1. Introduction
Herbal medicines continue to be the first line of treatment worldwide, with more than 80% of the population consuming them for major and minor illnesses [
1]. The ginger root (
Zingiber officinale Roscoe) has been used as a medicinal herb for more than 2000 years and is one of the most widely consumed dietary supplements in the world [
1]. Ginger is used primarily as a remedy for digestive disorders including dyspepsia, colic, nausea, vomiting, gastritis, and diarrhea [
2,
3]. Studies using animal models have shown that ginger and its phenolic constituents (i.e., 6-gingerol) suppress carcinogenesis in the skin [
4,
5,
6,
7,
8], gastrointestinal tract [
9], colon [
10], and breast [
11]. Ginger extracts have been tested for both anti-tumor promotion and apoptotic potential in several
invitro cancer cell lines, including leukemia [
12], gastric [
13], prostate [
14], ovarian [
15], and lung carcinoma [
16].
The chemopreventive and chemotherapeutic mechanisms of ginger are not well understood but are thought to involve upregulation of carcinogen detoxifying enzymes, anti-oxidants, and anti-inflammatory [
17,
18,
19] activity. Ginger also inhibits nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) activation induced by a variety of agents that have been shown to downregulate NF-κB regulated gene products involved in cellular proliferation and angiogenesis [
