.; Xue, C.C.; Zhou, S.F. Clinical drugs that interact with St. John’s wort and implication in drug development. Curr. Pharm. Des. 2008, 14, 1723–1742. [Google Scholar] [CrossRef] [PubMed]
- Haslam, I.S.; Jones, K.; Coleman, T.; Simmons, N.L. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem. Pharmacol. 2008, 76, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Malekshah, O.M.; Lage, H.; Bahrami, A.R.; Afshari, J.T.; Behravan, J. PXR and NF-κB correlate with the inducing effects of IL-1β and TNF-α on ABCG2 expression in breast cancer cell lines. Eur. J. Pharm. Sci. 2012, 47, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Sterz, K.; Mollmann, L.; Jacobs, A.; Baumert, D.; Wiese, M. Activators of P-glycoprotein: Structure-activity relationships and investigation of their mode of action. ChemMedChem 2009, 4, 1897–1911. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J.; Duarte, J.A.; Remiao, F.; Sanchez-Navarro, A.; Bastos, M.L.; Carvalho, F. Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 2006, 227, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Bastos, M.L.; Remião, F. Hypericin-mediated P-glycoprotein induction protects caco-2 cells against paraquat toxicity: In vitro and in silico studies. Toxicol. Lett. 2015, 238 (Suppl. 2), S317. [Google Scholar] [CrossRef]
- Arias, A.; Rigalli, J.P.; Villanueva, S.S.; Ruiz, M.L.; Luquita, M.G.; Perdomo, V.G.; Vore, M.; Catania, V.A.; Mottino, A.D. Regulation of expression and activity of multidrug resistance proteins MRP2 and MDR1 by estrogenic compounds in Caco-2 cells. Role in prevention of xenobiotic-induced cytotoxicity. Toxicology 2014, 320, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Zerin, T.; Kim, Y.S.; Hong, S.Y.; Song, H.Y. Protective effect of methylprednisolone on paraquat-induced A549 cell cytotoxicity via induction of efflux transporter, P-glycoprotein expression. Toxicol. Lett. 2012, 208, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Haslam, I.S.; Pitre, A.; Schuetz, J.D.; Paus, R. Protection against chemotherapy-induced alopecia: Targeting ATP-binding cassette transporters in the hair follicle? Trends Pharmacol. Sci. 2013, 34, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Haslam, I.S.; El-Chami, C.; Faruqi, H.; Shahmalak, A.; O’Neill, C.A.; Paus, R. Differential expression and functionality of ATP-binding cassette transporters in the human hair follicle. Br. J. Dermatol. 2015, 172, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, G.M.; Kurban, M.; Anyane-Yeboa, K.; Dall’Armi, C.; Di Paolo, G.; Feenstra, H.; Silverberg, N.; Rohena, L.; Lopez-Cepeda, L.D.; Jobanputra, V.; et al. Mutations in the cholesterol transporter gene ABCA5 are associated with excessive hair overgrowth. PLoS Genet. 2014, 10, e1004333. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Berardi, F.; Cantore, M.; Contino, M.; Inglese, C.; Niso, M.; Perrone, R. Perspectives of P-glycoprotein modulating agents in oncology and neurodegenerative diseases: Pharmaceutical, biological, and diagnostic potentials. J. Med. Chem. 2010, 53, 1883–1897. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L. Blood-brain barrier P-glycoprotein function in neurodegenerative disease. Curr. Pharm. Des. 2011, 17, 2771–2777. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid β-Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Bello, I.; Salerno, M. Evidence against a role of P-glycoprotein in the clearance of the Alzheimer’s disease Abeta1–42 peptides. Cell Stress Chaperones 2015, 20, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Aβ1–42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef] [PubMed]
- Pahnke, J.; Langer, O.; Krohn, M. Alzheimer’s and ABC transporters—New opportunities for diagnostics and treatment. Neurobiol. Dis. 2014, 72 Pt A, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, S.; Jedlitschky, G.; Brenn, A.; Walker, L.C. The role of the ATP-binding cassette transporter P-glycoprotein in the transport of beta-amyloid across the blood-brain barrier. Curr. Pharm. Des. 2011, 17, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Vogelgesang, S.; Kroemer, H.K. MDR1-P-glycoprotein (ABCB1)-mediated disposition of amyloid-beta peptides: Implications for the pathogenesis and therapy of Alzheimer’s disease. Clin. Pharmacol. Ther. 2010, 88, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s β-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.; van Berckel, B.N. Blood-Brain Barrier ABC-transporter P-glycoprotein in Alzheimer’s Disease: Still a Suspect? Curr. Pharm. Des. 2016, 22, 5808–5816. [Google Scholar] [CrossRef] [PubMed]
- Abuznait, A.H.; Cain, C.; Ingram, D.; Burk, D.; Kaddoumi, A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: Investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J. Pharm. Pharmacol. 2011, 63, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Aβ1–40 peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Li, G.J.; Wang, S.; Zheng, W. Use of Z310 cells as an in vitro blood-cerebrospinal fluid barrier model: Tight junction proteins and transport properties. Toxicol. In Vitro 2008, 22, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.V.; Dahlheimer, J.L.; Bardgett, M.E.; Snyder, A.Z.; Finch, R.A.; Sartorelli, A.C.; Piwnica-Worms, D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 3900–3905. [Google Scholar] [CrossRef] [PubMed]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A.; et al. Discovery of a marine-derived bis-indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure-activity relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Padala, A.K.; Wani, A.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. Functional induction of P-glycoprotein efflux pump by phenyl benzenesulfonamides: Synthesis and biological evaluation of T0901317 analogs. Eur. J. Med. Chem. 2016, 122, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Berardi, F.; Perrone, M.G.; Capparelli, E.; Cantore, M.; Inglese, C.; Perrone, R. Substrates, inhibitors and activators of P-glycoprotein: Candidates for radiolabeling and imaging perspectives. Curr. Top. Med. Chem. 2010, 10, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Contino, M.; Cantore, M.; Capparelli, E.; Perrone, M.G.; Niso, M.; Inglese, C.; Berardi, F.; Leopoldo, M.; Perrone, R.; Colabufo, N.A. A benzopyrane derivative as a P-glycoprotein stimulator: A potential agent to decrease beta-amyloid accumulation in Alzheimer’s disease. ChemMedChem 2012, 7, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Peng, C.; Liu, Y.; Liu, X.; Chen, Q.; Huang, Z. Genetic association of NOS1 exon18, NOS1 exon29, ABCB1 1236C/T, and ABCB1 3435C/T polymorphisms with the risk of Parkinson’s disease: A meta-analysis. Medicine 2016, 95, e4982. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, M.; Belin, A.C.; Anvret, A.; Hakansson, A.; Nissbrandt, H.; Lind, C.; Sydow, O.; Olson, L.; Galter, D. Association of a polymorphism in the ABCB1 gene with Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Drozdzik, M.; Bialecka, M.; Honczarenko, K.; Klodowska-Duda, G.; Teo, Y.Y.; Tang, K.; Wong, L.P.; Chong, S.S.; Tan, C.; et al. Analysis of MDR1 haplotypes in Parkinson’s disease in a white population. Neurosci. Lett. 2004, 372, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F. The influence of MDR1 polymorphisms on P-glycoprotein expression and function in humans. Adv. Drug Deliv. Rev. 2002, 54, 1295–1310. [Google Scholar] [CrossRef]
