Abstract
The previously published structure of the fungal metabolite acremine P is revised by re-evaluation of chemical shift values and NOESY data, and by DFT calculations.
1. Introduction
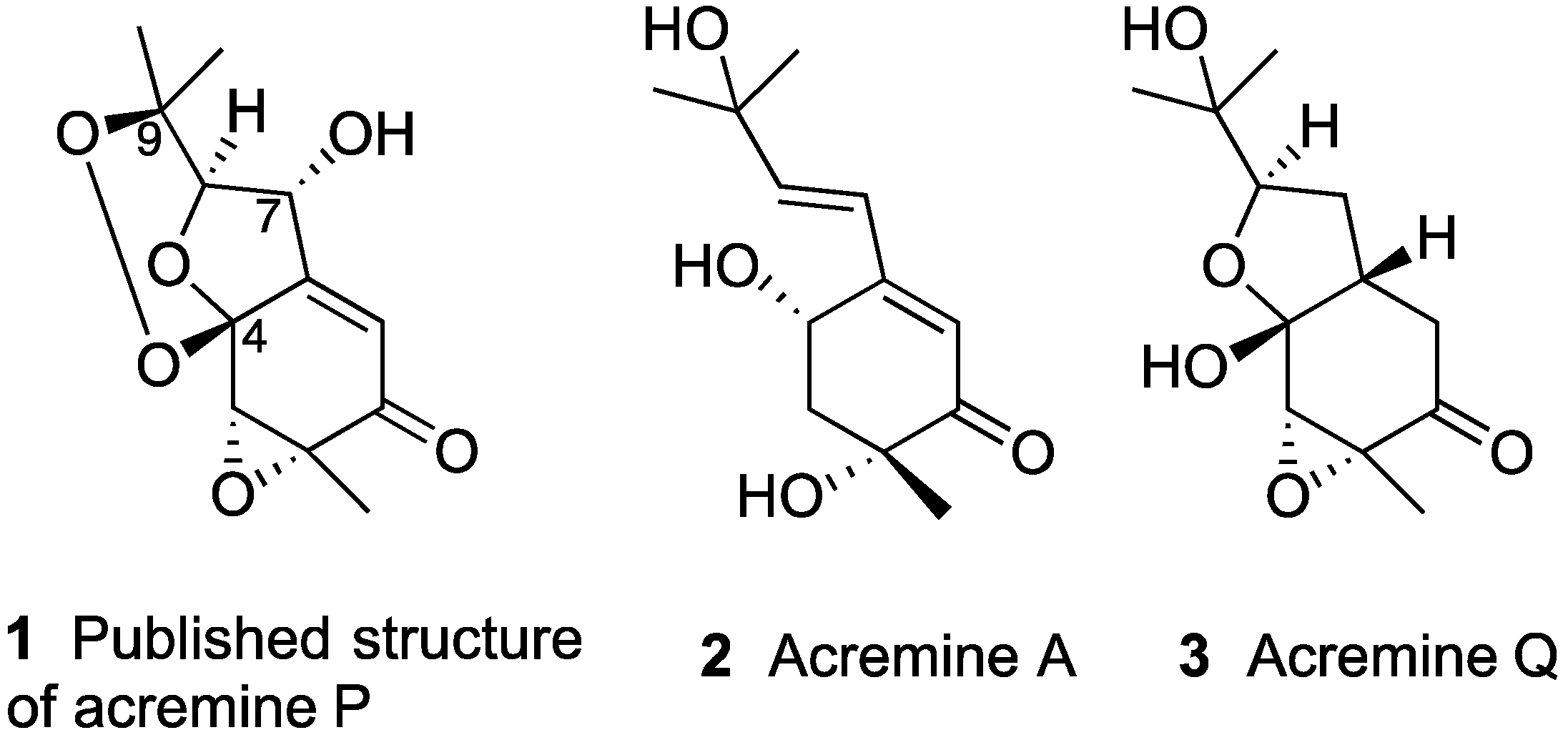
Fungi from the genus Acremonium isolated from both terrestrial or marine sources have been reported to produce meroterpenoids, alkaloids, peptides or oxygenated metabolites [1,2,3,4,5,6]. Recently, we reported the structure elucidation, including a stereochemical investigation, of the meroterpenoid acremine P (1) (Chart 1) from a strain of Acremonium persicinum isolated from the marine sponge Anomoianthella rubra, obtained offshore from Mooloolaba in southeast Queensland. The chemical correlation of acremine P with its co-metabolite acremine A (2) by catalytic hydrogenation under mild conditions (H2, Pd/C, 24 h) was instrumental in defining the carbon framework and partial relative configuration of acremine P, while the absolute configuration at C-6 was determined by NMR analysis of O-methylmandelate (MPA) ester derivatives. The remaining stereochemical elements were resolved by evaluation of NOESY data, molecular modeling and selected heteronuclear coupling constant values [7].