20], including interleukin-8 (IL-8) [
14] and vascular endothelial growth factor (VEGF) [
21]. Components of ginger extract (GE) at proapoptotic concentrations induce G
2/M phase cell cycle arrest and aberrant mitotic cell death associated with tubulin aggregation [
22]. It is also reported that GE can induce antioxidant response element (ARE)-reporter gene activity and nuclear factor erythroid 2 related factor 2 (NrF2) expression [
23].
While multiple mechanisms have been relatively well-studied on the remedial action of ginger on a spectrum of disease indications, the pharmacokinetic (PK) characteristics of GE phenolics and their correlation with the mechanism of action remains elusive. Only a handful of studies are available that have examined the absorption, bioavailability, metabolism, and elimination of GE phenolics [
24,
25,
26,
27]. In particular, only few of the pungent compounds like 6-gingerol (6G), 10-gingerol (10G), 6-shogaol (6S), and zingerone have been investigated [
28,
29,
30,
31,
32,
33]. In addition, reported studies demonstrate administration of 6G using an intravenous bolus route [
34,
35], which is unlikely to reflect conventional oral dosing for natural medicines. In humans, when ginger was administered up to two grams, and sub-therapeutic concentrations of GE phenolics were detected. Interestingly, glucuronide and sulfate conjugated forms of GE phenolics in plasma have been reported [
