
Metal-Free α-C(sp3)–H Functionalized Oxidative Cyclization of Tertiary N,N-Diaryl Amino Alcohols: Theoretical Approach for Mechanistic Pathway



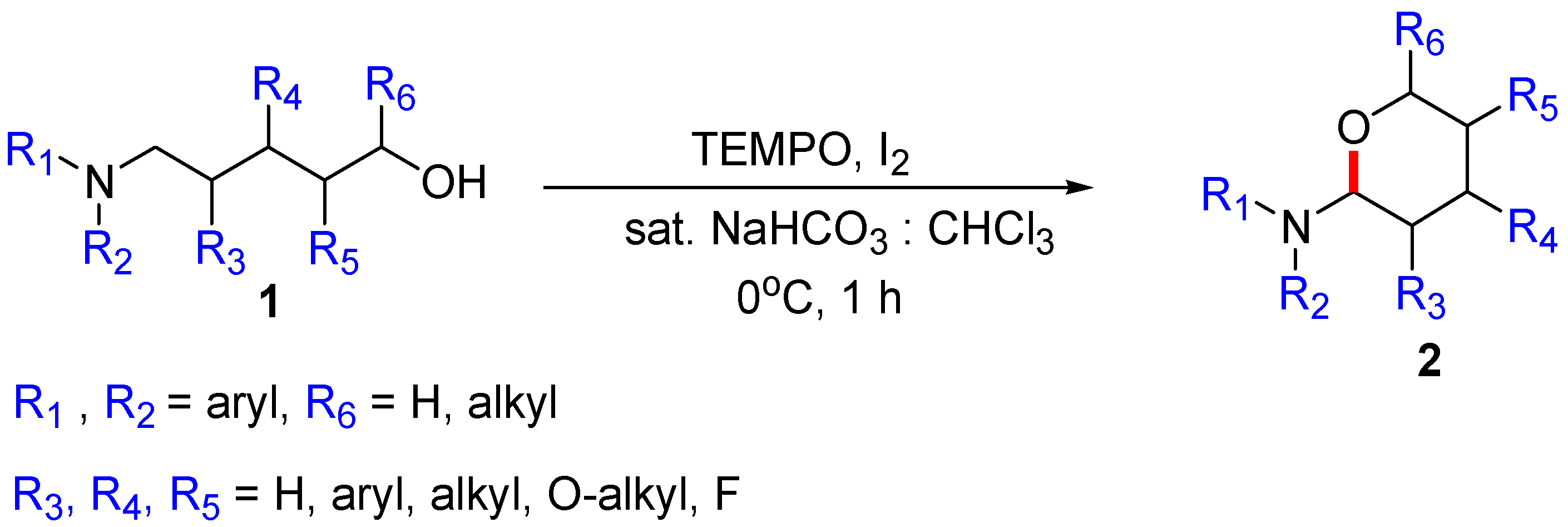{kind=link}
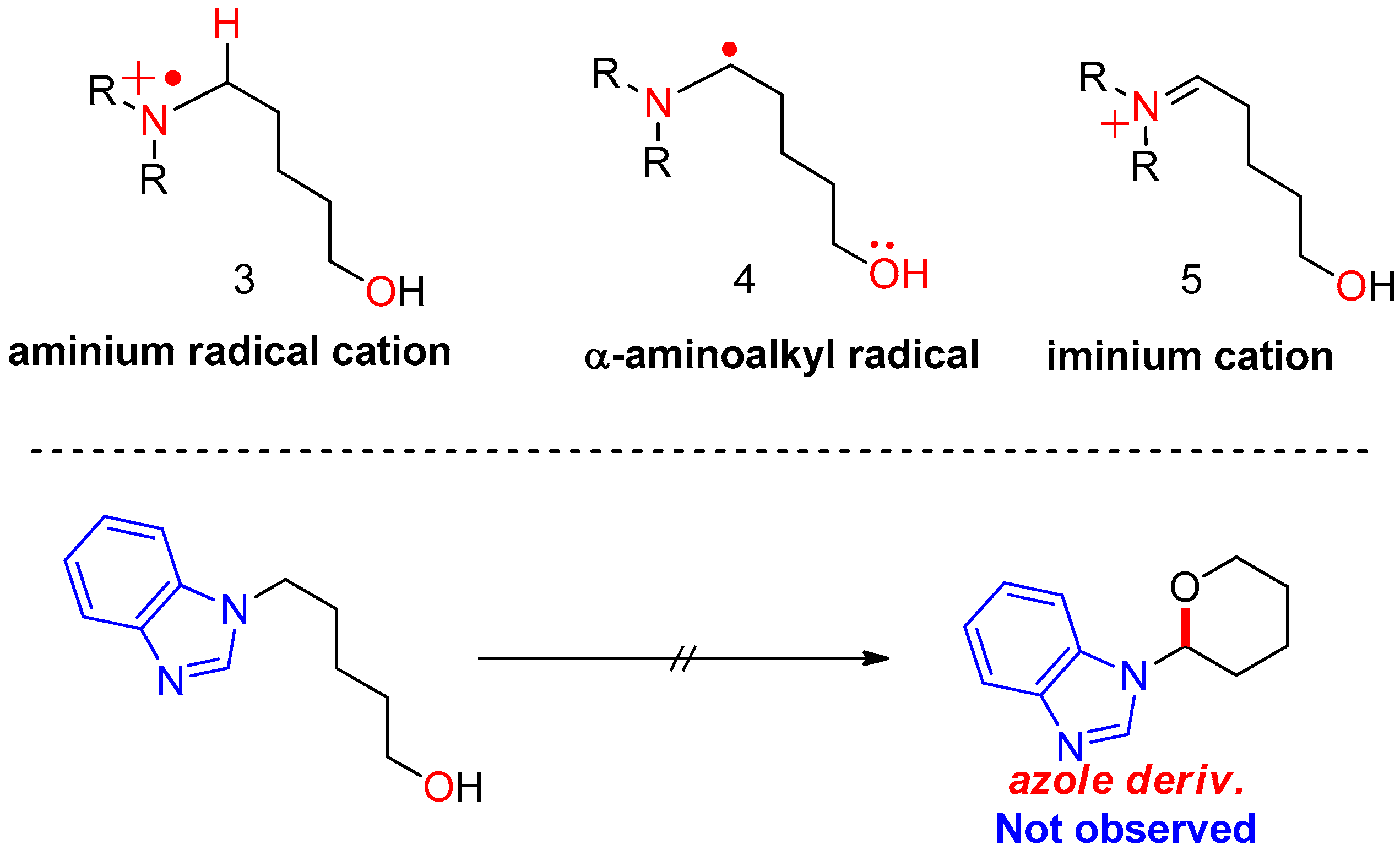{kind=link}
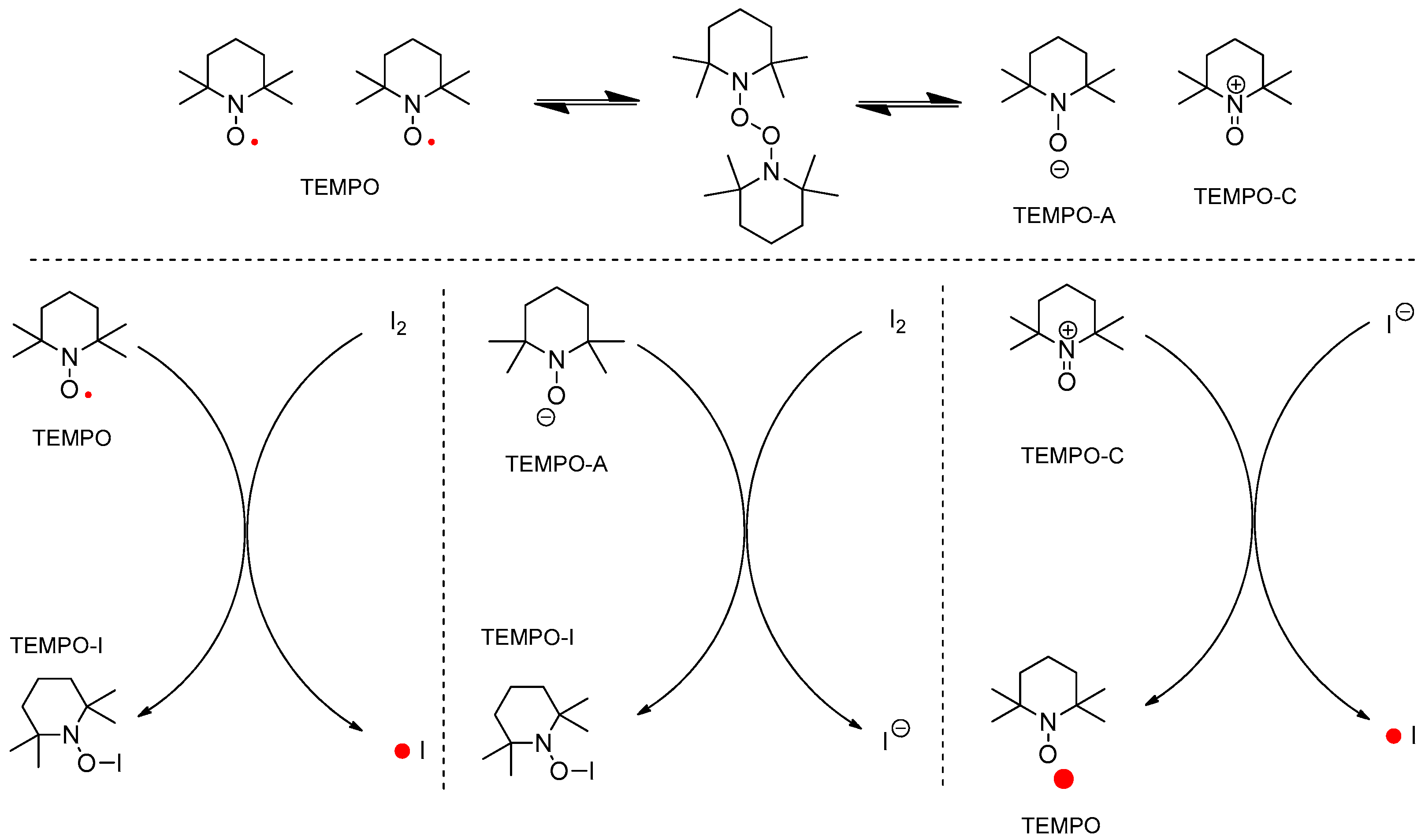{kind=link}
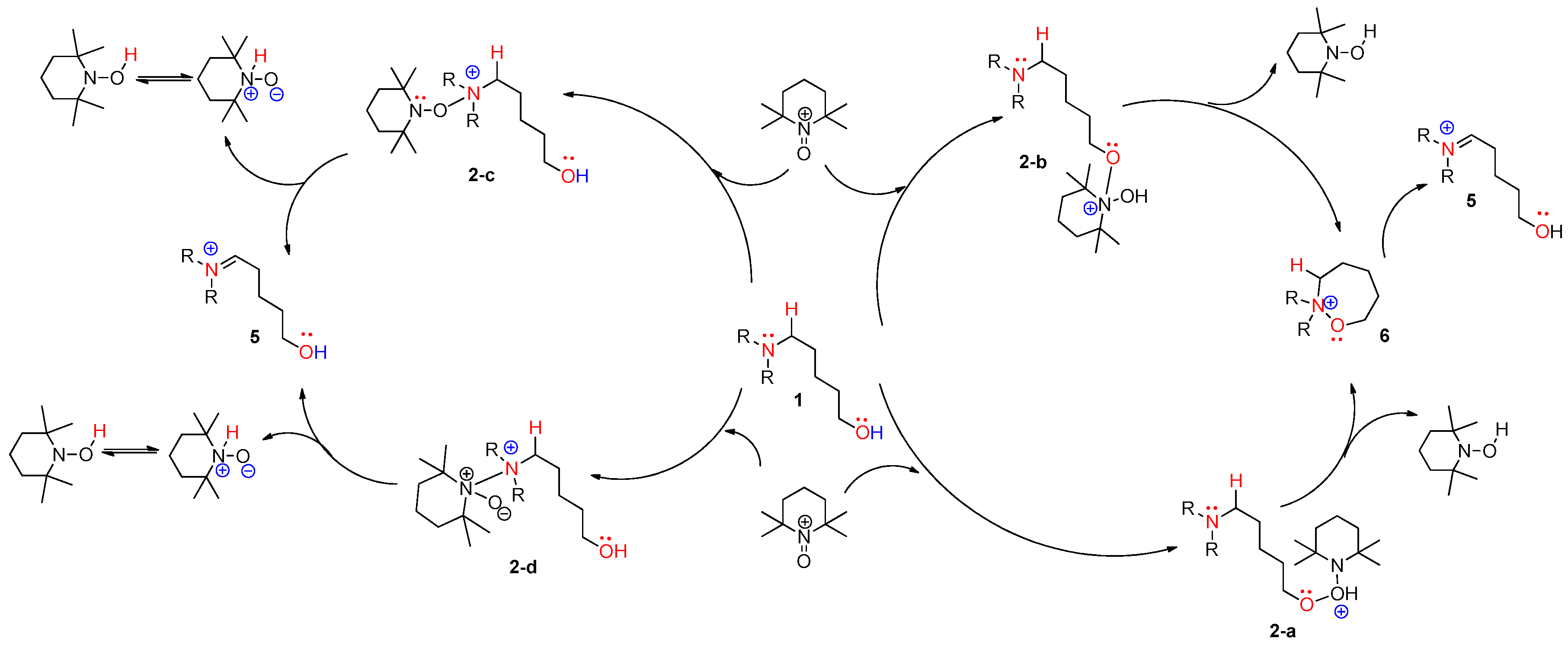{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
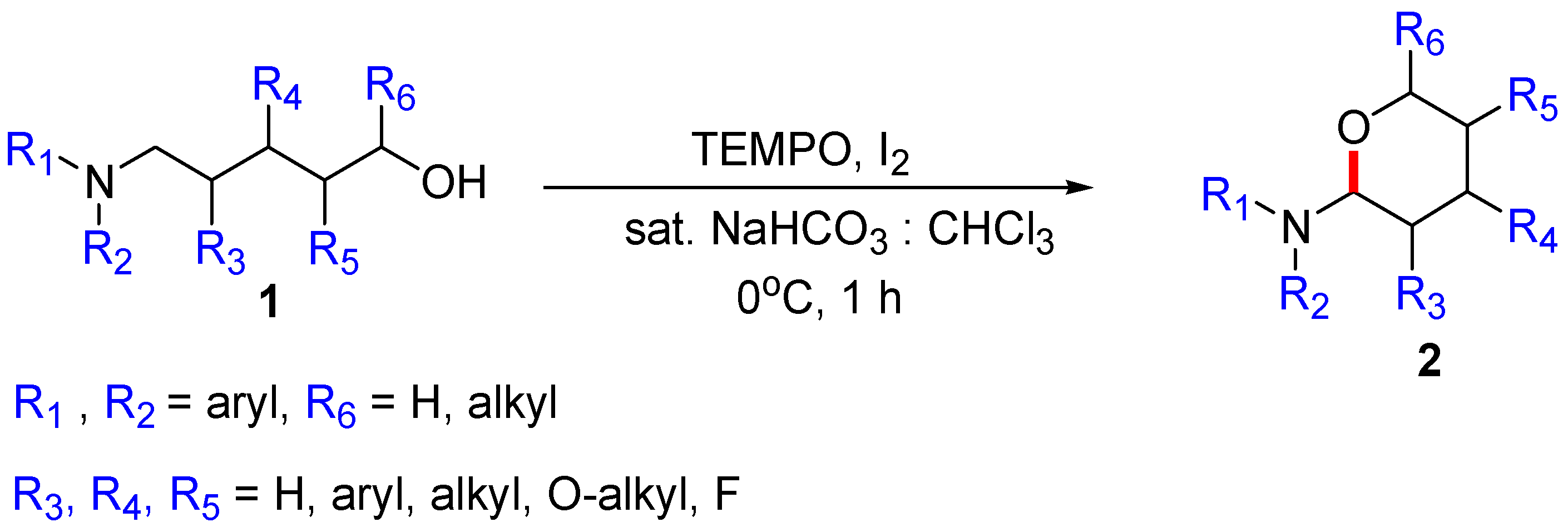
:1. Introduction
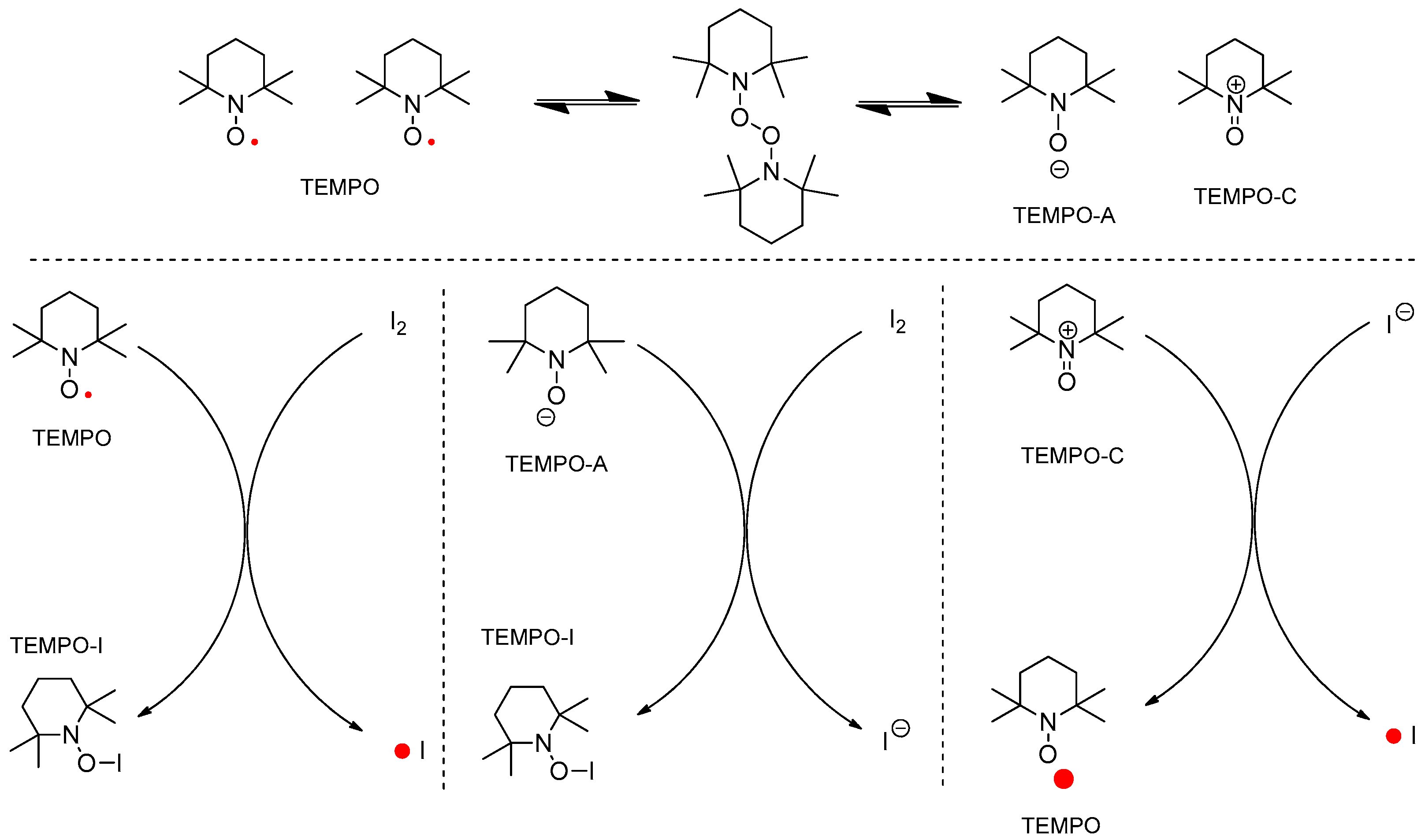
2. Initial Assumptions and Theoretical Background
3. Computational Methods
4. Results and Discussion
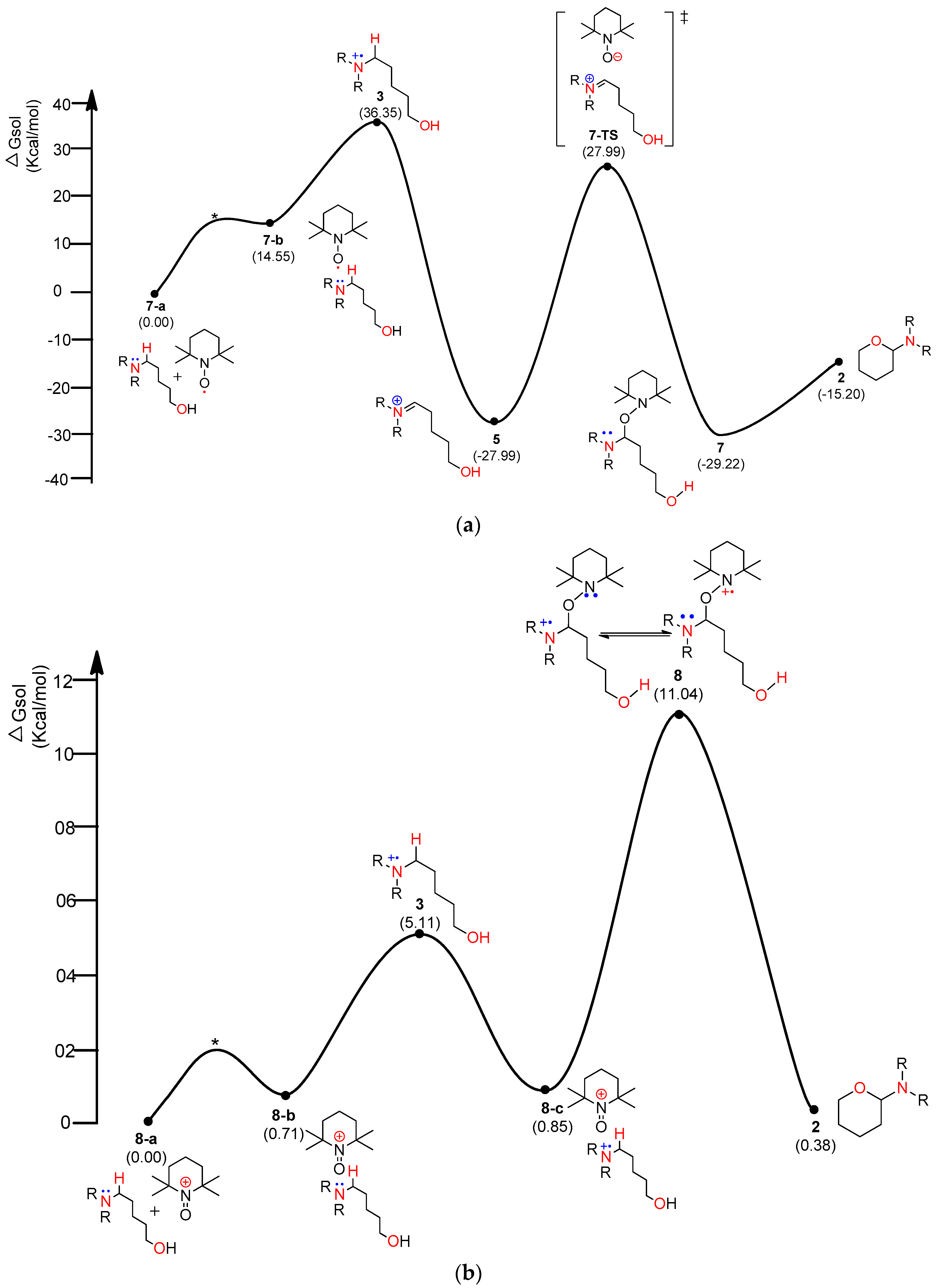
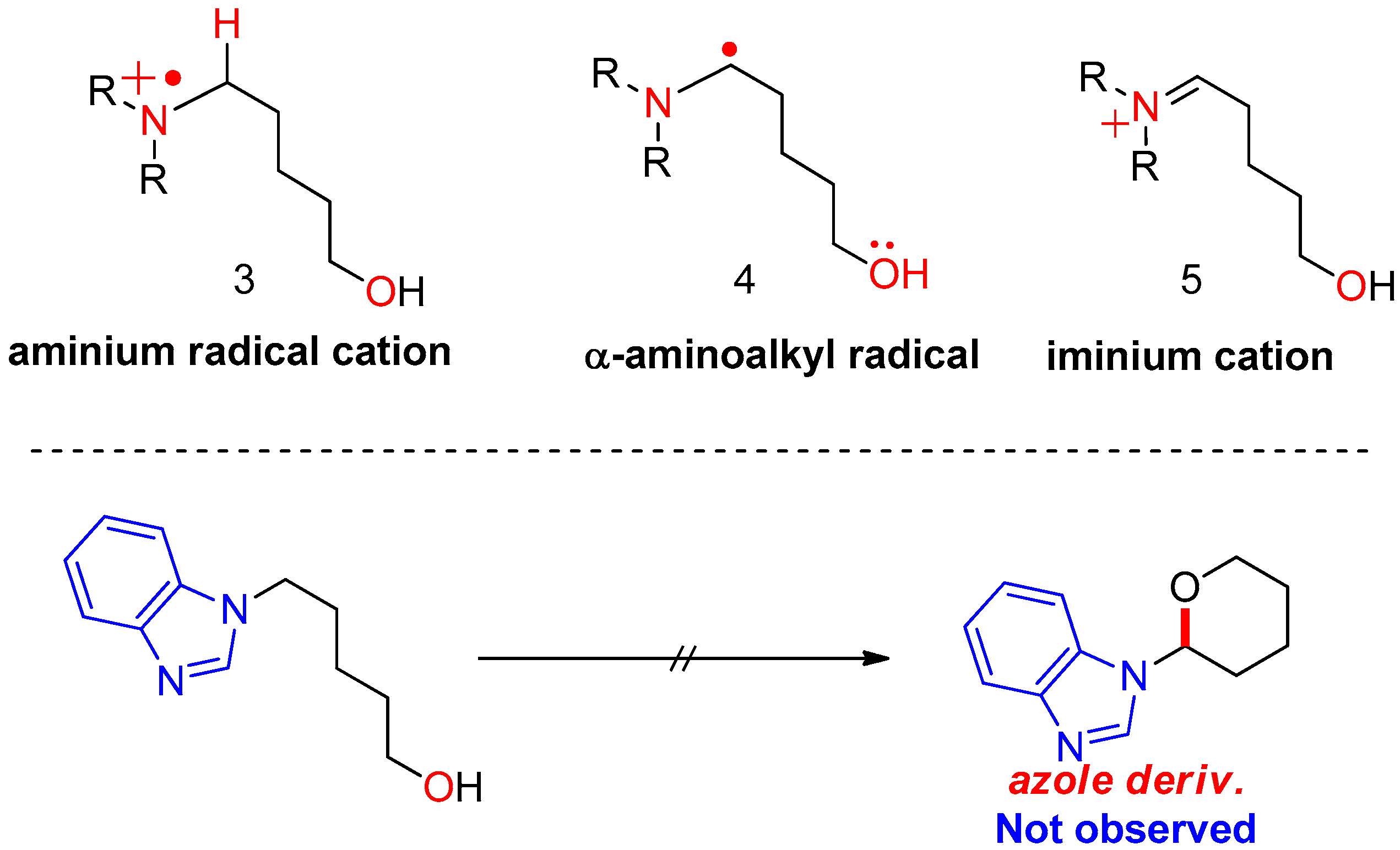
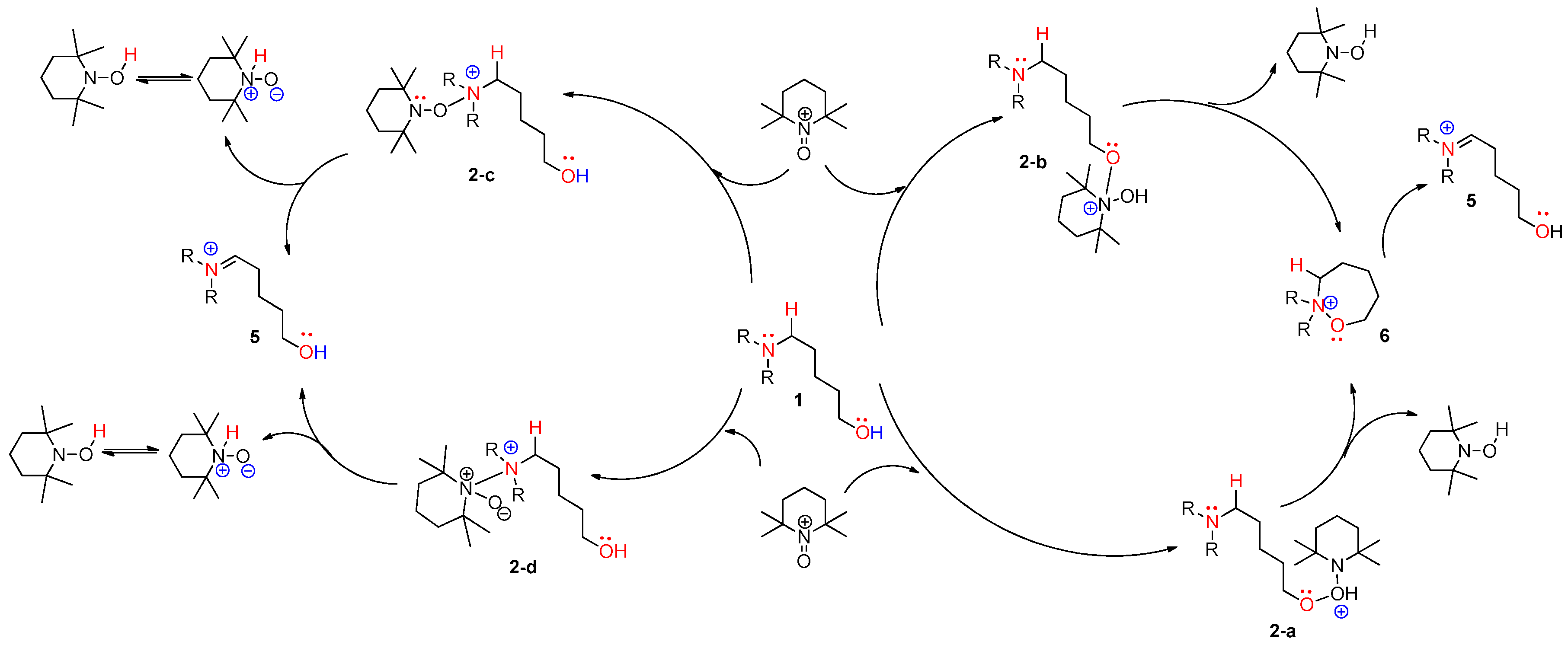
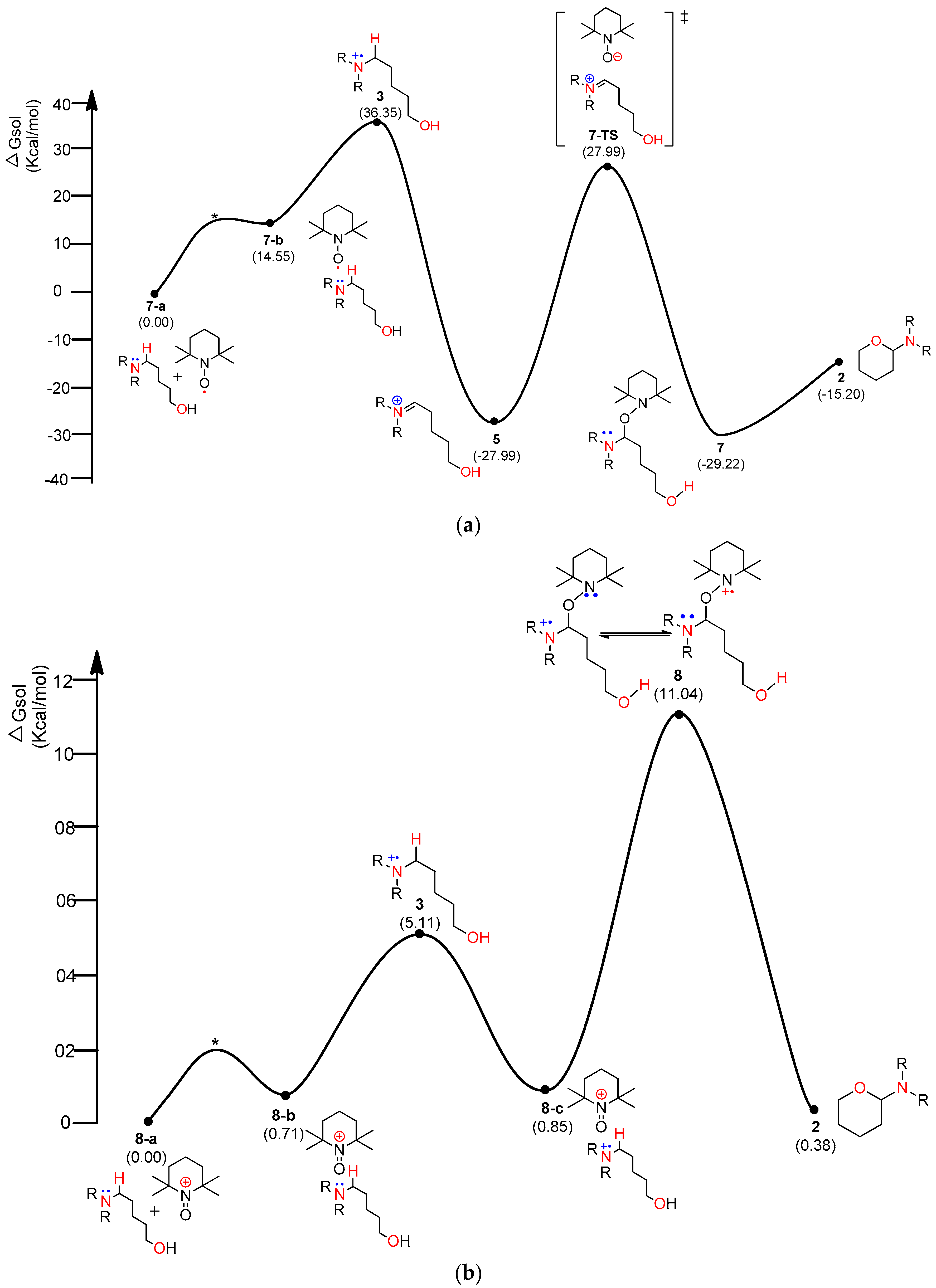
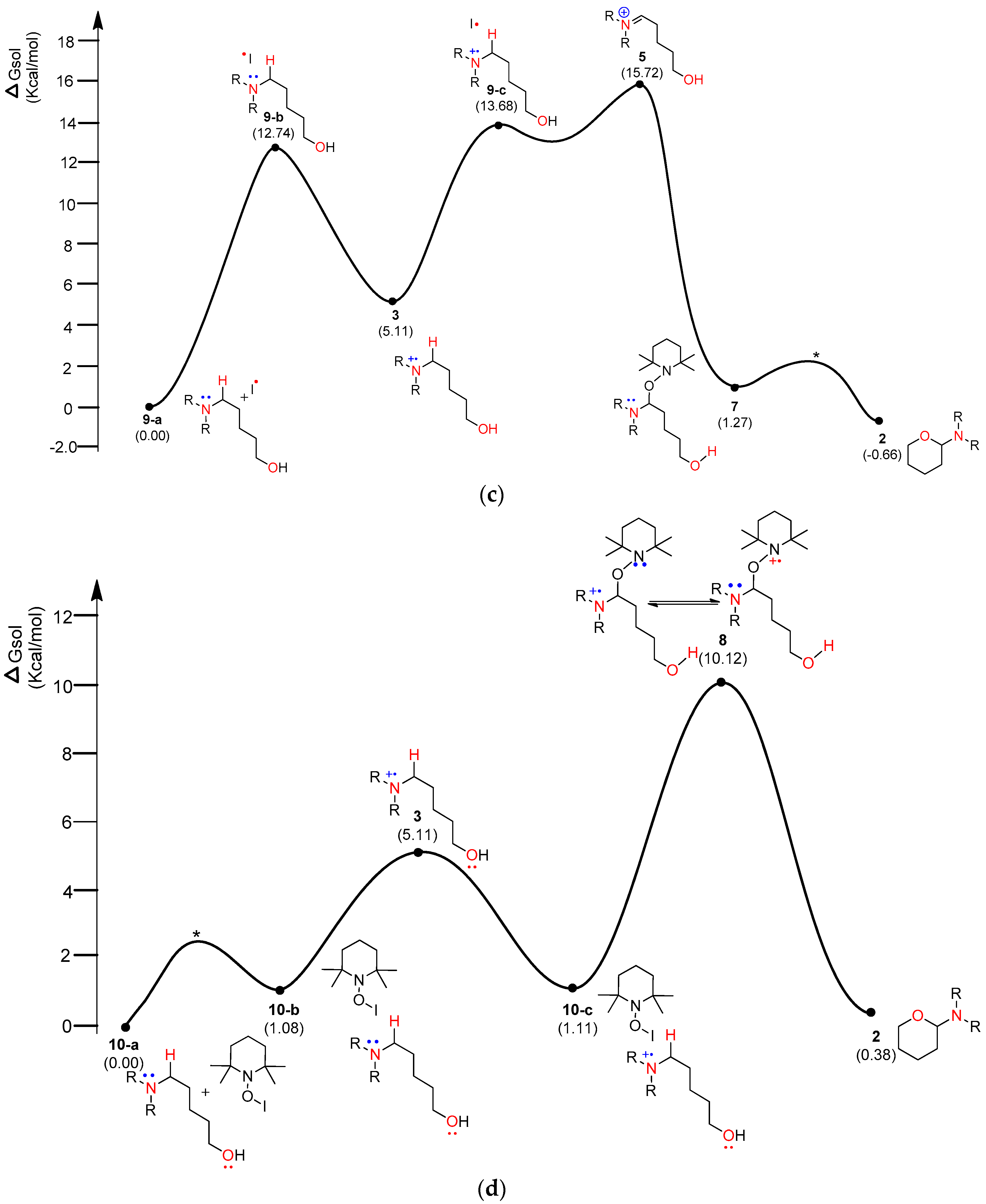
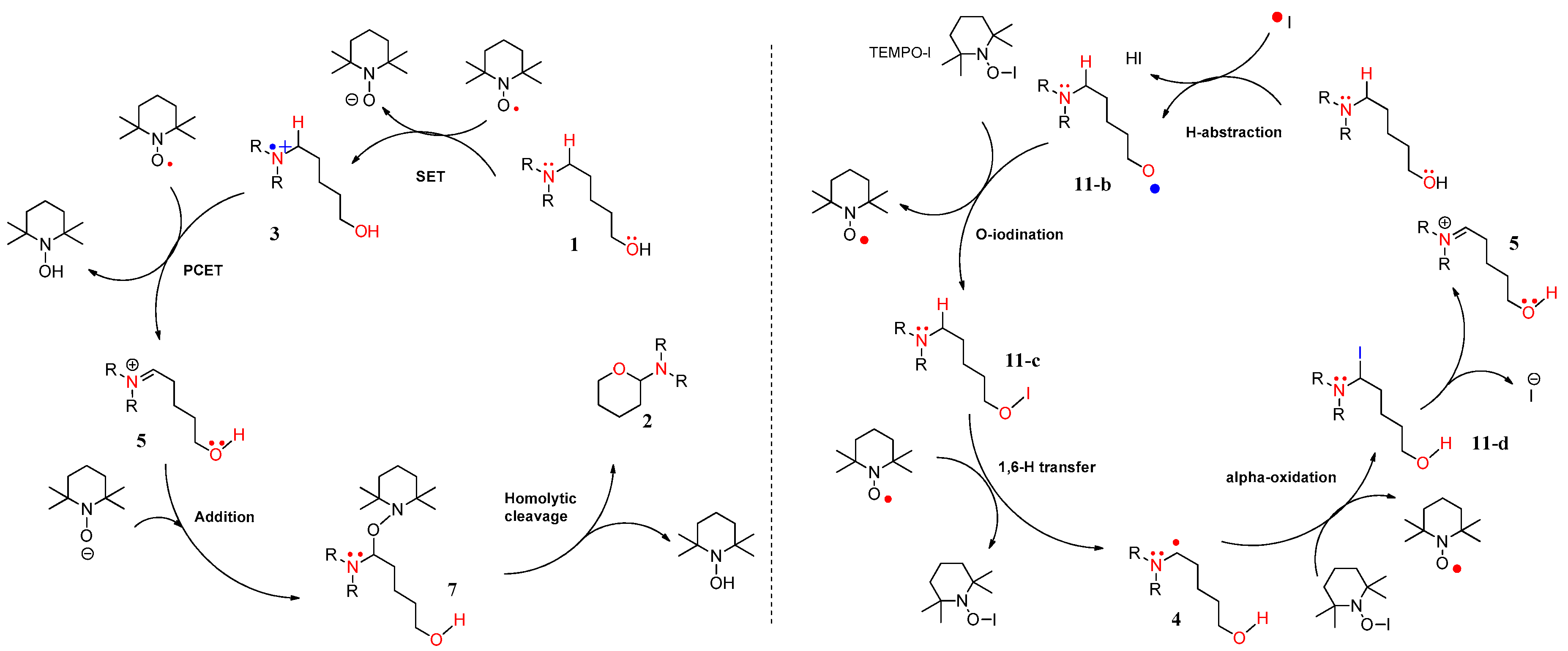
4.1. Pathway A: Aminium Radical Cation 3 and SET/PCET Mechanism
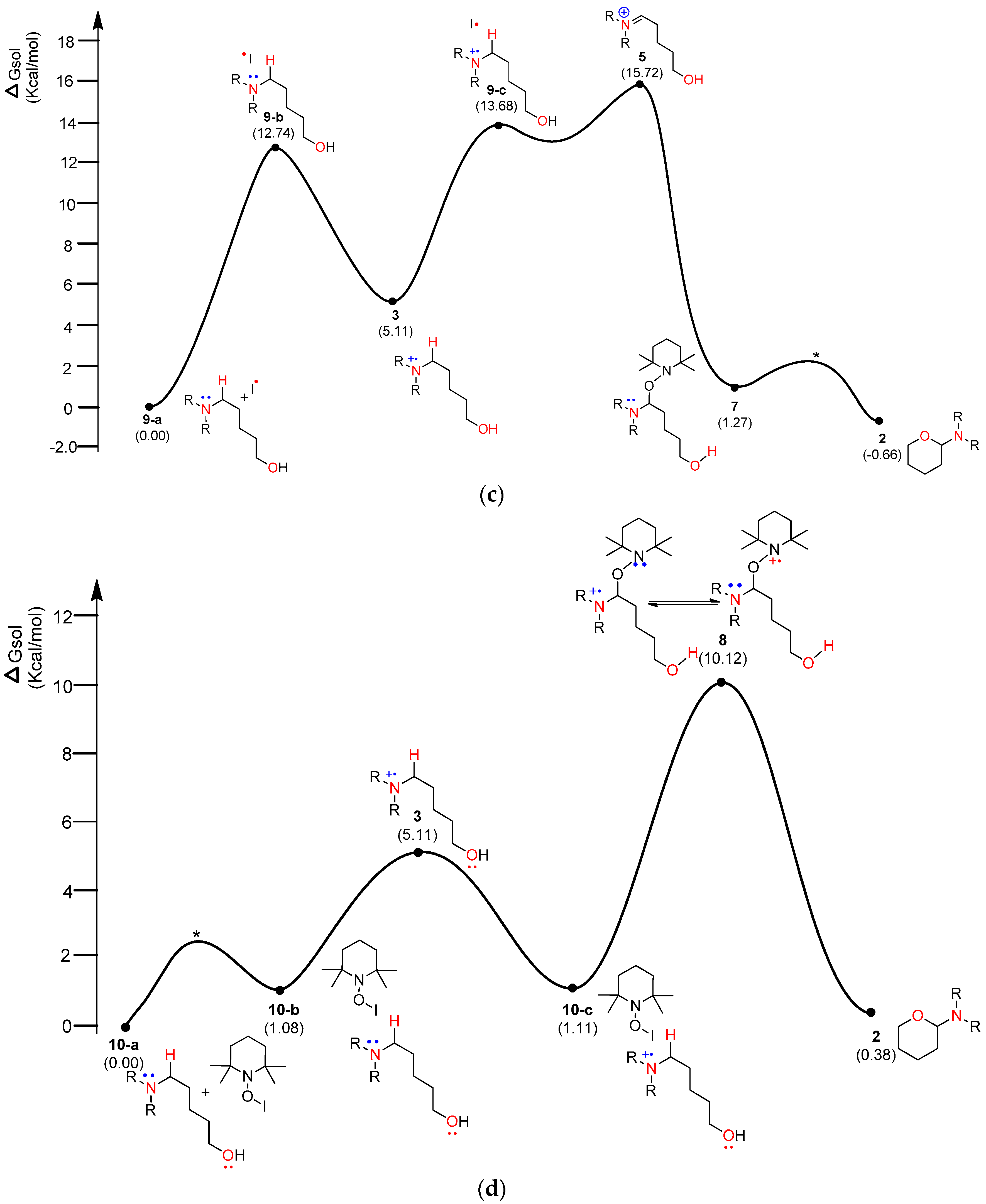