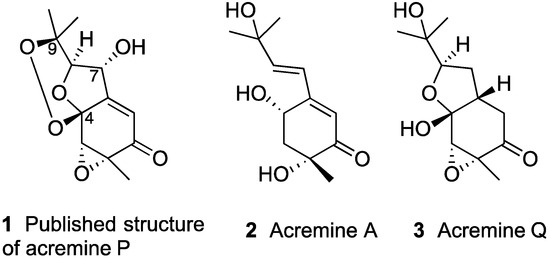
Chart 1.
Selected meroterpenoids from A. persicinum.
In this communication, we revise the carbon framework of acremine P based on a comparison of calculated and experimental NMR chemical shift data. The predictions of chemical shift values by quantum chemical methods have provided valuable insights into natural product structures, including guiding the choice of diastereomer for structure confirmation by total synthesis [8,9,10]. Recent examples which highlight the role of computed chemical shift values in the stereochemical evaluation of natural products include vannusal B [11] and nobilisitine [12]. The structural revision of acremine A likewise necessitated a review of its overall relative stereochemistry, and this was informed by nOe data as well as molecular modeling. Finally, a biosynthetic pathway based on oxidative cleavage of a didehydro analogue of acremine Q is proposed.
2. Results and Discussions
The structures of acremines P and Q were examined using a combination quantum chemical/empirical approach (Spartan, Wavefunction Inc., Irvine, CA, USA) [13] that provides 13C-NMR shifts with a mean absolute error (MAE) of ~1.6 ppm. The calculations involve Boltzmann averaging from a ωB97X-V/6-311+G(2df,2p) model using conformer geometries from the ωB97X-D/6-31G* model and chemical shifts from ωB97X-D/6-31G*, but are also empirically corrected to account for the local environment. There was close agreement between the calculated vs. experimental 13C-NMR shift values for acremine Q (3), thereby corroborating the structure and stereochemistry of the metabolite as previously published. For acremine P, originally assigned structure 2, the preliminary computational search for conformers revealed a single dominant conformer, with the closest alternative being upwards of 10 kJ/mol higher in energy and thus not contributing significantly to the equilibrium mixture. As shown in Table 1, there was a significant mismatching of the calculated vs. experimental 13C-NMR shift values for acremine P, with deviations of 20.4 ppm for the alkene carbon (C-2) and 23.0 ppm for the hydroxymethine carbon (C-7). Consequently, the published structure for acremine P could not be correct. While deviations of 3 ppm between the experimental and calculated values are considered acceptable, even up to 5–6 ppm for carbonyl groups, a deviation of >10 ppm between experimental and calculated values is diagnostic of an incorrect structure [8,14].

Table 1.