26].
To better understand and correlate the pharmacodynamic (PD) efficacy with PK characteristics of GE phenolics, we investigated the solubility and stability of GE phenolics in various pH buffers and simulated gastric fluids (SGF) and simulated intestinal fluids (SIF) to assess their suitability for oral dose administration. We assessed their metabolic stability in mouse, rat, dog, and human liver microsomes to gain insights into interspecies differences and their propensity to form phase I metabolites. We performed a mouse pharmacokinetic study with and without co-administration of ketoconazole (KTZ), a known inhibitor of cytochrome P450s and uridine glucuronosyl transferases (UGTs), to assess the effect of first pass metabolism on the exposure of GE phenolics. KTZ is considered a non-specific inhibitor of UGTs with potential effect on UGT1A1, 1A9, 1A6, 1A4, and 2B7 [
36,
37].
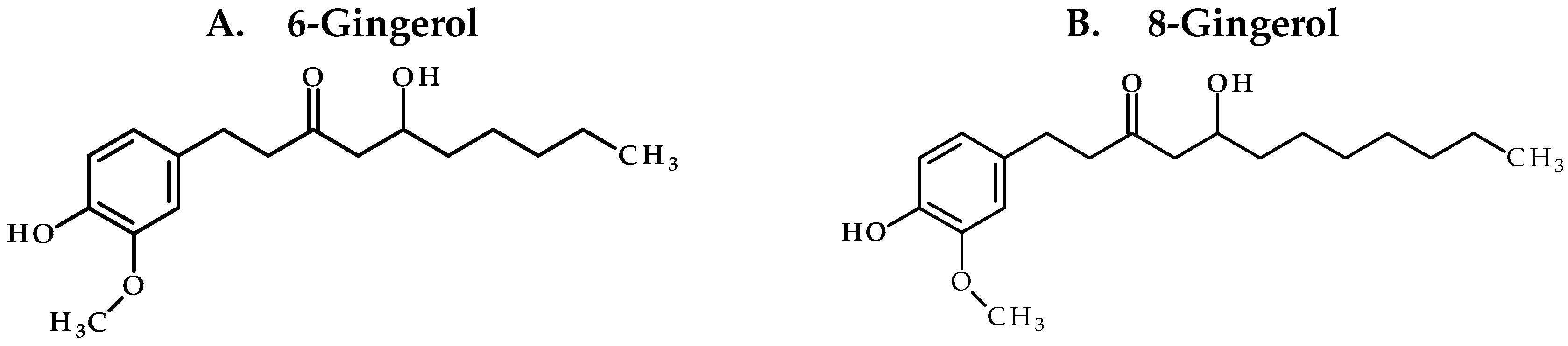
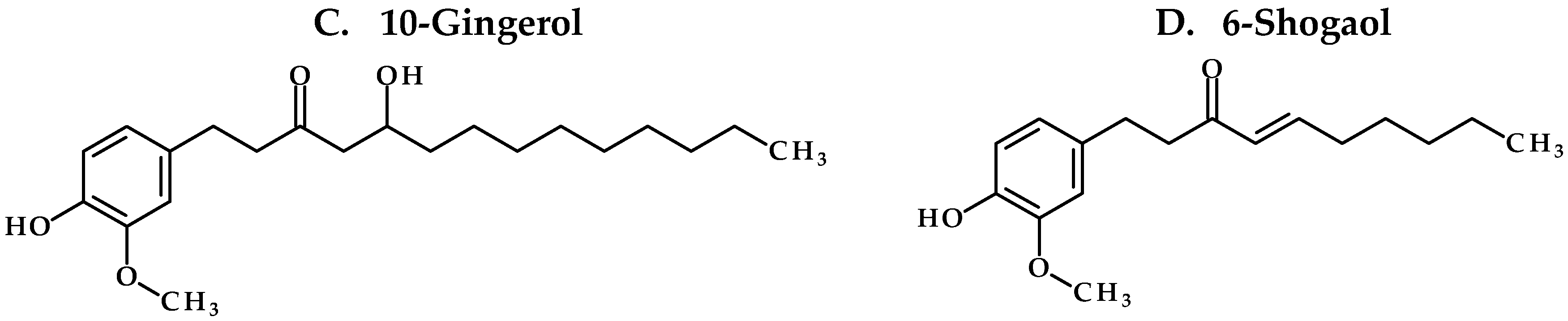
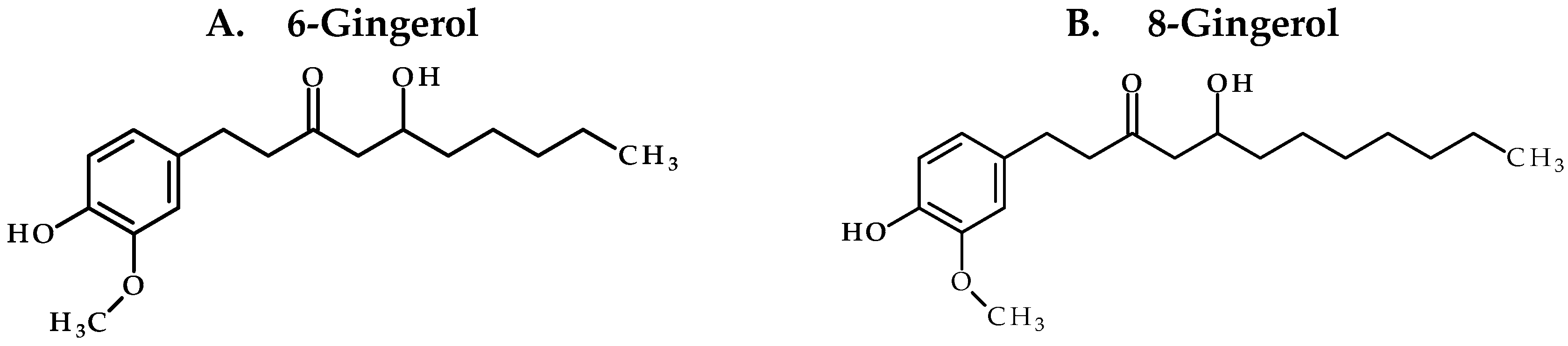
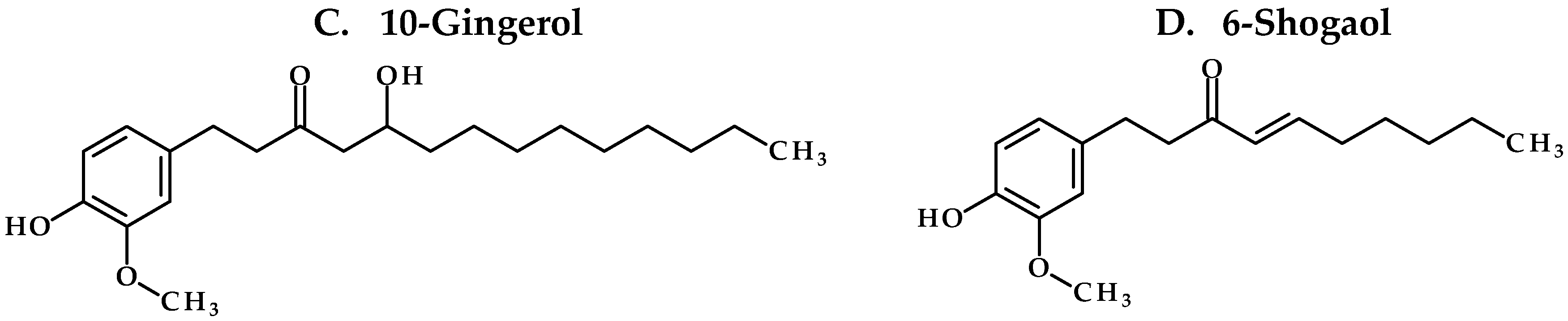
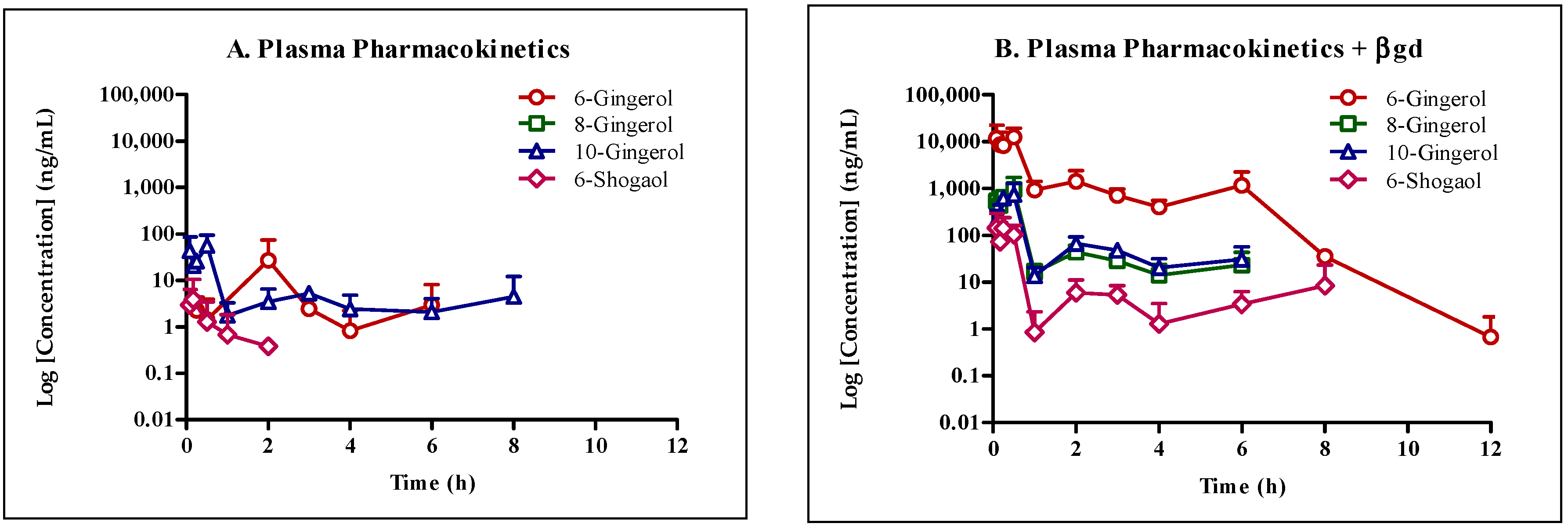
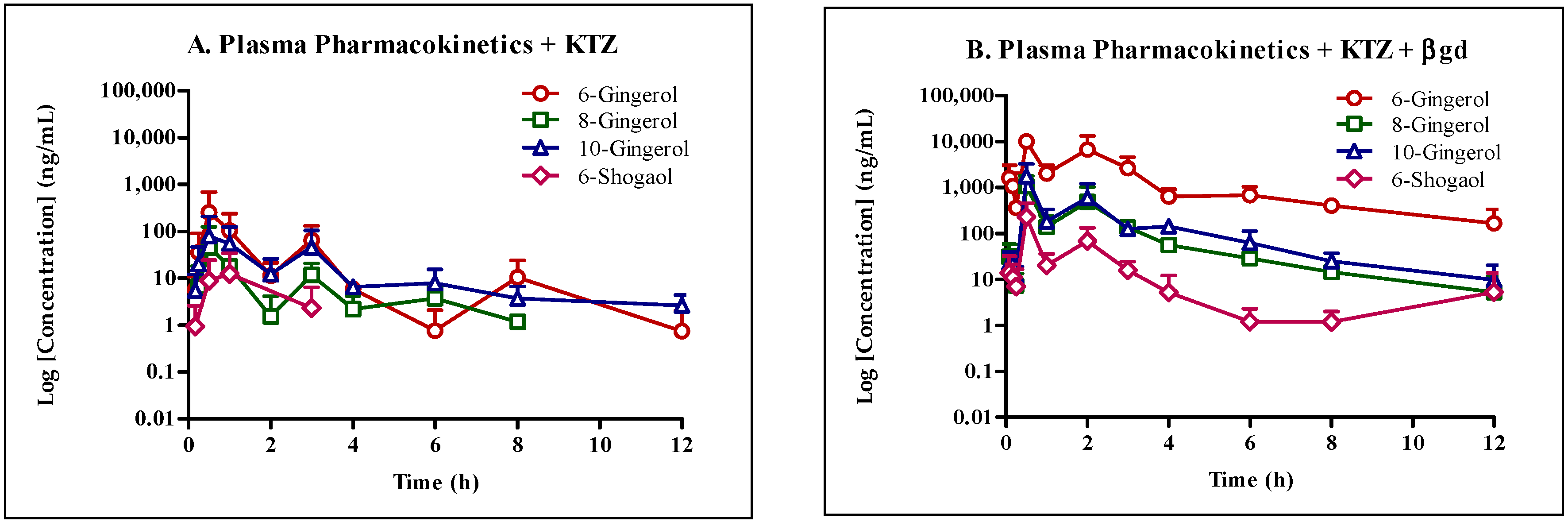
Our study confirms the suitability of whole ginger extract (GE) for oral dosing as the active GE phenolics (
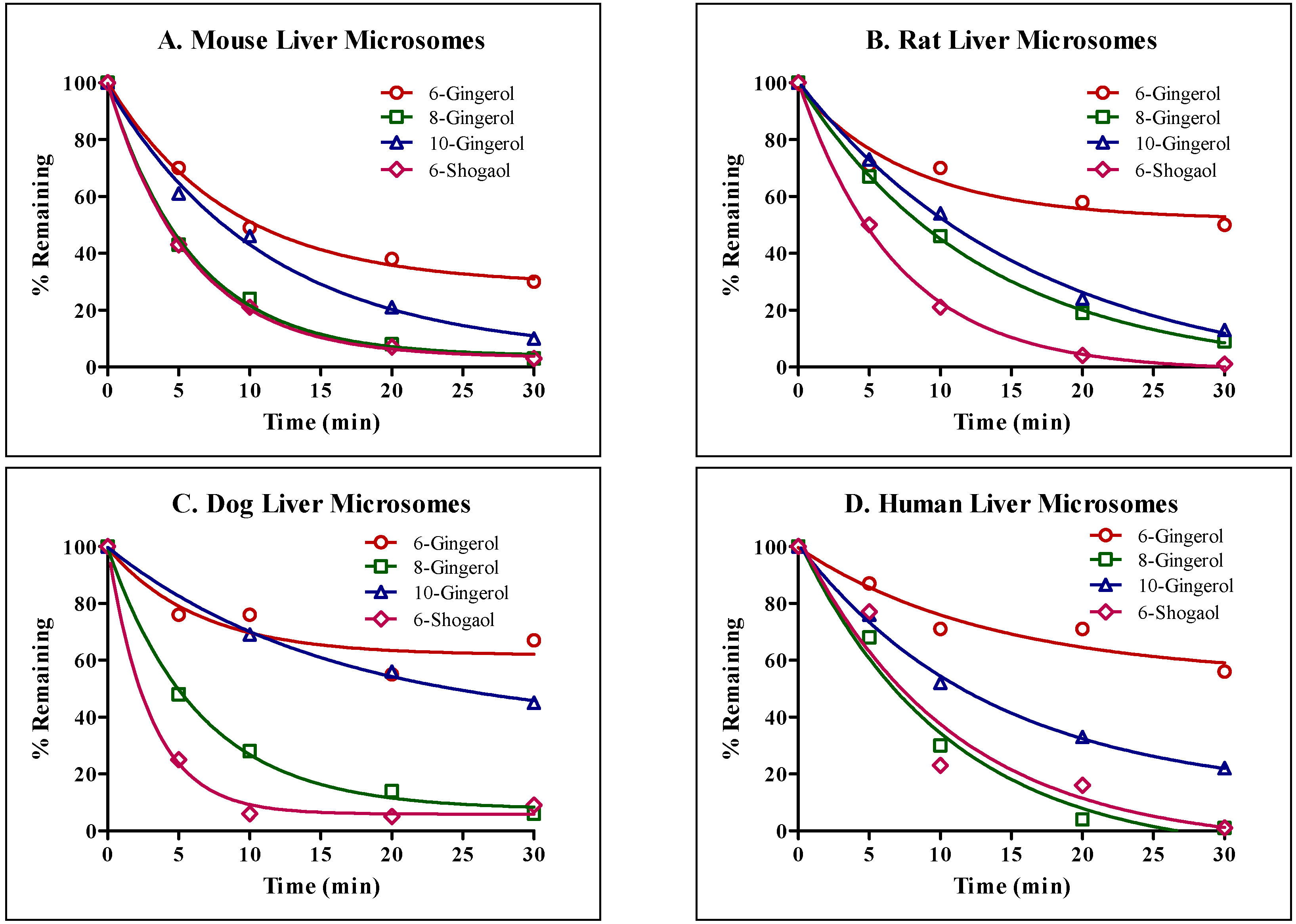
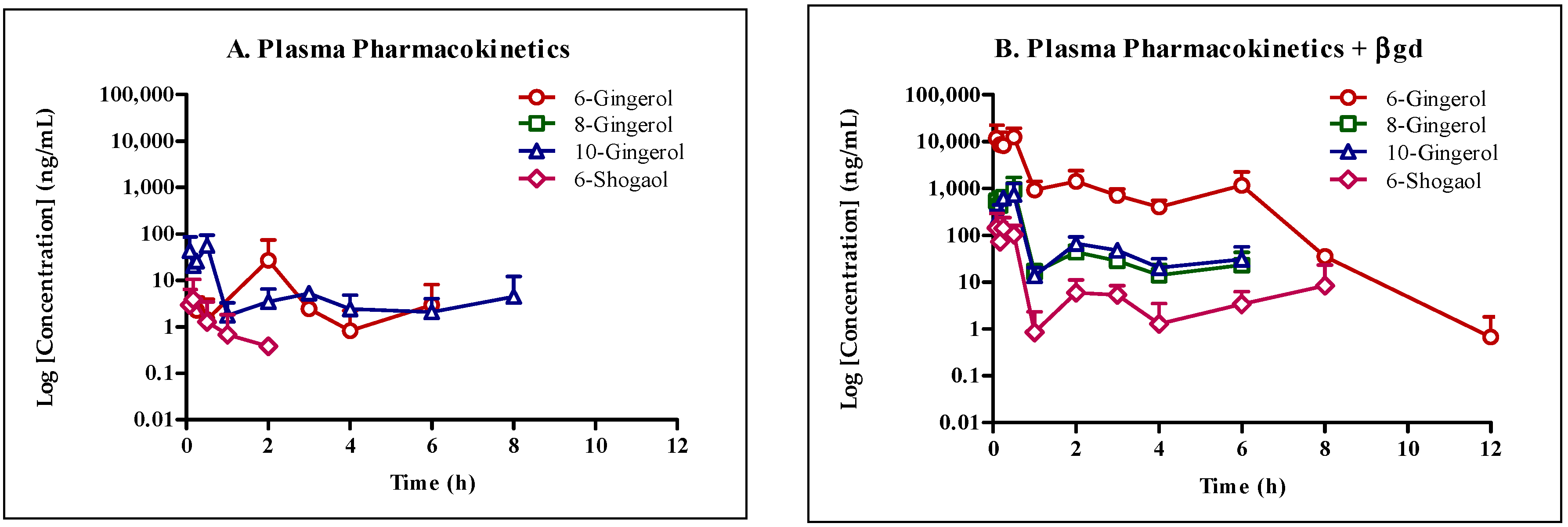