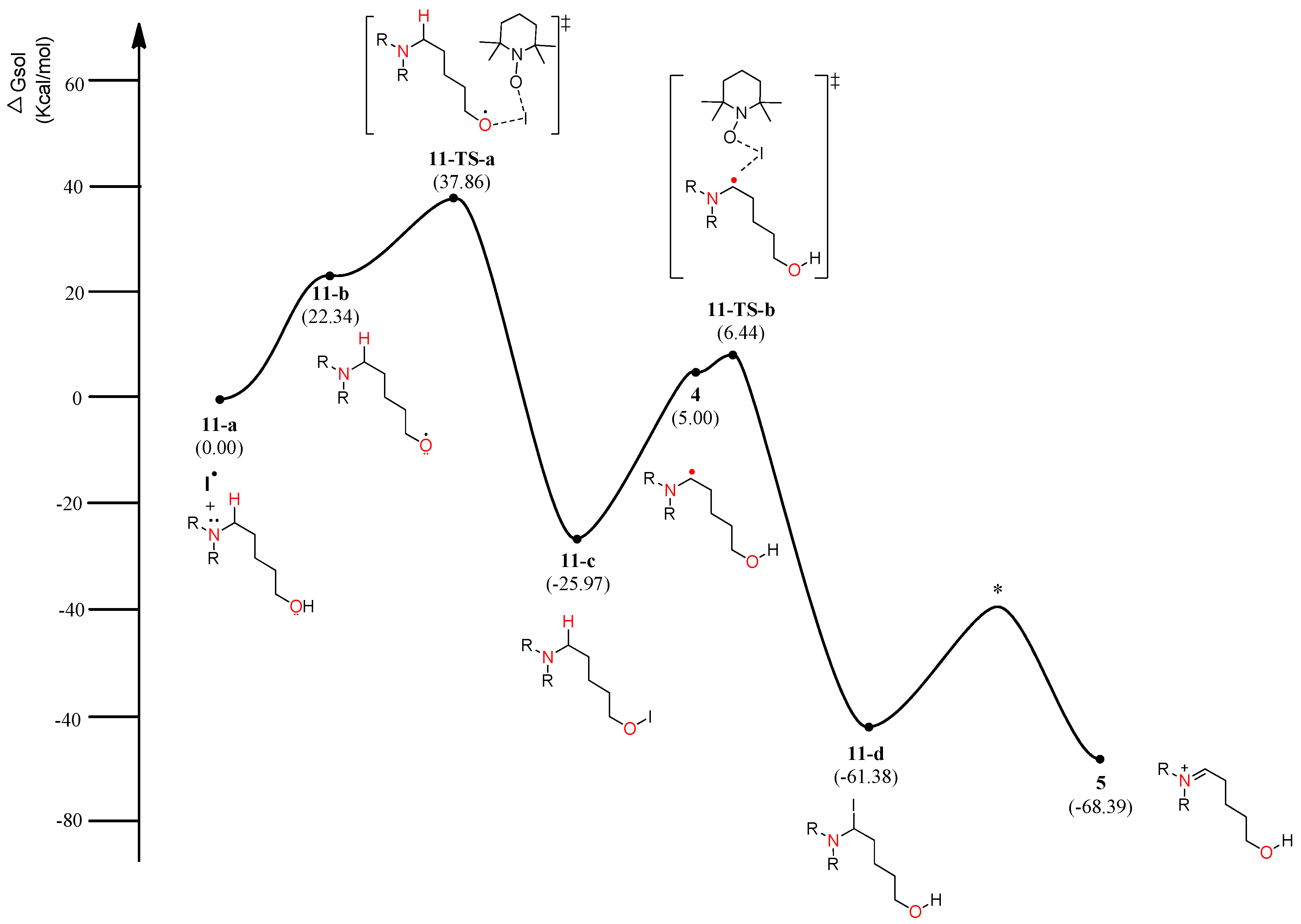
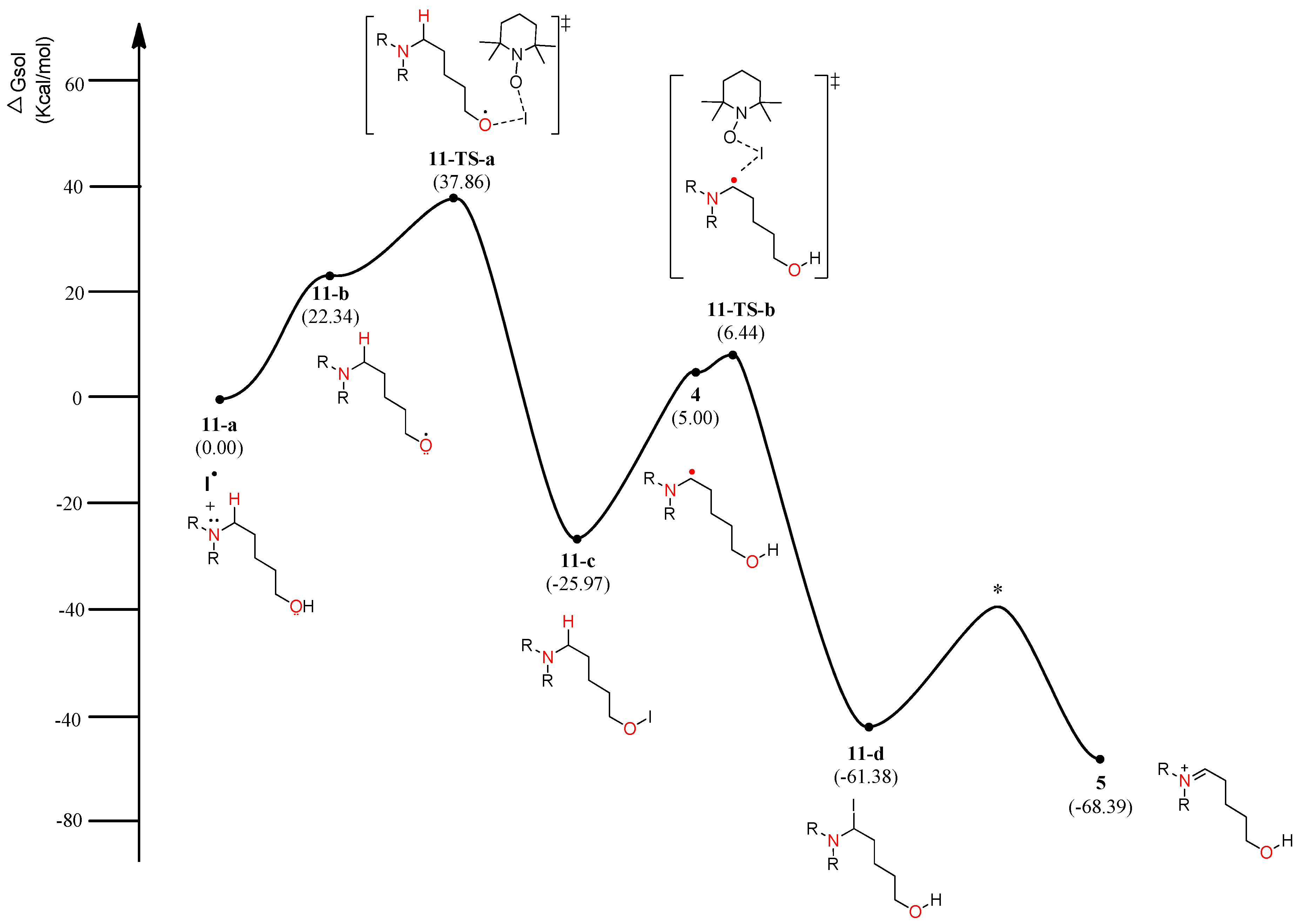
4.2. Pathway B: α-Aminoalkyl Radical and HAT Mechanism
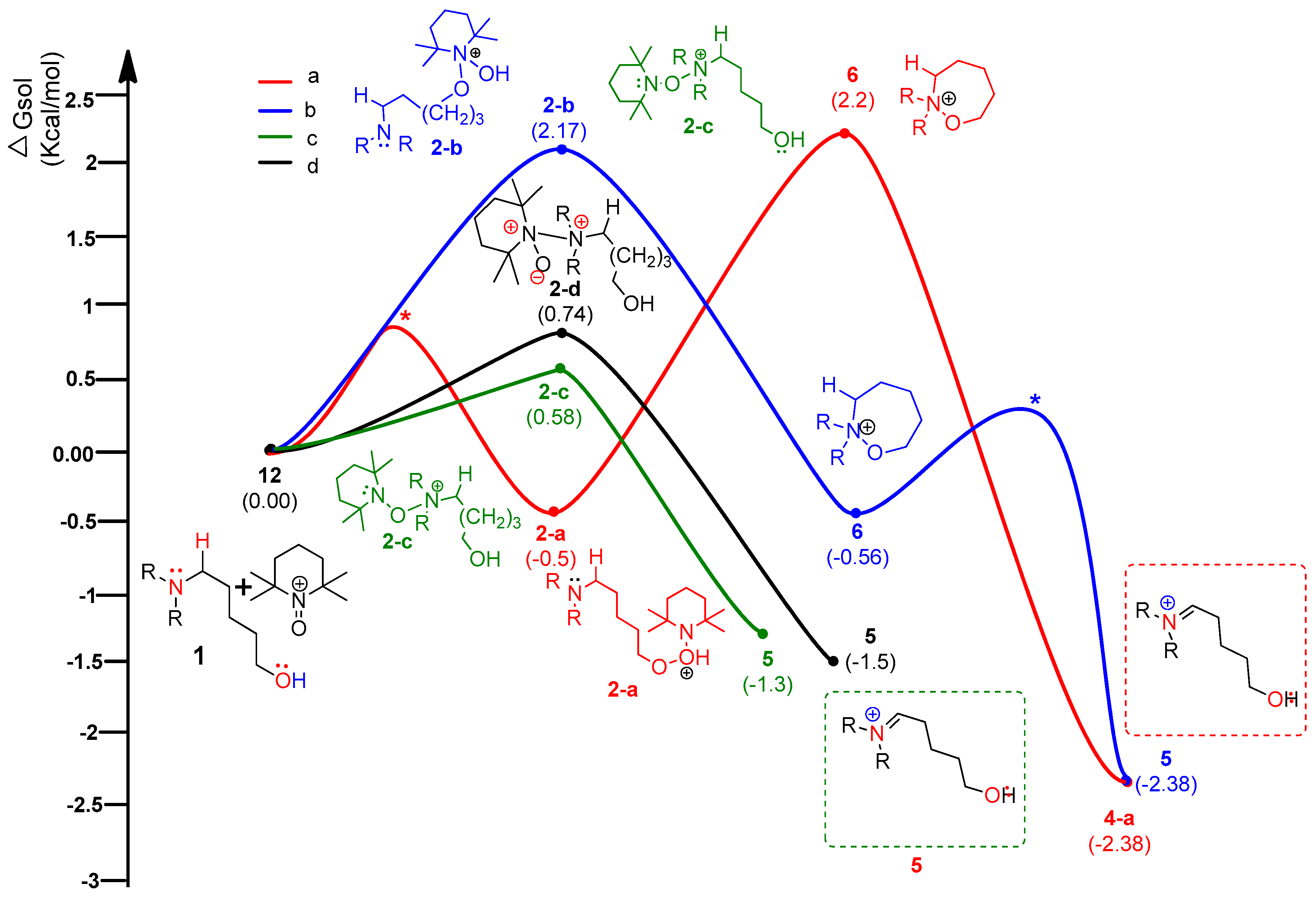
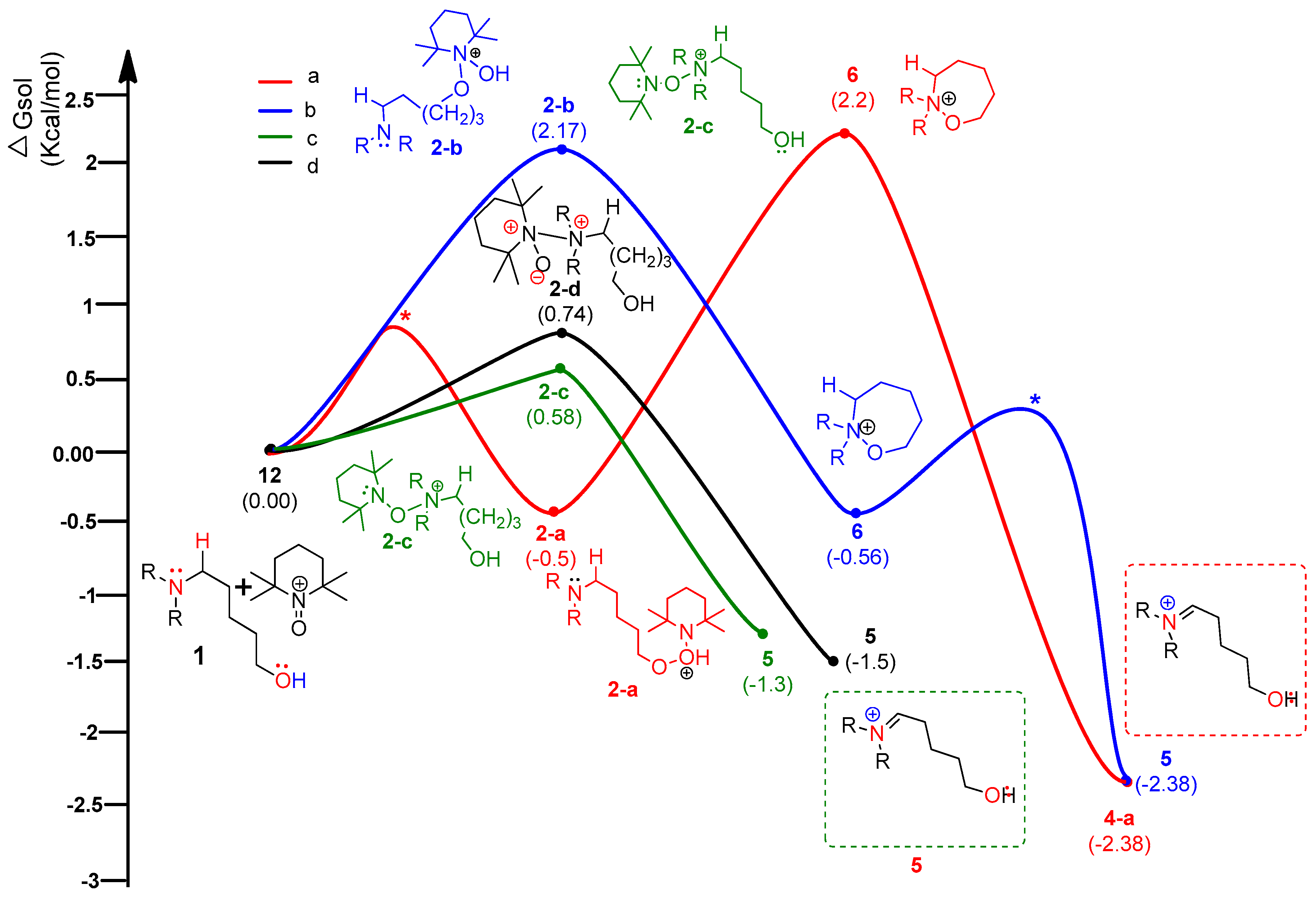
4.3. Pathway C: Conventional Ionic Mechanism
4.4. Combined Analysis of Mechanistic Study
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CDC | Cross-dehydrogenative coupling |
| PCET | Proton-coupled electron transfer |
| SET | Single-electron transfer |
| HAT | Hydrogen-atom transfer |
| GEA | Electron attachment free energy |
| SCF | Self-consistent field |
| ZPE | Zero-point energy |
| BDFE | Homolytic bond dissociation free energy |
References
- Narayan, R.; Matcha, K.; Antonchick, A.P. Metal-Free Oxidative C-C Bond Formation through C-H Bond Functionalization. Chem.-Eur. J. 2015, 21, 14678–14693. [Google Scholar] [CrossRef] [PubMed]