Comparison of experimental vs. calculated 13C-NMR chemical shifts for acremine P (4). 1
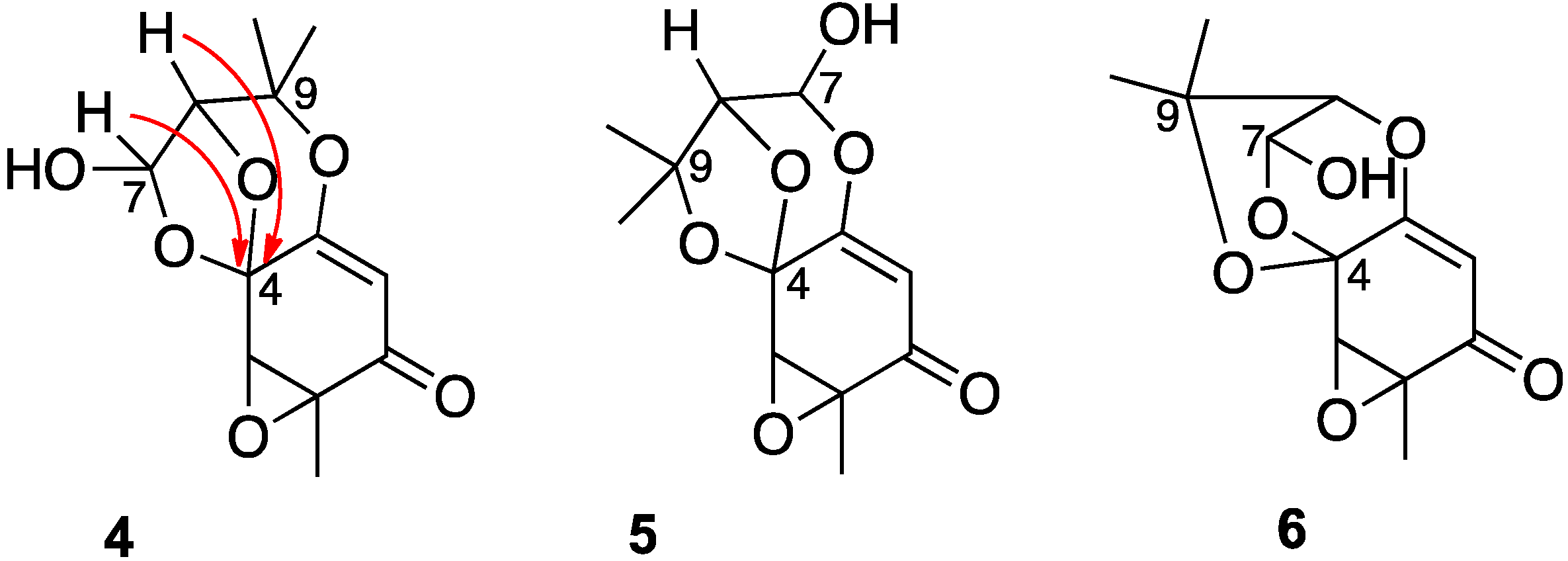
A re-evaluation of the 13C-NMR shift values suggested that the signal at 95.0 ppm (C-7), previously assigned to a secondary alcohol center, was instead associated with an acetal or lactol center. Furthermore, the alkene carbon signals (102.4 and 162.5 ppm) indicated a polarized double bond, likely enolized given the number of oxygen atoms in the molecule. With this information, three candidate structures (4)–(6) (Chart 2), each of which contained a lactol functionality, were assessed against the previously reported HMBC data. In acremine P, the lactol proton at δH 5.83 (d) and the signal at ™H 4.15 (s) for the hydroxymethine proton (H-8) each showed HMBC bond correlations to the acetal carbon at 99.0 ppm. In isomer 4, these correspond to three bond correlations, whereas in isomers 5 and 6, the lactol proton and the hydroxymethine proton, respectively, were each four bonds distant from the acetal center. Furthermore, in planar structure 5, an HMBC between the lactol proton and the alkene C-3 at 162.5 ppm would be anticipated.