Figure 1) were stable under various pH conditions and in SGF and SIF. However, microsomal stability studies uncovered that GE phenolics undergo extensive phase I metabolism. No species differences were observed, suggesting the appropriateness of animal models for extrapolating the data to humans. The mouse pharmacokinetic study with 250 mg/kg GE showed sub-therapeutic concentration of all GE phenolics. Treatment of plasma samples with β-glucuronidase (βgd) released free GE phenolics, confirming their conversion to glucuronide conjugates. Oral co-administration with 50 mg/kg KTZ did increase the exposure of GE phenolics but did not potently inhibit the phase II glucuronide conjugation reaction. Moreover, the exposure of glucuronide conjugates was far more compared to the increase in exposure due to inhibition of phase I metabolism by KTZ. These results suggest a major role of phase II glucuronide conjugation in clearance of GE phenolics.
3. Discussion
Ginger is being used as a therapy for many clinical conditions with no concrete mechanism of action proven. In human clinical studies, glucuronide and sulfate conjugates of GE phenolics were found in plasma with sub-therapeutic concentrations of free forms following a two-grams dose administered orally. We believe that there is a knowledge gap in tying the efficacy of GE to the analytes responsible for it. To address this gap, we systematically performed various in vitro studies with purified GE phenolics and a pharmacokinetic study in mouse using GE.
We first studied the structural properties of GE phenolics using the Lipinski rule of 5 and Veber rule for druglikeness. All GE phenolics followed the Lipinski and Veber rules confirming their suitability for oral dose administration. Next, we tried to classify GE phenolics according to the biopharmaceutical drug disposition classification system (BDDCS) and assessed their solubility at pH 2, 4, and 7.4. 6G and 8G were found to be soluble up to 100 µM at all three pH conditions. 10G and 6S showed solubility of less than 10 µM. Based on the solubility data and considering a dose of 2 g ginger taken along with 250 mL of water in humans, we classified GE phenolics as Class II or Class IV compounds. To understand the stability of GE phenolics in the gastro-intestinal tract (GIT), we assessed their stability in SGF and SIF. All GE phenolics were found to be stable in SGF and SIF confirming their suitability for oral dose administration. This also confirmed that the sub-therapeutic concentrations of free GE phenolics found in vivo were not due to stability issues in GIT. By using suitable formulation ingredients, solubility of GE phenolics can be improved to achieve desired exposure in vivo. We prepared a solution formulation for GE with various co-solvents for single dose administration.