- Marzolini, C.; Paus, E.; Buclin, T.; Kim, R.B. Polymorphisms in human MDR1 (P-glycoprotein): Recent advances and clinical relevance. Clin. Pharmacol. Ther. 2004, 75, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hou, J.; Chen, X.; Liu, G.; Zhang, D.; Sun, H.; Zhang, J. P-glycoprotein mediated efflux limits the transport of the novel anti-Parkinson’s disease candidate drug FLZ across the physiological and PD pathological in vitro BBB models. PLoS ONE 2014, 9, e102442. [Google Scholar] [CrossRef] [PubMed]
- Luurtsema, G.; Elsinga, P.; Dierckx, R.; Boellaard, R.; van Waarde, A. PET Tracers for Imaging of ABC Transporters at the Blood-Brain Barrier: Principles and Strategies. Curr. Pharm. Des. 2016, 22, 5779–5785. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Pekcec, A.; Soldner, E.L.; Zhong, Y.; Schlichtiger, J.; Bauer, B. P-gp protein expression and transport activity in rodent seizure models and human epilepsy. Mol. Pharm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, J.; Liu, J.; Shen, S.; Cao, Z.; Pan, J.; Zhou, S.; Pang, Z.; Geng, D.; Zhang, J. A multimodal Pepstatin A peptide-based nanoagent for the molecular imaging of P-glycoprotein in the brains of epilepsy rats. Biomaterials 2016, 76, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Zips, D.; Thames, H.D.; Baumann, M. New anticancer agents: In vitro and in vivo evaluation. In Vivo 2005, 19, 1–7. [Google Scholar] [PubMed]
- Loscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Girardin, F. Membrane transporter proteins: A challenge for CNS drug development. Dialogues Clin. Neurosci. 2006, 8, 311–321. [Google Scholar] [PubMed]
- Agarwal, S.; Hartz, A.M.; Elmquist, W.F.; Bauer, B. Breast cancer resistance protein and P-glycoprotein in brain cancer: Two gatekeepers team up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Dai, H.; Shaik, N.; Elmquist, W.F. Drug efflux transporters in the CNS. Adv. Drug Deliv. Rev. 2003, 55, 83–105. [Google Scholar] [CrossRef]
- ElAli, A.; Hermann, D.M. ATP-binding cassette transporters and their roles in protecting the brain. Neuroscientist 2011, 17, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, H.; Elmquist, W.F.; Miller, D.W. Expression of various multidrug resistance-associated protein (MRP) homologues in brain microvessel endothelial cells. Brain Res. 2000, 876, 148–153. [Google Scholar] [CrossRef]
- Miller, D.S.; Nobmann, S.N.; Gutmann, H.; Toeroek, M.; Drewe, J.; Fricker, G. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol. Pharmacol. 2000, 58, 1357–1367. [Google Scholar] [PubMed]
- Dombrowski, S.M.; Desai, S.Y.; Marroni, M.; Cucullo, L.; Goodrich, K.; Bingaman, W.; Mayberg, M.R.; Bengez, L.; Janigro, D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia 2001, 42, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Decleves, X.; Jacob, A.; Yousif, S.; Shawahna, R.; Potin, S.; Scherrmann, J.M. Interplay of drug metabolizing CYP450 enzymes and ABC transporters in the blood-brain barrier. Curr. Drug Metab. 2011, 12, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 2011, 71, 113–128. [Google Scholar]
- Chaves, C.; Shawahna, R.; Jacob, A.; Scherrmann, J.M.; Decleves, X. Human ABC transporters at blood-CNS interfaces as determinants of CNS drug penetration. Curr. Pharm. Des. 2014, 20, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. In vitro blood-brain barrier models: Current and perspective technologies. J. Pharm. Sci. 2012, 101, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Poller, B.; Gutmann, H.; Krahenbuhl, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Tuffin, G.; Drewe, J.; Huwyler, J. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 2008, 107, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Eigenmann, D.E.; Xue, G.; Kim, K.S.; Moses, A.V.; Hamburger, M.; Oufir, M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 2013, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Ikeda, C.; Uchida, Y.; Sakamoto, Y.; Miller, F.; Glacial, F.; Decleves, X.; Scherrmann, J.M.; Couraud, P.O.; Kubo, Y.; et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol. Pharm. 2013, 10, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, S.; Miller, F.; Couraud, P.O.; Weaver, R.J.; Weksler, B.; Romero, I.A.; Scherrmann, J.M.; de Waziers, I.; Decleves, X. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem. Pharmacol. 2009, 77, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Ketabi-Kiyanvash, N.; Herold-Mende, C.; Kashfi, F.; Caldeira, S.; Tommasino, M.; Haefeli, W.E.; Weiss, J. NKIM-6, a new immortalized human brain capillary endothelial cell line with conserved endothelial characteristics. Cell Tissue Res. 2007, 328, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Shimizu, F.; Abe, M.; Maeda, T.; Kashiwamura, Y.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; Kajiwara, K.; Fujii, M.; et al. Establishment of a new conditionally immortalized human brain microvascular endothelial cell line retaining an in vivo blood-brain barrier function. J. Cell. Physiol. 2010, 225, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Cianciolo, G.J.; Combes, V.; Grau, G.E. Inhibition of endothelial activation: A new way to treat cerebral malaria? PLoS Med. 2005, 2, e245. [Google Scholar] [CrossRef] [PubMed]
- Joo, F. The blood-brain barrier in vitro: Ten years of research on microvessels isolated from the brain. Neurochem. Int. 1985, 7, 1–25. [Google Scholar] [CrossRef]