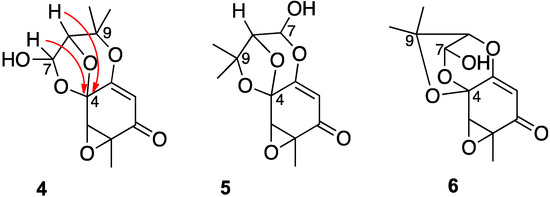
Chart 2.
Candidate structures for acremine P.
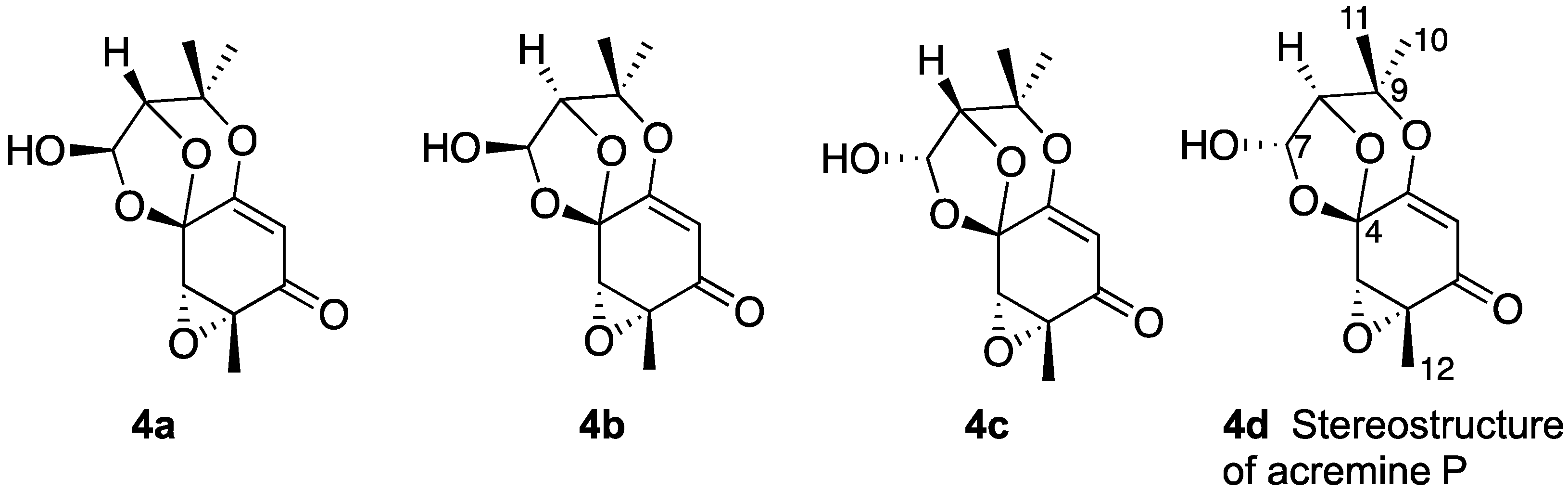
Therefore, planar structure 3 was considered further with DFT computations undertaken on four diastereomers (4a–4d) (Chart 3) following a conformational search on each of the four candidates. Diastereomers 4a and 4d had two contributing conformers, while 4b and 4c each had one contributing conformer. Three of the four candidates were reasonable matches to the 13C data, suggesting that the correct planar structure of acremine P had been elucidated; further, these data enabled diastereomer 4b, for which the C-4 chemical shift was calculated at 116.5 ppm compared to an experimental value of 99.0 ppm, to be eliminated. It was not feasible to use the computational data to distinguish between the three remaining diastereomers owing to the similarity of their 13C-NMR shift values, and also to the errors inevitably inherent to the quantum mechanical calculations. It was noted that the quantum mechanical calculations revealed stereoisomers 4a and 4d to be the lowest in energy.