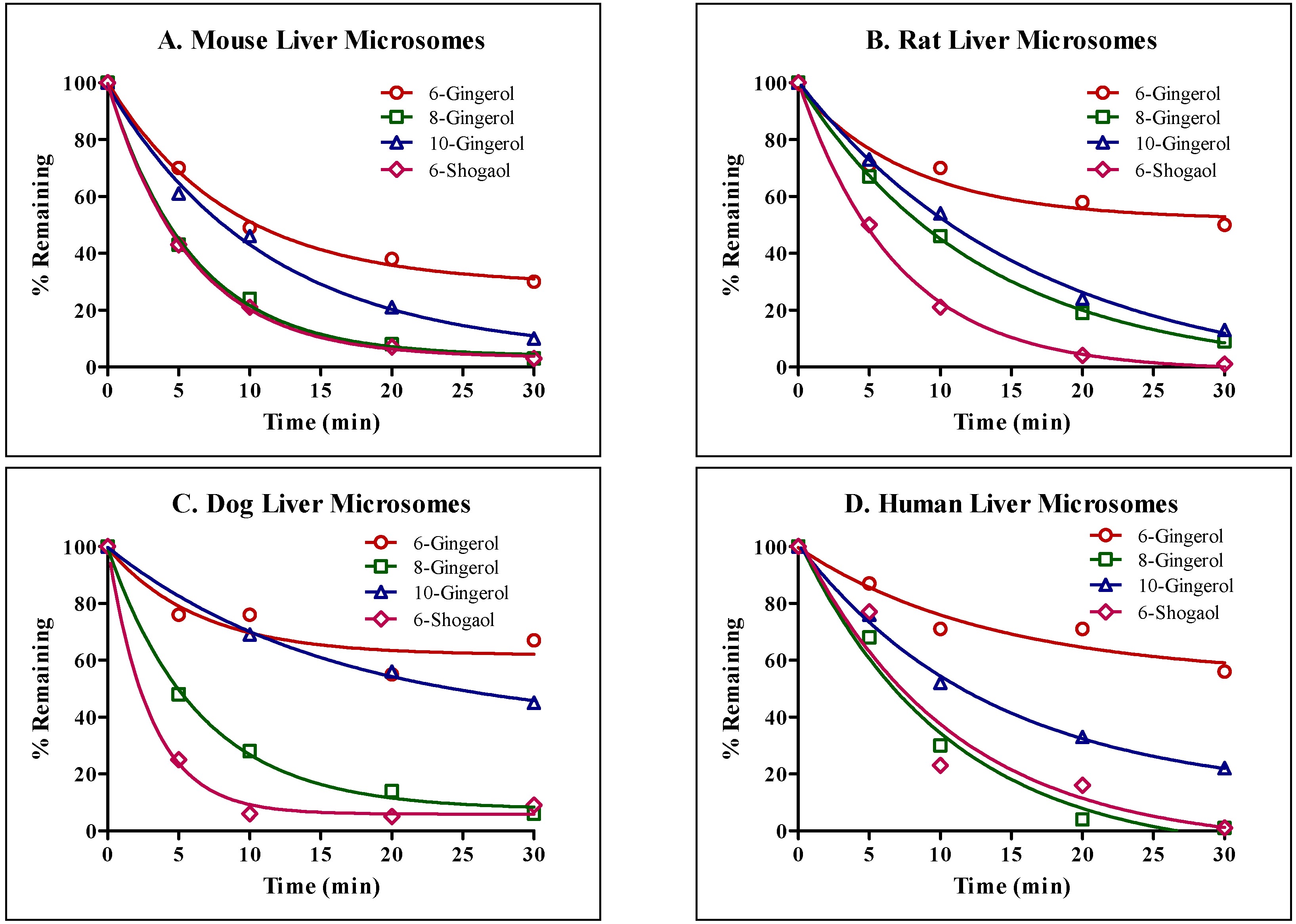
Next, we compared interspecies differences in phase I metabolism of GE phenolics individually. Liver microsomal stability studies were conducted using mouse, rat, dog and human liver microsomes. All GE phenolics were unstable and showed high intrinsic clearance, with values above 5 mL/min/g, and no interspecies differences in metabolism were observed.
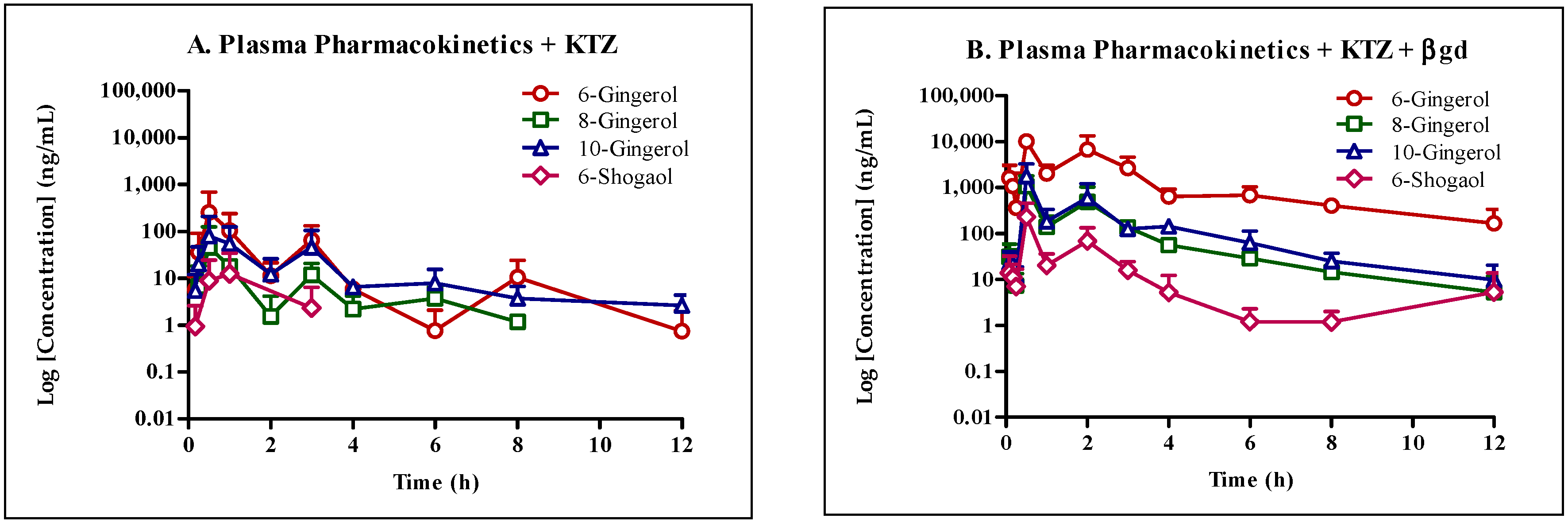
To understand the disposition of GE phenolics in vivo, we conducted a mouse pharmacokinetic study with and without KTZ co-administration and treatment of plasma samples with βgd. The concentration of all GE phenolics increased following treatment with βgd with 6G (361-fold) and 8G (698-fold) showing the highest fold increase in exposure (AUClast) followed by 6S (66-fold) and 10G (10-fold). Same or earlier Tmax of GE phenolics following βgd treatment compared to the non-treated group in the presence or absence of KTZ co-administration confirmed the propensity of GE phenolics to undergo phase II glucuronidation reaction.