- Emmert, D.; Campos, C.R.; Ward, D.; Lu, P.; Namanja, H.A.; Bohn, K.; Miller, D.S.; Sharom, F.J.; Chmielewski, J.; Hrycyna, C.A. Reversible dimers of the atypical antipsychotic quetiapine inhibit P-glycoprotein-mediated efflux in vitro with increased binding affinity and in situ at the blood-brain barrier. ACS Chem. Neurosci. 2014, 5, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Luna-Munguia, H.; Salvamoser, J.D.; Pascher, B.; Pieper, T.; Getzinger, T.; Kudernatsch, M.; Kluger, G.; Potschka, H. Glutamate-mediated upregulation of the multidrug resistance protein 2 in porcine and human brain capillaries. J. Pharmacol. Exp. Ther. 2015, 352, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Kooij, G.; Kroon, J.; Paul, D.; Reijerkerk, A.; Geerts, D.; van der Pol, S.M.; van Het Hof, B.; Drexhage, J.A.; van Vliet, S.J.; Hekking, L.H.; et al. P-glycoprotein regulates trafficking of CD8(+) T cells to the brain parenchyma. Acta Neuropathol. 2014, 127, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Solbach, T.F.; Konig, J.; Fromm, M.F.; Zolk, O. ATP-binding cassette transporters in the heart. Trends Cardiovasc. Med. 2006, 16, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Meissner, K.; Sperker, B.; Karsten, C.; Meyer Zu Schwabedissen, H.; Seeland, U.; Bohm, M.; Bien, S.; Dazert, P.; Kunert-Keil, C.; Vogelgesang, S.; et al. Expression and localization of P-glycoprotein in human heart: Effects of cardiomyopathy. J. Histochem. Cytochem. 2002, 50, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Meissner, K.; Heydrich, B.; Jedlitschky, G.; Meyer Zu Schwabedissen, H.; Mosyagin, I.; Dazert, P.; Eckel, L.; Vogelgesang, S.; Warzok, R.W.; Bohm, M.; et al. The ATP-binding cassette transporter ABCG2 (BCRP), a marker for side population stem cells, is expressed in human heart. J. Histochem. Cytochem. 2006, 54, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F.; Kim, R.B.; Stein, C.M.; Wilkinson, G.R.; Roden, D.M. Inhibition of P-glycoprotein-mediated drug transport: A unifying mechanism to explain the interaction between digoxin and quinidine. Circulation 1999, 99, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, E.; Demeule, M.; Ghitescu, L.; Beliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326 Pt. 2, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Estevez, M.D.; Wolf, A.; Schramm, U. Effect of PSC 833, verapamil and amiodarone on adriamycin toxicity in cultured rat cardiomyocytes. Toxicology In Vitro 2000, 14, 17–23. [Google Scholar] [CrossRef]
- Flens, M.J.; Zaman, G.J.; van der Valk, P.; Izquierdo, M.A.; Schroeijers, A.B.; Scheffer, G.L.; van der Groep, P.; de Haas, M.; Meijer, C.J.; Scheper, R.J. Tissue distribution of the multidrug resistance protein. Am. J. Pathol. 1996, 148, 1237–1247. [Google Scholar] [PubMed]
- Rosati, A.; Maniori, S.; Decorti, G.; Candussio, L.; Giraldi, T.; Bartoli, F. Physiological regulation of P-glycoprotein, MRP1, MRP2 and cytochrome P450 3A2 during rat ontogeny. Dev. Growth Differ. 2003, 45, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Dazert, P.; Meissner, K.; Vogelgesang, S.; Heydrich, B.; Eckel, L.; Bohm, M.; Warzok, R.; Kerb, R.; Brinkmann, U.; Schaeffeler, E.; et al. Expression and localization of the multidrug resistance protein 5 (MRP5/ABCC5), a cellular export pump for cyclic nucleotides, in human heart. Am. J. Pathol. 2003, 163, 1567–1577. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S.; Yokoi, T. Tissue-specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab. Pharmacokinet. 2004, 19, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Redeker, S.; van Vliet, E.A.; Ramkema, M.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Leenstra, S.; Baayen, J.C.; et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia 2005, 46, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.A.; Vaidya, S.S.; St-Pierre, M.V.; Mikheev, A.M.; Desino, K.E.; Nyandege, A.N.; Audus, K.L.; Unadkat, J.D.; Gerk, P.M. Placental ABC Transporters: Biological Impact and Pharmaceutical Significance. Pharm. Res. 2016, 33, 2847–2878. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q. BCRP/ABCG2 in the placenta: Expression, function and regulation. Pharm. Res. 2008, 25, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Meeson, A.P.; Robertson, S.M.; Hawke, T.J.; Richardson, J.A.; Bates, S.; Goetsch, S.C.; Gallardo, T.D.; Garry, D.J. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev. Biol. 2004, 265, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Roy, U.; Mondal, D. MRP (ABCC) transporters-mediated efflux of anti-HIV drugs, saquinavir and zidovudine, from human endothelial cells. Exp. Biol. Med. 2008, 233, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Higashikuni, Y.; Sainz, J.; Nakamura, K.; Takaoka, M.; Enomoto, S.; Iwata, H.; Tanaka, K.; Sahara, M.; Hirata, Y.; Nagai, R.; et al. The ATP-binding cassette transporter ABCG2 protects against pressure overload-induced cardiac hypertrophy and heart failure by promoting angiogenesis and antioxidant response. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Kock, K.; Brouwer, K.L. A perspective on efflux transport proteins in the liver. Clin. Pharmacol. Ther. 2012, 92, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Wang, L.L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Stedman, C.A.; Liddle, C.; Downes, M. Hepatobiliary ABC transporters: Physiology, regulation and implications for disease. Front. Biosci. 2009, 14, 4904–4920. [Google Scholar] [CrossRef]
- Muller, F.; Fromm, M.F. Transporter-mediated drug-drug interactions. Pharmacogenomics 2011, 12, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Inui, K.I.; Masuda, S.; Saito, H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000, 58, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Mottino, A.D.; Catania, V.A. Hepatic drug transporters and nuclear receptors: Regulation by therapeutic agents. World J. Gastroenterol. 2008, 14, 7068–7074. [Google Scholar] [CrossRef] [PubMed]
- Kipp, H.; Arias, I.M. Intracellular trafficking and regulation of canalicular ATP-binding cassette transporters. Semin. Liver Dis. 2000, 20, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B. Drug uptake systems in liver and kidney: A historic perspective. Clin. Pharmacol. Ther. 2010, 87, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Van Montfoort, J.E.; Hagenbuch, B.; Groothuis, G.M.; Koepsell, H.; Meier, P.J.; Meijer, D.K. Drug uptake systems in liver and kidney. Curr. Drug Metab. 2003, 4, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Le Vee, M.; Jigorel, E.; Glaise, D.; Gripon, P.; Guguen-Guillouzo, C.; Fardel, O. Functional expression of sinusoidal and canalicular hepatic drug transporters in the differentiated human hepatoma HepaRG cell line. Eur. J. Pharm. Sci. 2006, 28, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Hoffmaster, K.A.; Nezasa, K.; Tallman, M.N.; Brouwer, K.L. Integration of hepatic drug transporters and phase II metabolizing enzymes: Mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur. J. Pharm. Sci. 2006, 27, 447–486. [Google Scholar] [CrossRef] [PubMed]