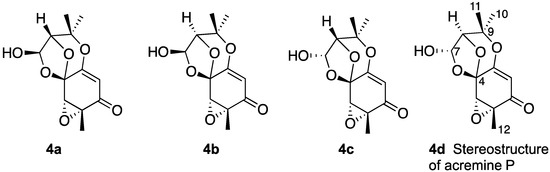
Chart 3.
Candidate diastereomers of acremine P.
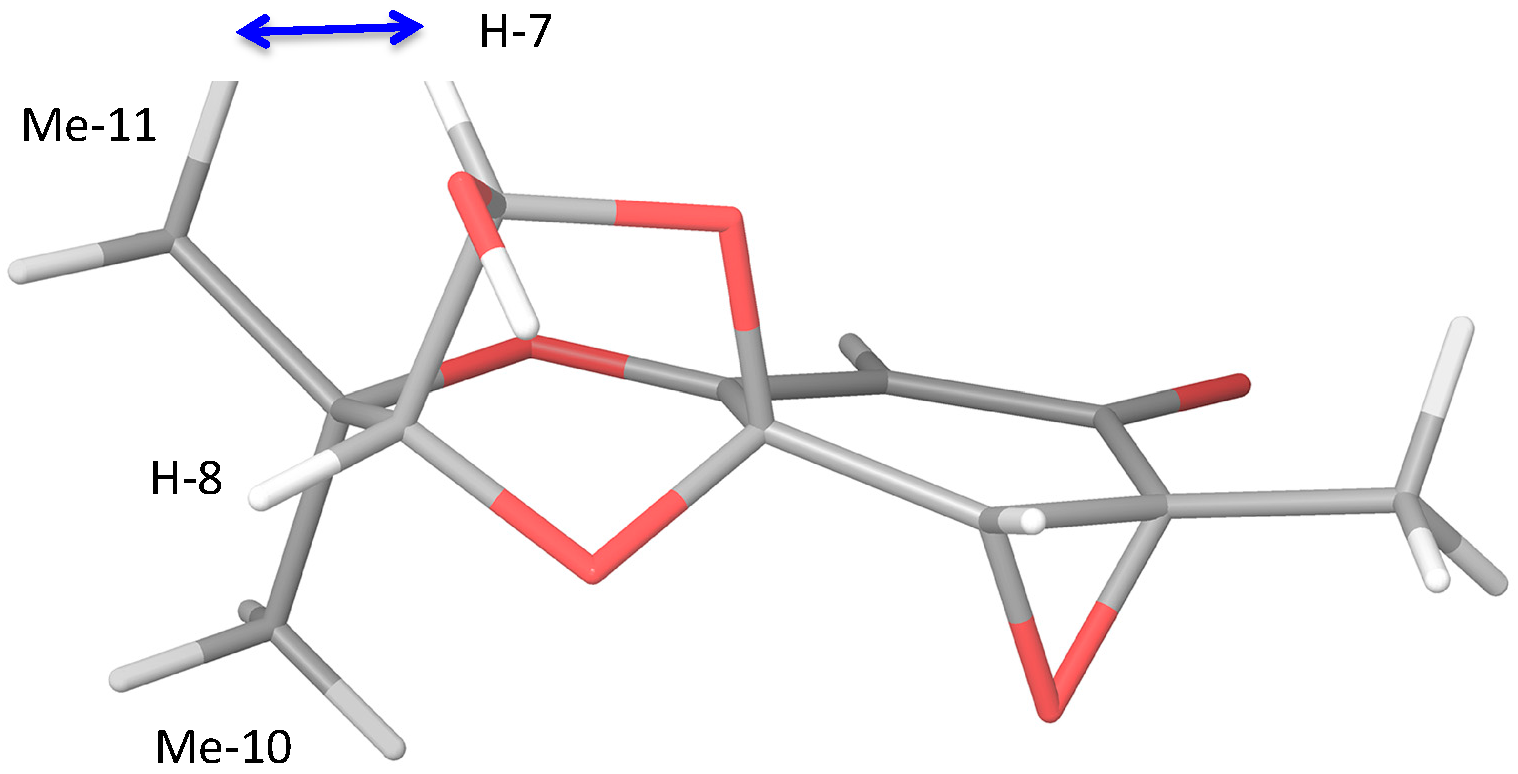
The three remaining stereoisomers could be further distinguished by the zero coupling between the vicinal lactol and hydroxymethine protons, which necessitated a bond angle close to 90°. Selected homonuclear (H7–H8) couplings were calculated using the methods of Kutateladze et al. [15], yielding values of 0.2, 4.1, 3.9 and 0.3 Hz for JH7–H8 in stereoisomers 4a–4b, respectively. The two stereoisomers (4a, 4d) thus fitted the coupling data. It was noted that all four stereoisomers showed a heteronuclear (C4-H8) J value close to 5 Hz, and so these data did not distinguish 4a from 4d. NOe data also supported the choice of stereoisomer, in that from their 3D stereostructures both isomers 4a and 4d were expected to show an nOe between the lactol proton and one only of the two methyl groups; in contrast, isomers 4b and 4c were each expected to show nOes between the lactol proton and both methyl groups. In acremine P, the lactol proton at δH 5.83 shows an nOe to the methyl signal at δH 1.43 for H-11, but not to the methyl signal at δH 1.47 (Figure 1). The calculated chemical shifts were further examined using the DP4+ computational approach developed by Sarotti et al. to assign the most probable diastereomer [16]. Using the 13C-NMR data alone, the probability was 99.7% that 4d was the correct diastereomer.
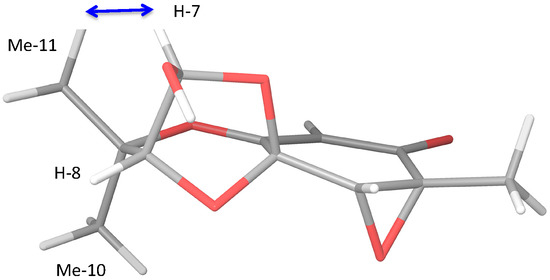
Figure 1.
Three-dimensional image of stereoisomer 4d showing nOe to Me-11.
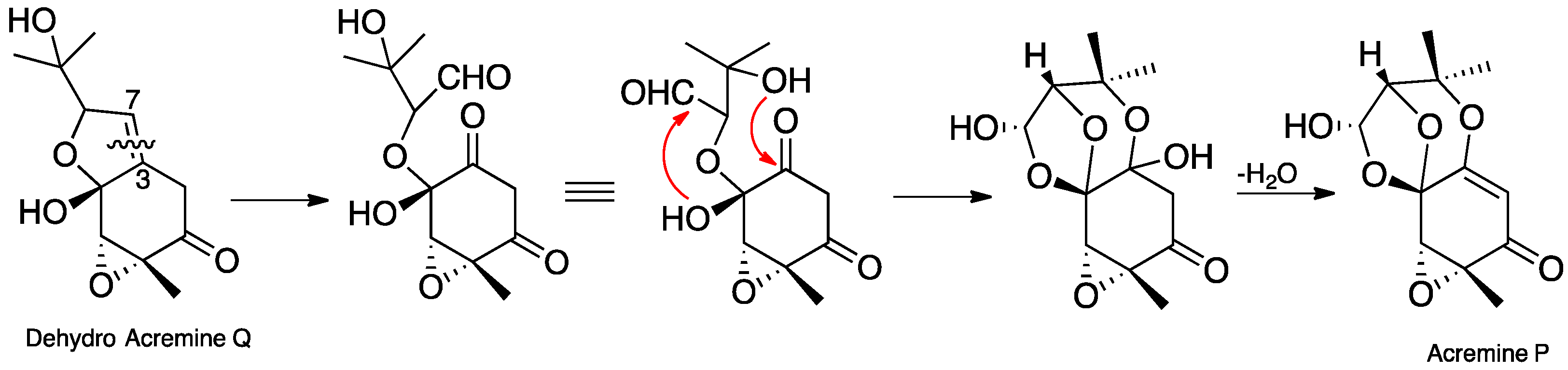