Following co-administration with KTZ, the exposure (AUC
last) of all GE phenolics increased by 3 to 60-fold confirming inhibition of phase I and phase II enzymes. Following treatment of plasma samples with βgd to release free GE phenolics (+KTZ +βgd), the exposure of GE phenolics increased and was within 2-fold of the non KTZ co-administered GE group treated with βgd (−KTZ +βgd,
Table 8). In addition, as there was no increase in T
max values between the βgd treated and untreated groups, thus it is reasonable to conclude that KTZ did not inhibit the phase II glucuronidation pathway. No significant inhibition seen in the KTZ administered group for glucuronide conjugation could also be due to the lower concentrations achieved in vivo but enough to inhibit phase I biotransformation. This assumption needs to be further confirmed by assessing the concentration of KTZ in vivo and subsequent in vitro phase II glucuronidation inhibition studies in microsomes.
Based on low to moderate solubility and metabolic stability being the major route of elimination and in vivo PK data supporting this, GE phenolics are BCS Class II compounds. To circumvent the sub-therapeutic concentrations seen in vivo, we propose a suitable formulation recipe which can solubilize the GE phenolics to attain desired systemic exposure. Another strategy to increase the systemic exposure of GE phenolics will be to use specific and selective inhibitor(s) for UGTs.
4. Materials and Methods
4.1. Chemicals
The ginger extract (GE) was a generous gift from Sabinsa Corporation (East Windsor, NJ, USA). The active ginger constituents, 6G, 8G, 10G, and 6S, were isolated from GE by column chromatography and were characterized for >98% purity by high performance liquid chromatography. Acetonitrile (ACN) and methanol were purchased from Fisher Scientific (Pittsburgh, PA, USA). Nicotinamide adenine dinucleotide phosphate (NADPH), Carboxyl methylcellulose (CMC), dihydrocapsaicin, and β-glucuronidase were purchased from Sigma (St. Louis, MO, USA). Mouse, rat, dog, and human liver microsomes were purchased from Xenotech LLC (Kansas City, KS, USA). All other reagents used in the study were of analytical grade.
4.2. pH Dependent Solubility of Ginger Extract Phenolics
Kinetic solubility of GE phenolics was assessed from 10 to 100 μM by spiking dimethylsulfoxide (DMSO, Sigma-Aldrich) stock solutions (5 μL, in duplicate) into 995 μL buffer (pH 2.0—hydrochloride, 4.0–100 mM citrate buffer and 7.4–100 mM phosphate buffer) in a 96-well plate and placing at room temperature (22–24 °C) for 2 h. Calibration standards were prepared by spiking 5 μL of DMSO stock solutions into 995 μL acetonitrile/buffer (1:1) mixture. Following centrifugation (10,000 rpm, 10 min, 25 °C) the reaction samples were diluted 1:1 with acetonitrile.
4.3. Solubility and Stability in Simulated Gastric Fluid and Intestinal Stability
SGF and SIF were prepared as per USP method. Briefly, SGF was prepared by adding 2 g of sodium chloride and 3.2 g of pepsin with water and pH was adjusted to 1.2 ± 0.1 using hydrochloric acid (7 mL) and volume made up to 1 L. SIF was prepared by dissolving 6.8 g monobasic potassium phosphate and 10 g pancreatin in Milli-Q water (Millipore, Billerica, MA, USA). pH was adjusted to 6.8 ± 0.1 with sodium hydroxide solution and volume was made upto 1 L. Reactions were initiated by spiking DMSO stock solutions of GE phenolics 10 μL into 1990 μL of SGF and SIF incubated at 37 °C for 10 min. Samples (200 μL) were withdrawn at 0, 15, 30, and 60 min for SGF and at 0, 30, 60, and 120 min for SIF and quenched with 200 μL acetonitrile containing internal standard.
4.4. Microsomal Stability
Pooled liver microsomes from CD1 mouse, IGS Sprague-Dawley rat, Beagle dog, and mixed gender human were used for assays. Incubations (1 mL) consisted of liver microsomes (0.5 mg/mL), NADPH (1 mM) and 100 mM sodium phosphate buffer (pH 7.4). Following pre-incubation (10 min, 37 °C), reactions were initiated by spiking ACN/DMSO (80:20) stock solutions of GE phenolics (5 μL, 10 μM). Samples (100 μL) were withdrawn at 0, 5, 10, 15, 20 and 30 min and quenched with 200 μL acetonitrile containing internal standard. Concomitant NADPH-free control incubations were prepared similarly.