- Funk, C. The role of hepatic transporters in drug elimination. Expert Opin. Drug Metab. Toxicol. 2008, 4, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, P.; Englund, G.; Skogastierna, C.; Loof, J.; Johansson, J.; Hoogstraate, J.; Afzelius, L.; Andersson, T.B. Functional ATP-binding cassette drug efflux transporters in isolated human and rat hepatocytes significantly affect assessment of drug disposition. Drug Metab. Dispos. 2014, 42, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Schwabedissen, H.E.; Kroemer, H.K. In vitro and in vivo evidence for the importance of breast cancer resistance protein transporters (BCRP/MXR/ABCP/ABCG2). In Drug Transporters; Fromm, M.F., Kim, R.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 325–371. [Google Scholar]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. In Drug Transporters; Fromm, M.F., Kim, R.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 105–167. [Google Scholar]
- Soldatow, V.Y.; Lecluyse, E.L.; Griffith, L.G.; Rusyn, I. In vitro models for liver toxicity testing. Toxicol. Res. 2013, 2, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Guguen-Guillouzo, C.; Guillouzo, A. General review on in vitro hepatocyte models and their applications. Methods Mol. Biol. 2010, 640, 1–40. [Google Scholar] [PubMed]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouet, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Decaens, C.; Durand, M.; Grosse, B.; Cassio, D. Which in vitro models could be best used to study hepatocyte polarity? Biol. Cell 2008, 100, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- LeCluyse, E.L.; Bullock, P.L.; Parkinson, A.; Hochman, J.H. Cultured rat hepatocytes. Pharm. Biotechnol. 1996, 8, 121–159. [Google Scholar] [PubMed]
- Dunn, J.C.; Yarmush, M.L.; Koebe, H.G.; Tompkins, R.G. Hepatocyte function and extracellular matrix geometry: Long-term culture in a sandwich configuration. FASEB J. 1989, 3, 174–177. [Google Scholar] [PubMed]
- Dunn, J.C.; Tompkins, R.G.; Yarmush, M.L. Long-term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol. Prog. 1991, 7, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Swift, B.; Pfeifer, N.D.; Brouwer, K.L. Sandwich-cultured hepatocytes: An in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev. 2010, 42, 446–471. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, T.; Chatterjee, S.; Fattah, S.; Keemink, J.; Nicolai, J.; Augustijns, P.; Annaert, P. Sandwich-cultured hepatocytes: Utility for in vitro exploration of hepatobiliary drug disposition and drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2013, 9, 589–616. [Google Scholar] [CrossRef] [PubMed]
- Barton, H.A.; Lai, Y.; Goosen, T.C.; Jones, H.M.; El-Kattan, A.F.; Gosset, J.R.; Lin, J.; Varma, M.V. Model-based approaches to predict drug-drug interactions associated with hepatic uptake transporters: Preclinical, clinical and beyond. Expert Opin. Drug Metab. Toxicol. 2013, 9, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, K.; Kienhuis, A.S.; Brauers, K.J.; Jennen, D.G.; Lahoz, A.; Kleinjans, J.C.; van Delft, J.H. Assessing the metabolic competence of sandwich-cultured mouse primary hepatocytes. Drug Metab. Dispos. 2009, 37, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Le Vee, M.; Jouan, E.; Noel, G.; Stieger, B.; Fardel, O. Polarized location of SLC and ABC drug transporters in monolayer-cultured human hepatocytes. Toxicology In Vitro 2015, 29, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, O.; Ohtsuki, S.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Sakamoto, A.; Ishiguro, N.; Matsumaru, T.; Terasaki, T.; et al. Absolute quantification and differential expression of drug transporters, cytochrome P450 enzymes, and UDP-glucuronosyltransferases in cultured primary human hepatocytes. Drug Metab. Dispos. 2012, 40, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Achilli, T.M.; McCalla, S.; Meyer, J.; Tripathi, A.; Morgan, J.R. Multilayer spheroids to quantify drug uptake and diffusion in 3D. Mol. Pharm. 2014, 11, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, R.B. Transporters and renal drug elimination. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 137–166. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Russel, F.G.; Masereeuw, R.; van Aubel, R.A. Molecular aspects of renal anionic drug transport. Annu. Rev. Physiol. 2002, 64, 563–594. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Endou, H. Renal drug transporters and nephrotoxicity. AATEX J. 2007, 14, 447–452. [Google Scholar]
- Smeets, P.H.; van Aubel, R.A.; Wouterse, A.C.; van den Heuvel, J.J.; Russel, F.G. Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J. Am. Soc. Nephrol. 2004, 15, 2828–2835. [Google Scholar] [CrossRef] [PubMed]
- Romiti, N.; Tramonti, G.; Donati, A.; Chieli, E. Effects of grapefruit juice on the multidrug transporter P-glycoprotein in the human proximal tubular cell line HK-2. Life Sci. 2004, 76, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Nieri, P.; Romiti, N.; Adinolfi, B.; Chicca, A.; Massarelli, I.; Chieli, E. Modulation of P-glycoprotein activity by cannabinoid molecules in HK-2 renal cells. Bri. J. Pharmacol. 2006, 148, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Chieli, E.; Romiti, N. Kidney proximal human tubule HK-2 cell line as a tool for the investigation of P-glycoprotein modulation by natural compounds. Bol. Latinoam. Caribe Plantas Med. Aromat. 2008, 7, 281–295. [Google Scholar]
- Wang, Q.; Lu, Y.; Yuan, M.; Darling, I.M.; Repasky, E.A.; Morris, M.E. Characterization of monocarboxylate transport in human kidney HK-2 cells. Mol. Pharm. 2006, 3, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, S.E.; Chung, G.W.; van Loon, E.; Bakar, N.S.; Dalzell, A.M.; Brown, C.D. The limitations of renal epithelial cell line HK-2 as a model of drug transporter expression and function in the proximal tubule. Pflugers Arch. Eur. J. Physiol. 2012, 464, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Tramonti, G.; Romiti, N.; Norpoth, M.; Chieli, E. P-glycoprotein in HK-2 proximal tubule cell line. Ren. Fail. 2001, 23, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Matsunami, T.; Kobayashi, K.; Uchibayashi, T.; Koshida, K.; Tanaka, M.; Namiki, M.; Mizuhara, Y.; Akiba, T.; Yokogawa, K.; et al. Involvement of ABC transporters in chemosensitivity of human renal cell carcinoma, and regulation of MRP2 expression by conjugated bilirubin. Anticancer Res. 2005, 25, 2729–2735. [Google Scholar] [PubMed]
- Pang, K.S. Modeling of intestinal drug absorption: Roles of transporters and metabolic enzymes (for the Gillette Review Series). Drug Metab. Dispos. 2003, 31, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Maeda, K.; Sugiyama, Y. Hepatic and intestinal drug transporters: Prediction of pharmacokinetic effects caused by drug-drug interactions and genetic polymorphisms. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 581–612. [Google Scholar] [CrossRef] [PubMed]