In our earlier publication [7], we reported that the hydrogenation of acremine P yielded acremine A (2) as the sole product, and used this information to specify the absolute configuration at C-6 of acremine P; clearly this information must be in error since the dioxolane ring of the revised structure is incompatible with the tetrahydrofuran ring previously ascribed to acremine P. Nevertheless, we anticipated that acremine P would have the same 6R configuration as its co-metabolite acremine A. Scheme 1 shows a plausible biosynthetic route to the revised structure of acremine P. A didehydro derivative, generated by the action of a P450 enzyme on the co-metabolite acremine Q, could undergo oxidative ring cleavage of the C-3/C7 double bond [17], thereby generating an aldehyde group at the original C-7 position. The 1,3-dioxolane ring is formed by cyclization of the C-4 hydroxy group onto the aldehyde, and would be anticipated to provide the thermodynamically most stable lactol product. Finally, nucleophilic attack of the C-9 hydroxy group onto the C-3 carbonyl generates a hemiacetal intermediate; the subsequent dehydration step which generates an enol ether is facilitated by the associated formation of an α,β-unsaturated carbonyl.
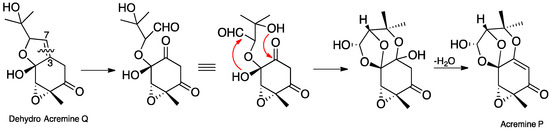
Scheme 1.
Putative biosynthetic pathway to acremine P (4d) from a dehydro derivative of acremine Q.
3. Materials and Methods
The isolation and characterization of acremine P has been previously reported [7]. Details of computational calculations are given in the supplementary materials.
4. Conclusions
In conclusion, our revision of the structure of acremine P illustrates the valuable role of computational studies in evaluating the structures and stereochemistry of stereochemically complex natural products. At the same time, a single piece of data in the original study, i.e., the suggested conversion of acremine P into acremine A by hydrogenation, compromised the structural study and thereby incorrectly informed the structure determination.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