4.5. Plasma Stability Assay
Mouse plasma (995 µL, C57BL/6) was spiked with 5 µL of individual stock solutions of GE phenolics (200 µM stock solution prepared in ACN/DMSO (80:20). After gentle inversion, samples (100 μL) were withdrawn at 0, 15, 30, and 60 min and quenched with 400 μL of ice cold acetonitrile containing internal standard. Assay was performed in duplicate.
4.6. Mouse Pharmacokinetic Study
Pharmacokinetic study in male mice (C57BL6J, Harlan Laboratories, Indianapolis, IN, USA) was run with 36 animals in each group at a GE dose of 250 mg/kg with a dose volume of 10 mL/kg. One group was given GE alone and in the second group GE was co-administered with KTZ (50 mg/kg). A terminal sampling design was used to collect blood samples at 0 (pre-dose), 0.08, 0.16, 0.25, 0.5, 1, 2, 3, 4, 6, 8, and 12 h. At each time, 1 mL of blood was collected in ethylenediaminetetraacetic acid di-potassium salt (K2EDTA) tubes (BD Biosciences, Franklin Lakes, NJ, USA). Plasma was separated by centrifugation of samples at 4000 rpm for 10 min and stored below −60 °C till bioanalysis. All experiments were performed with approval from the Institutional Animal Care and Use Committee (IACUC, approval code: A14031). All animals were acclimatized for five days before dosing in the experimental area. Food and water was provided ad libitum throughout the study period. Animals were marked and housed (three per cage, n = 36 per group) in polypropylene cages and maintained in controlled environmental conditions with 12 h light and dark cycles. The temperature and humidity of the room was maintained between 22 ± 3 °C and 30% to 50%, respectively, and approximately 10–12 fresh air change cycles per hour.
4.7. Enzymatic Hydrolysis of Ginger Extract Phenolic Conjugates
To confirm the presence of GE glucuronide conjugates, plasma (100 μL) was treated with βgd (20 μL, 500 units) and incubated at 37 °C for 1 h. Each sample was analyzed twice with and without β-gd treatment. Samples without gde treatment were diluted with 20 μL of buffer. Total (conjugated + free) and free ginger phenolics data were reported following bioanalysis.
4.8. Bioanalysis
All in vitro and in vivo samples were processed by protein precipitation method. An aliquot (100 μL) of the sample was spiked with 200 μL of acetonitrile containing dihydrocapsaicin as internal standard (IS) and vortex mixed for 3 min. The tubes were centrifuged at 12,000 rpm for 10 min and an aliquot of supernatant was transferred into autosampler vials for analysis. The stock solutions of 6G, 8G, 10G, and 6S were prepared in ACN/water (70:30) at 1 mg/mL. A calibration curve (CC) range of 2–2000 ng/mL was employed for the quantification of analytes and IS concentration was 150 ng/mL for each analysis. The CC consisted of blank, blank with IS and seven non-zero calibration standards. The calibration standards were within ±15% of the nominal concentration and lower limit of quantification was within ±20% of nominal. All samples were analyzed using liquid chromatography tandem mass spectrometry (LC-MS/MS) method (Agilent 6410 series, Santa Clara, CA, USA). A positive ionization mode with multiple reaction monitoring (MRM, m/z Q1/Q3) of 6G (m/z 277.1/177.1, retention time (RT) 2.0 min), 8G (m/z 305.1/177.1, RT 4.0 min), 10G (m/z 333.1/177.1, RT 7.7 min), 6S (m/z 277.1/137.1, RT 5.1 min), and IS (m/z 308.2/137.1, RT 3.4 min) was employed. The ion spray voltage was set at 3500 V, ionization temperature was set to 300 °C and drying gas flow rate was 10 L/min. Data acquisition and quantitation were performed using Mass Hunter software (Agilent Technologies, Wilmington, DE, USA). Separation was achieved using HP1100 series LC (Agilent Technologies) with an Agilent Zorbax reversed-phase (SB-C18, 2.1 × 50 mm, 5.0 μm) column. A gradient method was employed to separate the individual GE phenolics using mobile phase A (0.1% formic acid in water) and mobile phase B (Acetonitrile). The gradient elution method with 50% B at 0 min, 90% B at 5 min, held for 2.1 min, back to 50% B at 7.1 min with a flow rate of 0.2 mL/min. An injection volume of 2 μL was used for analysis.
4.9. Pharmacokinetic Analysis
The pharmacokinetic parameters were calculated from the concentration-time data using the non-compartmental analysis tool of Phoenix WinNonlin software (version 6.3, Pharsight, St. Louis, MO, USA). The area under the concentration-time curve (AUClast) was calculated using the linear trapezoidal rule. Following oral administration, peak concentration (Cmax) and time for the peak concentration (Tmax) were the observed values.
4.10. Statistical Analysis
Concentration-time profiles were represented as mean ± standard deviation (SD).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}