- Oostendorp, R.L.; Beijnen, J.H.; Schellens, J.H. The biological and clinical role of drug transporters at the intestinal barrier. Cancer Treat. Rev. 2009, 35, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Han, T.K.; Everett, R.S.; Proctor, W.R.; Ng, C.M.; Costales, C.L.; Brouwer, K.L.; Thakker, D.R. Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco-2 cell monolayers and enterocytes. Mol. Pharmacol. 2013, 84, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Mooij, M.G.; de Koning, B.E.; Lindenbergh-Kortleve, D.J.; Simons-Oosterhuis, Y.; van Groen, B.D.; Tibboel, D.; Samsom, J.N.; de Wildt, S.N. Human Intestinal PEPT1 Transporter Expression and Localization in Preterm and Term Infants. Drug Metab. Dispos. 2016, 44, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B. Transporters and xenobiotic disposition. Toxicology 2002, 181–182, 291–297. [Google Scholar] [CrossRef]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.L.; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Takano, M. Intestinal efflux transporters and drug absorption. Expert Opin. Drug Metab. Toxicol. 2008, 4, 923–939. [Google Scholar] [CrossRef] [PubMed]
- Grandvuinet, A.S.; Steffansen, B. Interactions between organic anions on multiple transporters in Caco-2 cells. J. Pharm. Sci. 2011, 100, 3817–3830. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Arias, I.M. Intracellular trafficking of P-glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Kis, O.; Zastre, J.A.; Hoque, M.T.; Walmsley, S.L.; Bendayan, R. Role of drug efflux and uptake transporters in atazanavir intestinal permeability and drug-drug interactions. Pharm. Res. 2013, 30, 1050–1064. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Keiser, M.; Drozdzik, M.; Oswald, S. Expression, regulation and function of intestinal drug transporters: An update. Biol. Chem. 2017, 398, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suzuki, H.; Horie, T.; Sugiyama, Y. Apical/basolateral surface expression of drug transporters and its role in vectorial drug transport. Pharm. Res. 2005, 22, 1559–1577. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.M.; Charman, W.N. The mucosa of the small intestine: How clinically relevant as an organ of drug metabolism? Clin. Pharmacokinet. 2002, 41, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F.; Kauffmann, H.M.; Fritz, P.; Burk, O.; Kroemer, H.K.; Warzok, R.W.; Eichelbaum, M.; Siegmund, W.; Schrenk, D. The effect of rifampin treatment on intestinal expression of human MRP transporters. Am. J. Pathol. 2000, 157, 1575–1580. [Google Scholar] [CrossRef]
- Xiao, L.; Yi, T.; Chen, M.; Lam, C.W.; Zhou, H. A new mechanism for increasing the oral bioavailability of scutellarin with Cremophor EL: Activation of MRP3 with concurrent inhibition of MRP2 and BCRP. Eur. J. Pharm. Sci. 2016, 93, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Hirouchi, M.; Kusuhara, H.; Schuetz, J.D.; Sugiyama, Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3). J. Pharmacol. Exp. Ther. 2008, 327, 465–473. [Google Scholar] [CrossRef] [PubMed]
- De Waart, D.R.; van de Wetering, K.; Kunne, C.; Duijst, S.; Paulusma, C.C.; Oude Elferink, R.P. Oral availability of cefadroxil depends on ABCC3 and ABCC4. Drug Metab. Dispos. 2012, 40, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Kunta, J.R.; Sinko, P.J. Intestinal drug transporters: In vivo function and clinical importance. Curr. Drug Metab. 2004, 5, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.R.; Goncalves, P.; Catarino, T.; Araujo, J.R.; Correia-Branco, A.; Martel, F. The effect of oxidative stress upon the intestinal uptake of folic acid: In vitro studies with Caco-2 cells. Cell Biol. Toxicol. 2012, 28, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Kawasaki, M.; Sakane, T.; Omatsu, H.; Moriya, Y.; Nakamura, T.; Sakaeda, T.; Okumura, K.; Langguth, P.; Yamashita, S. Induction of human P-glycoprotein in Caco-2 cells: Development of a highly sensitive assay system for P-glycoprotein-mediated drug transport. Drug Metab. Pharmacokinet. 2006, 21, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Zweibaum, A.; Laburthe, M.; Grasset, E.; Louvard, D. Intestinal absorption and secretion. In Handbook of Physiolgy; Oxford University Press: Oxford, MI, USA, 1991; Volume IV. [Google Scholar]
- Sun, H.; Pang, K.S. Permeability, transport, and metabolism of solutes in Caco-2 cell monolayers: A theoretical study. Drug Metab. Dispos. 2008, 36, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Brandon, E.F.; Bosch, T.M.; Deenen, M.J.; Levink, R.; van der Wal, E.; van Meerveld, J.B.; Bijl, M.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. Validation of in vitro cell models used in drug metabolism and transport studies; genotyping of cytochrome P450, phase II enzymes and drug transporter polymorphisms in the human hepatoma (HepG2), ovarian carcinoma (IGROV-1) and colon carcinoma (CaCo-2, LS180) cell lines. Toxicol. Appl. Pharmacol. 2006, 211, 1–10. [Google Scholar] [PubMed]
- Gutmann, H.; Fricker, G.; Torok, M.; Michael, S.; Beglinger, C.; Drewe, J. Evidence for different ABC-transporters in Caco-2 cells modulating drug uptake. Pharm. Res. 1999, 16, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Taipalensuu, J.; Tornblom, H.; Lindberg, G.; Einarsson, C.; Sjoqvist, F.; Melhus, H.; Garberg, P.; Sjostrom, B.; Lundgren, B.; Artursson, P. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 2001, 299, 164–170. [Google Scholar] [PubMed]
- Van Breemen, R.B.; Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, A.L.; Gyurdieva, A.V.; Mabus, J.R.; Ferguson, C.; Yan, Z.; Hornby, P.J. Alternative functional in vitro models of human intestinal epithelia. Front. Pharmacol. 2013, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bachmeier, C.; Miller, D.W. In vitro and in vivo models for assessing drug efflux transporter activity. Adv. Drug Deliv. Rev. 2003, 55, 31–51. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Mehn, D.; Jani, M.; Oosterhuis, B.; Heredi-Szabo, K.; Krajcsi, P. Utilization of membrane vesicle preparations to study drug-ABC transporter interactions. Expert Opin. Drug Metab. Toxicol. 2008, 4, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Al-Shawi, M.K.; Urbatsch, I.L.; Senior, A.E. Covalent inhibitors of P-glycoprotein ATPase activity. J. Biol. Chem. 1994, 269, 8986–8992. [Google Scholar] [PubMed]
- Langmann, T.; Mauerer, R.; Zahn, A.; Moehle, C.; Probst, M.; Stremmel, W.; Schmitz, G. Real-time reverse transcription-PCR expression profiling of the complete human ATP-binding cassette transporter superfamily in various tissues. Clin. Chem. 2003, 49, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Gottesman, M.M. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr. Pharm. Biotechnol. 2011, 12, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Wittwer, C. Flow cytometry: Principles and clinical applications in hematology. Clin. Chem. 2000, 46 Pt. 2, 1221–1229. [Google Scholar] [PubMed]