- Samanta, R.; Matcha, K.; Antonchick, A.P. Metal-Free Oxidative Carbon-Heteroatom Bond Formation through C-H Bond Functionalization. Eur. J. Org. Chem. 2013, 2013, 5769–5804. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Mathison, C.J.; Montagnon, T. o-Iodoxybenzoic acid (IBX) as a viable reagent in the manipulation of nitrogen- and sulfur-containing substrates: Scope, generality, and mechanism of IBX-mediated amine oxidations and dithiane deprotections. J. Am. Chem. Soc. 2004, 126, 5192–5201. [Google Scholar] [CrossRef] [PubMed]
- Funes-Ardoiz, I.; Sameera, W.M.; Romero, R.M.; Martinez, C.; Souto, J.A.; Sampedro, D.; Muniz, K.; Maseras, F. DFT Rationalization of the Diverse Outcomes of the Iodine(III)-Mediated Oxidative Amination of Alkenes. Chem.-Eur. J. 2016, 22, 7545–7553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Cheng, R.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Hypervalent iodine-mediated oxygenation of N,N-diaryl tertiary amines: Intramolecular functionalization of sp3 C-H bonds adjacent to nitrogen. J. Org. Chem. 2014, 79, 10581–10587. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Liu, K.; Long, Y.; Gao, X.; Gao, M.; Lei, A. Iodine-catalyzed radical oxidative annulation for the construction of dihydrofurans and indolizines. Org. Lett. 2015, 17, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Shibata, H.; Akazome, M. alpha-Diketone formation accompanied by oxidation of sulfur functional group by the reaction of o-alkynylarenesulfoxide with iodine. J. Org. Chem. 2013, 78, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Venkanna, A.; Park, H.G.; Kim, M.H. Metal-Free α-C(sp(3))-H Functionalized Oxidative Cyclization of Tertiary N,N-Diarylamino Alcohols: Construction of N,N-Diarylaminotetrahydropyran Scaffolds. Asian J. Org. Chem. 2016, 5, 232–239. [Google Scholar] [CrossRef]
- Nijhuis, W.H.N.; Verboom, W.; Abu El-Fadl, A.; Van Hummel, G.J.; Reinhoudt, D.N. Stereochemical aspects of the “tert-amino effect”. 2. Enantio- and diastereoselectivity in the synthesis of quinolines, pyrrolo[1,2-a]quinolines, and [1,4]oxazino[4,3-a]quinolines. J. Org. Chem. 1989, 54, 209–216. [Google Scholar] [CrossRef]