- Nolan, J.P.; Yang, L. The flow of cytometry into systems biology. Brief. Funct. Genom. Proteom. 2007, 6, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Lin, B.; Qiu, J.; Chan, L.L.; Bates, S.E. Rapid detection of ABC transporter interaction: Potential utility in pharmacology. J. Pharmacol. Toxicol. Methods 2011, 63, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Silva, R.; Gaio, A.R.; Martins, A.M.; Lima, S.C.; Cordeiro-da-Silva, A.; de Lourdes Bastos, M.; Remiao, F. P-glycoprotein activity in human Caucasian male lymphocytes does not follow its increased expression during aging. Cytometry A 2011, 79, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; Bastos Mde, L.; Remiao, F. Colchicine effect on P-glycoprotein expression and activity: In silico and in vitro studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.V.; Pande, P.; Patton, W.F. Sensitive and specific fluorescent probes for functional analysis of the three major types of mammalian ABC transporters. PLoS ONE 2011, 6, e22429. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J. Pharm. Pharm. Sci. 2009, 12, 46–78. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.M.; Valizadeh, H.; Hamishehkar, H.; Zakeri-Milani, P. Inhibition of P-glycoprotein expression and function by anti-diabetic drugs gliclazide, metformin, and pioglitazone in vitro and in situ. Res. Pharm. Sci. 2016, 11, 177–186. [Google Scholar] [PubMed]
- Cantore, M.; Leopoldo, M.; Berardi, F.; Perrone, R.; Colabufo, N.A. Design and Synthesis of New Selective P-gp Substrates and Inhibitors. Curr. Pharm. Des. 2016, 22, 5774–5778. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Mei, H.; Qu, S.; Huang, S.; Sun, J.; Yang, L.; Chen, H. Prediction and characterization of P-glycoprotein substrates potentially bound to different sites by emerging chemical pattern and hierarchical cluster analysis. Int. J. Pharm. 2016, 502, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C.; Hersom, M.; Kuhlmann, L.B.; Badolo, L.; Nielsen, C.U.; Brodin, B. An electrically tight in vitro blood-brain barrier model displays net brain-to-blood efflux of substrates for the ABC transporters, P-gp, Bcrp and Mrp-1. AAPS J. 2014, 16, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Cisternino, S.; Rousselle, C.; Lorico, A.; Rappa, G.; Scherrmann, J.M. Apparent lack of Mrp1-mediated efflux at the luminal side of mouse blood-brain barrier endothelial cells. Pharm. Res. 2003, 20, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, Y.Z.; Seigel, G.M.; Hu, Z.H.; Huang, M.; Yu, A.M. Breast cancer resistance protein BCRP/ABCG2 regulatory microRNAs (hsa-miR-328, -519c and -520h) and their differential expression in stem-like ABCG2+ cancer cells. Biochem. Pharmacol. 2011, 81, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, C.I.; Arias, A.; Novak, A.; Carpini, G.D.; Villanueva, S.; Blazquez, A.G.; Marin, J.J.; Mottino, A.D.; Rubio, M.C. Acetaminophen-induced stimulation of MDR1 expression and activity in rat intestine and in LS 174T human intestinal cell line. Biochem. Pharmacol. 2011, 81, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- del Amo, E.M.; Heikkinen, A.T.; Monkkonen, J. In vitro-in vivo correlation in P-glycoprotein mediated transport in intestinal absorption. Eur. J. Pharm. Sci. 2009, 36, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F. Importance of P-glycoprotein for drug disposition in humans. Eur. J. Clin. Investig. 2003, 33, 6–9. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; He, Z.G.; Cheng, G.; Wang, S.J.; Hao, X.H.; Zou, M.J. Multidrug resistance P-glycoprotein: Crucial significance in drug disposition and interaction. Med. Sci. Monit. 2004, 10, RA5–A14. [Google Scholar] [PubMed]
- Novak, A.; Godoy, Y.C.; Martinez, S.A.; Ghanem, C.I.; Celuch, S.M. Fructose-induced metabolic syndrome decreases protein expression and activity of intestinal P-glycoprotein. Nutrition 2015, 31, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Carpini, G.D.; Ruiz, M.L.; Luquita, M.G.; Rubio, M.C.; Mottino, A.D.; Ghanem, C.I. Acetaminophen inhibits intestinal p-glycoprotein transport activity. J. Pharm. Sci. 2013, 102, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Zhang, X.-Y.; Zhang, G.-N.; Wang, Y.-J.; Xu, H.; Zhang, D.; Shukla, S.; Liu, L.; Yang, D.-H.; Ambudkar, S.V.; Chen, Z.-S. Selective reversal of BCRP-mediated MDR by VEGFR-2 inhibitor ZM323881. Biochem. Pharmacol. 2017. [CrossRef] [PubMed]
- László, L.; Sarkadi, B.; Hegedűs, T. Jump into a new fold—A homology based model for the ABCG2/BCRP multidrug transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef] [PubMed]
- Talha, S.; Ishrat, J. Grid-independent Descriptors (GRIND) analysis and SAR guided molecular docking studies to probe selectivity profiles of inhibitors of multidrug resistance transporters ABCB1 and ABCG2. Curr. Cancer Drug Targets 2017, 17, 177–190. [Google Scholar]
- Qiu, J.-G.; Zhang, Y.-J.; Li, Y.; Zhao, J.-M.; Zhang, W.-J.; Jiang, Q.-W.; Mei, X.-L.; Xue, Y.-Q.; Qin, W.-M.; Yang, Y.; et al. Trametinib modulates cancer multidrug resistance by targeting ABCB1 transporter. Oncotarget 2015, 6, 15494–15509. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, S.; Ravichandran, S.; Vetrivel, U.; Krishnakumar, S. In vitro and In silico studies on inhibitory effects of curcumin on multi drug resistance associated protein (MRP1) in retinoblastoma cells. Bioinformation 2012, 8, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Koeda, A.; Morikawa, H.; Satoh, T.; Narimatsu, S.; Naito, S. Comparison of Inducibility of Multidrug Resistance (MDR)1, Multidrug Resistance-Associated Protein (MRP)1, and MRP2 mRNAs by prototypical microsomal enzyme inducers in primary cultures of human and cynomolgus monkey hepatocytes. Biol. Pharm. Bull. 2008, 31, 2068–2072. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Koeda, A.; Suzuki, E.; Kawano, Y.; Nakayama, M.; Satoh, T.; Narimatsu, S.; Naito, S. Regulation of mRNA expression of MDR1, MRP1, MRP2 and MRP3 by prototypical microsomal enzyme inducers in primary cultures of human and rat hepatocytes. Drug Metab. Pharmacokinet. 2006, 21, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, H.-M.; Pfannschmidt, S.; Zöller, H.; Benz, A.; Vorderstemann, B.; Webster, J.I.; Schrenk, D. Influence of redox-active compounds and PXR-activators on human MRP1 and MRP2 gene expression. Toxicology 2002, 171, 137–146. [Google Scholar] [CrossRef]
- Schrenk, D.; Baus, P.R.; Ermel, N.; Klein, C.; Vorderstemann, B.; Kauffmann, H.-M. Up-regulation of transporters of the MRP family by drugs and toxins. Toxicol. Lett. 2001, 120, 51–57. [Google Scholar] [CrossRef]
- Allen, J.D.; van Dort, S.C.; Buitelaar, M.; van Tellingen, O.; Schinkel, A.H. Mouse breast cancer resistance protein (Bcrp1/Abcg2) mediates etoposide resistance and transport, but etoposide oral availability is limited primarily by P-glycoprotein. Cancer Res. 2003, 63, 1339–1344. [Google Scholar] [PubMed]
- Kaur, M.; Badhan, R.K. Phytochemical mediated-modulation of the expression and transporter function of breast cancer resistance protein at the blood-brain barrier: An in vitro study. Brain Res. 2017, 1654, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Badolo, L.; Jensen, B.; Säll, C.; Norinder, U.; Kallunki, P.; Montanari, D. Evaluation of 309 molecules as inducers of CYP3A4, CYP2B6, CYP1A2, OATP1B1, OCT1, MDR1, MRP2, MRP3 and BCRP in cryopreserved human hepatocytes in sandwich culture. Xenobiotica 2015, 45, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Parmentier, Y.; Fardel, O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 2006, 34, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.; Seidel, A.; Lampen, A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005, 26, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Brok, M.; Wiemer, E.A.; de Bruijn, E.A.; Guetens, G.; de Boeck, G.; Sparreboom, A.; Verweij, J.; Nooter, K. Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Biol. Ther. 2005, 4, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, L.M.; Li, H.; Li, L.; Lynch, C.; Xie, Y.; Nakanishi, T.; Ross, D.D.; Wang, H. A novel xenobiotic responsive element regulated by Aryl Hydrocarbon Receptor is involved in the induction of BCRP/ABCG2 in LS174T cells. Biochem. Pharmacol. 2010, 80, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.; Seidel, A.; Lampen, A. Phytochemicals induce breast cancer resistance protein in Caco-2 cells and enhance the transport of Benzo[a]pyrene-3-sulfate. Toxicol. Sci. 2007, 96, 227–236. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein and Gene Name | Tissue Distribution | Substrates | Inhibitors | Inducers | Activators |
|---|---|---|---|---|---|
| P-gp (ABCB1 or MDR1) | Brain, liver, kidney, intestine, uterus, ovary, testes, placenta, adrenal gland, cancer cells [1,3,14] | Anticancer drugs (vinca alkaloids (vinblastine, vincristine, catharanthine), anthracyclines (doxorubicin, daunorubicin), taxanes (paclitaxel and docetaxel), epipodophyllotoxins (etoposide, teniposide), camptothecins (topotecan, methotrexate), anthracenes (bisantrene, mitoxantrone)) HIV protease inhibitors (ritonavir, saquinavir, nelfinavir, amprenavir, indinavir, maraviroc, darunavir) Analgesics (morphine) Antidepressants (amitryptiline, nortryptiline, doxepin, venlafaxine, paroxetine) Antihistamines (terfenadine, fexofenadine) Histamine H2-receptor antagonists (cimetidine) Antidiarrheal agents (loperamide) Immunosuppressive agents (sirolimus, valspodar, cyclosporin A, tacrolimus (FK506) Antiarrhythmics (quinidine, amiodarone, propafenone) Antiepileptics (phenytoin, felbamate, topiramate, carbamazepine, lamotrigine, phenobarbital, gabapentin, topiramate) Fluorescent compounds (calcein-AM, Hoechst 33342, rhodamine 123) HMG-CoA reductase inhibitors (lovastatin, simvastatin) Antiemetics (ondansetron, domperidone) Antimicrobial agents (doxycycline, erythromycin, itraconazole, ketoconazole, levofloxacin, rifampin, sparfloxacin, tetracycline, grepafloxacin), Antihelminthics (abamectin, ivermectin) Tyrosine kinase inhibitors (imatinib mesylate, gefitinib, nilotinib, tandutinib) Cardiac glycosides (digitoxin, digoxin, quinidine) Calcium-channel blockers (verapamil, nifedipine, azidopine, diltiazem, nicardipine) Calmodulin antagonists (trifluoperazine, chlorpromazine, trans-flupentixol) Anti-tuberculous agent (rifampin) Anti-hypertensives (debrisoquine, reserpine, propranolol, celiprolol, diltiazem, losartan, talinolol, prazosin) Anticonvulsants (phenobarbital, phenytoin) Antibiotics (actinomycind, clarithromycin, amoxicillin, erythromycin, gramicidin A, valinomycin, tetracyclines, fluoroquinolines) Anti-gout agents (colchicine) Anti-tuberculous agent (erythromycin) Thrombin inhibitors (dabigatran) Steroids (dexamethasone, hydrocortisone, corticosterone, cortisol, aldosterone, methylprednisolone) Opioids (loperamide, morphine, pentazocine) Pesticides (methylparathion, endosulfan, paraquat, cypermethrin, fenvalerate) Nature products (flavonoids, curcuminoids, Rhei Rhizoma extract) Antialcoholism drug (disulfiram) [3,5,10,14,99,102,174,191] | 1st generation inhibitors: (verapamil, cyclosporin A, nifedipine, quinidine, quinine, amiodarone, tamoxifen detergents (cremophore EL)) 2nd generation inhibitors: (R-verapamil, PSC 833, dexniguldipine, valspodar, elacridar, biricodar, dexverapamil, dofequine fumarate) 3rd generation inhibitors: (ontogen (OC 144-093), zosuquidar, tariquidar, elacridar laniquidar, biricodar) 4th generation inhibitors: surfactants (e.g., sodium dodecyl sulphate, Tween-20 and Span-80) and lipids; compounds extracted from natural origins and their derivatives (flavonoids, such as tangeretin, sinensetin, baicalein and quercetin; alkaloids, such as ellipticine ; coumarins such as cnidiadin and praeruptorin; among others); peptidomimetics (e.g., reversins) and agents combining transport inhibition with another beneficial biological activity, also called dual ligands (aminated thioxanthones, that act as dual inhibitors of cell growth and P-gp (e.g., 1-[2-(1H-benzimidazol-2yl)ethanamine]-4-propoxy-9H-thioxanthen-9-one), which also demonstrated to be a potent inhibitor of other ABC transporters, such as BCRP, MRP-1 and MRP-3). [17,99,226] | The MDR1 gene responds to a vast diversity of internal or external chemical stimuli (e.g., drugs, cytokines, oxygen free radicals, tumor suppressor genes and heat shock) and to other environmental factors, such as X-irradiation, UV-irradiation. Some of the reported P-gp inducers comprise (listed alphabetically): Abacavir, Actinomycin D, Aflatoxin B1, Aldosterone, Ambrisentan, Amiodarone, amprenavir, m-amsacrine, apigenin, artemisinin, asiatic acid, atazanavir, atorvastatin, avermectin, beclomethasone, benzo(a)pyrene, benzo(e)pyrene, berberine, betamethasone, bilirubin, bosentan, bromocriptine, budesonide, caffeine, cadmium chloride, capsaicin, carbamazepine, catechin, celiprolol, cembratriene, R-cetirizine, CITCO, chlorambucil, cholate, chrysin, ciclesonide, cisplatin, clotrimazole, colchicine, corticosterone, curcuma extracts (extracts of Curcuma longa, Curcuma zedoaria and Curcuma aromatica), curcumin, cyanidin, cyclophosphamide, cyclosporine A, cytarabine, daidzein daunorubicin, daurunavir, depsipeptide (FR901228, FK228, NSC630176), desvenlafaxine, dexamethasone, diclofenac, digoxin, dihydroxylated xanthones (e.g., 1,2-dihydroxy-9H-xanthen-9-one (X2)), 1α,25-dihydroxyvitamin D3, diltiazem, dimethylformamide, 6,16α-dimethylpregnenolone, dimethylsulfoxide, docetaxel, doxorubicin, doxycycline, efavirenz, emetined, epigallocatechin-3-gallate, epirubicin, eriodictyol, erythromycin, β-estradiol, ethinylestradiol, etoposide, fenbufen, flavone, 5-fluorouracil, flutamide, fluticasone, genistein, ginkgolides