- Haidasz, E.A.; Meng, D.; Amorati, R.; Baschieri, A.; Ingold, K.U.; Valgimigli, L.; Pratt, D.A. Acid Is Key to the Radical-Trapping Antioxidant Activity of Nitroxides. J. Am. Chem. Soc. 2016, 138, 5290–5298. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiao, N. TEMPO-catalyzed aerobic oxygenation and nitrogenation of olefins via C horizontal lineC double-bond cleavage. J. Am. Chem. Soc. 2013, 135, 11692–11695. [Google Scholar] [CrossRef] [PubMed]
- Bogart, J.A.; Lee, H.B.; Boreen, M.A.; Jun, M.; Schelter, E.J. Fine-tuning the oxidative ability of persistent radicals: Electrochemical and computational studies of substituted 2-pyridylhydroxylamines. J. Org. Chem. 2013, 78, 6344–6349. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Rong, G.; Liu, D.; Zheng, Y.; Chen, J.; Mao, J. Stereoselective intermolecular nitroaminoxylation of terminal aromatic alkynes: Trapping alkenyl radicals by TEMPO. Org. Lett. 2014, 16, 6306–6309. [Google Scholar] [PubMed]
- Shibuya, M.; Osada, Y.; Sasano, Y.; Tomizawa, M.; Iwabuchi, Y. Highly efficient, organocatalytic aerobic alcohol oxidation. J. Am. Chem. Soc. 2011, 133, 6497–6500. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Jaguar. Version 8.1 Schrödinger; LLC: New York, NY, USA, 2013. [Google Scholar]
- Slater, J.C. Quantum Theory of Molecules and Solids: The Self-Consistent Field for Molecules and Solids; McGraw-Hill: New York, NY, USA, 1974; Volume 4. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Becke, A.D. Densityfunctional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New Model for Calculation of Solvation Free Energies: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Friedrichs, M.; Zhou, R.; Edinger, S.R.; Friesner, R.A. Poisson-Boltzmann analytical gradients for molecular modeling calculations. J. Phys. Chem. B 1999, 103, 3057–3061. [Google Scholar] [CrossRef]