a and b, hydroxyurea, hyperforin, hypericin, Hypericum perforatum extracts (Saint John’s wort), idarubicin, ifosfamide, indinavir, indomethacin, insulin, isosafrole, isoxanthohumol, ivermectin, lopinavir, LY191401, mangiferin, meloxicam, mepirizole, methotrexate, methylprednisolone, midazolam, mifepristone, mitoxantrone, morphine, mx2, myricetin, naringenin, nefazodone, nelfinavir, nevirapine, nicardipine, nifedipine, nimesulide, norathyriol, oleocanthal, ouabain, oxycodone, paclitaxel, parthenolide, pentylenetetrazole, phenobarbital, phenothiazine, phenytoin, phorbol 12-myristate 13-acetate, piperine, platelet-activating factor, prednisolone, 5β-pregnane-3,20-dione, pregnenolone-16α-carbonitrile, probenecid, propranolol, quercetin, quinidine, rapamycin or sirolimus, reduced rifampicin derivative (RedRif), rescinnamine, reserpine, retinoic acid, rhinacanthin-C, rifampicin, rilpivirinem, ritonavir, saquinavir, small molecule tyrosine kinase inhibitors (erlotinib, gefitinib, nilotinib, sorafenib, vandetanib), sildenafil, sodium arsenite, sodium butyrate, spironolactone, SR12813, sulindac, tacrolimus, tadalafil, tamoxifen, tangeretin, taurocholate, taxifolin, TCDD, tetrahydrocurcumin, thioxanthonic derivatives (e.g., 1-(propan-2-ylamino)-4-propoxy-9H-thioxanthen-9-one (TX 5)), γ-tocotrienol, topotecan, trazodone, triactyloleandomycin, trichostatin A, trimethoxybenzoylyohimbine, venlafaxine, verapamil, vinblastine, vincristine [3]. | A synthetic derivative of rifampicin (a reduced derivative, RedRif) [20] Dihydroxylated xanthones: 3,4-Dihydroxy-9H-xanthen-9-one (X1), 1,2-Dihydroxy-9H-xanthen-9-one (X2), 1,3-Dihydroxy-9H-xanthen-9-one (X3), 2,3-dihydroxy-9H-xanthen-9-one (X4) and 3,6-dihydroxy-9H-xanthen-9-one (X5) [21] Thioxanthonic derivatives: 1-[(3-hydroxypropyl)amino]-4-propoxy-9H-thioxanthen-9-one (TX 1), 1-chloro-4-hydroxy-9H-thioxanthen-9-one (TX 2), 1-{[2-(1,3-benzodioxol-5-yl) ethyl]amino}-4-propoxy-9H-thioxanthen-9-one (TX 3), 1-[(2-methylpropyl) amino]-4-propoxy-9H-thioxanthen-9-one (TX 4) and 1-(propan-2-ylamino)-4-propoxy-9H-thioxanthen-9-one (TX 5) [22] |
| MRP1 (ABCC1) | Brain, kidney, lung, intestine and testis [14] | Anticancer drugs (vinca alkaloids (vinblastine, vincristine), anthracyclines (doxorubicin, daunorubicin), taxanes (paclitaxel), epipodophyllotoxins (etoposide, teniposide), camptothecins (topotecan, methotrexate, irinotecan)) Glucuronide, sulfate and glutathione conjugates, and unconjugated compounds (glucuronosylbilirubin, estradiol-17-β-d-glucuronide, etoposide-glucuronide, SN-38-glucuronide, leukotrienes C4, D4 and E4, glutathione disulfide, prostaglandin A2-SG, hydroxynonenal-SG, aflatoxin B1-epoxide-SG, cyclophosphamide-SG, doxorubicin-SG, estrone-3-sulfate, dehydroepiandrosterone-3-sulfate, sulfatolithocholyl taurine) Antibiotics (difloxacin, grepafloxicin) Folates (folic acid, l-leucovorin) Anthracenes (mitoxantrone) Natural products (curcuminoids) Metalloids (sodium arsenate, sodium arsenite, potassium antimonite) Pesticides (fenitrothion, methoxychlor) Toxins (aflatoxin B1) Fluorescent compounds (calcein, fluorescein, Fluo-3, BCECF) HIV protease inhibitors (indinavir, adefovir) [1,5,10,14,99,102] | Sulfinpyrazone, biricodar, probenecid, MK571, LTC4, cyclosporin A, verapamil, PSC 833, benzbromarone, indomethacin, probenecid, agosterol A and analogs, verapamil derivatives, flavonoids derivatives (genistein and flavopiridol), raloxifene-based inhibitors (LY117018, LY329146 and indomethacin), piperazine and piperidine-based compounds as dual MRP1/P-gp inhibitors (N,N-disubstituted piperazines), isoxazole-based compounds (LY402913), quinazoline- and quinaxoline-derived molecules [5,10,99] | Dexamethasone [248] Rifampicin [248,249] Sulindac [53] TBHQ and quercetin [250] Vinblastine and TBHQ [251] | NR |
| BCRP (ABCG2) | Brain, breast, liver, intestine, placenta and cancer cells [1] | Anticancer drugs (anthracyclines (doxorubicin, daunorubicin), epipodophyllotoxins (etoposide, teniposide), camptothecins (topotecan, methotrexate, irinotecan, diflomotecan), anthracenes (mitoxantrone, bisantrene)) Anti-hypertensives (prazosin) Porphyrins (pheophorbide a, protoporphyrin IX, hematoporphyrin) Tyrosine kinase inhibitors (imatinib mesylate, gefitinib, tandutinib, lapatinib) Natural products (flavonoids (quercetin, genestein), curcuminoids, phytoestrogens) Carcinogens (aflatoxin B, PhiP) Toxins (fumitremorgin C, Ko143) HMG CoA reductase inhibitors (rosuvastatin, pravastatin, cerivastatin, pitavastatin, atorvastatin, rosuvastatin, simvastatin, fluvastatin) Metabolite conjugates (acetaminophen sulfate, estrone-3-sulfate, dehydroepiandrosterone sulfate, estradiol-17-β-d-glucuronide, dinitrophenyl-S-glutathione) Antihypertensives (reserpine) Antibiotics (ciprofloxacin, norfloxacin) Fluorescent compounds (Hoechst 33342, BODIPY-prazosin, rhodamine 123) Antiviral drugs (zidovudine, lamivudine) [5,10,14,99,174] | ABCG2-specific inhibitors (fumitremorgin C and analogues (Ko143, Ko132, Ko134, Cl1033), eltrombopag) Compounds also inhibit P-gp and/or MRP1 (elacridar, atazanavir, ritonavir, tariquidar, cyclosporine) Natural products (flavonoids, tryprostatin A and novobiocin) Tyrosine kinase inhibitors (gefitinib and imatinib) [10,23,99,252] | Efavirenz [10] Quercetin and naringin [253] Oxotremorine, Phenobarbital, Cetylpyridinium chloride, Promazine, Benzetimide, Ifenprodil, Etoposide, Perphenazine and venlafaxine [254] phenobarbital, rifampicin, and omeprazole [255] benzopyrene conjugates [256] Imatinib [257] 3-methylcholanthrene (3MC), troglitazone, resveratrol and omeprazole [258] TBHQ, dibenzoylmethane, quercetin, chrysin, flavone, indole-3-carbinol, Curcumin and resveratrol [259] | NR |
| Advantages | Disadvantages | ||
|---|---|---|---|
| In vitro assays | Cell-based assays |
|
|
| Membrane-based assays |
|
| |
| Ex vivo assays |
|
| |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.D.L.; Remião, F. Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP. Molecules 2017, 22, 600. https://doi.org/10.3390/molecules22040600
Gameiro M, Silva R, Rocha-Pereira C, Carmo H, Carvalho F, Bastos MDL, Remião F. Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP. Molecules. 2017; 22(4):600. https://doi.org/10.3390/molecules22040600
Chicago/Turabian StyleGameiro, Mariline, Renata Silva, Carolina Rocha-Pereira, Helena Carmo, Félix Carvalho, Maria De Lourdes Bastos, and Fernando Remião. 2017. "Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP" Molecules 22, no. 4: 600. https://doi.org/10.3390/molecules22040600
APA StyleGameiro, M., Silva, R., Rocha-Pereira, C., Carmo, H., Carvalho, F., Bastos, M. D. L., & Remião, F. (2017). Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP. Molecules, 22(4), 600. https://doi.org/10.3390/molecules22040600