- Edinger, S.R.; Cortis, C.; Shenkin, P.S.; Friesner, R.A. Solvation free energies of peptides: Comparison of approximate continuum solvation models with accurate solution of the Poisson-Boltzmann equation. J. Phys. Chem. B 1997, 101, 1190–1197. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar]
- Haidasz, E.A.; Shah, R.; Pratt, D.A. The catalytic mechanism of diarylamine radical-trapping antioxidants. J. Am. Chem. Soc. 2014, 136, 16643–16650. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Fuchs, P.L. An efficient C-H oxidation protocol for alpha-hydroxylation of cyclic steroidal ethers. Org. Lett. 2004, 6, 1437–1440. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Visible-Light-Mediated Utilization of alpha-Aminoalkyl Radicals: Addition to Electron-Deficient Alkenes Using Photoredox Catalysts. J. Am. Chem. Soc. 2012, 134, 3338–3341. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, Z.; Kim, M. Metal-Free α-C(sp3)–H Functionalized Oxidative Cyclization of Tertiary N,N-Diaryl Amino Alcohols: Theoretical Approach for Mechanistic Pathway. Molecules 2017, 22, 547. https://doi.org/10.3390/molecules22040547
Ullah Z, Kim M. Metal-Free α-C(sp3)–H Functionalized Oxidative Cyclization of Tertiary N,N-Diaryl Amino Alcohols: Theoretical Approach for Mechanistic Pathway. Molecules. 2017; 22(4):547. https://doi.org/10.3390/molecules22040547
Chicago/Turabian StyleUllah, Zakir, and Mihyun Kim. 2017. "Metal-Free α-C(sp3)–H Functionalized Oxidative Cyclization of Tertiary N,N-Diaryl Amino Alcohols: Theoretical Approach for Mechanistic Pathway" Molecules 22, no. 4: 547. https://doi.org/10.3390/molecules22040547
APA StyleUllah, Z., & Kim, M. (2017). Metal-Free α-C(sp3)–H Functionalized Oxidative Cyclization of Tertiary N,N-Diaryl Amino Alcohols: Theoretical Approach for Mechanistic Pathway. Molecules, 22(4), 547. https://doi.org/10.3390/molecules22040547