We thank the University of Queensland for financial support.
Author Contributions
M.J.G., W.H. and G.K.P. analyzed the data; S. contributed the sample of acremine P; M.J.G. and G.K.P. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Patterson, E.L.; Vanmeter, J.C.; Bohonos, N. Isolation of cephalosporin C. J. Med. Chem. 1964, 7, 689. [Google Scholar] [CrossRef] [PubMed]
- Assante, G.; Dallavalle, S.; Malpezzi, L.; Burruano, S.; Torta, L. Acremines A-F, novel secondary metabolites produced by a strain of an endophytic Acremonium, isolated from sporangiophores of Plasmopara viticola in grapevine leaves. Tetrahedron 2005, 61, 7686–7692. [Google Scholar] [CrossRef]
- Arnone, A.; Assante, G.; Bava, A.; Dallavalle, S.; Nasini, G. Acremines H-N, novel prenylated polyketide metabolites produced by a strain of Acremonium byssoides. Tetrahedron 2009, 65, 786–791. [Google Scholar] [CrossRef]
- Januar, L.A.; Molinski, T.F. Acremolin from Acremonium strictum is 2,3-etheno-2′-isopropyl-1-methylguanine, not an 1H-azirine. Synthesis and structural revision. Org. Lett. 2013, 15, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Boot, C.M.; Tenney, K.; Valeriote, F.A.; Crews, P. Highly N-methylated linear peptides produced by an atypical sponge-derived Acremonium sp. J. Nat. Prod. 2006, 69, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C-E from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.A.; Lambert, L.K.; Pierens, G.K.; Bernhardt, P.V.; Garson, M.J. Secondary metabolites of the sponge-derived fungus Acremonium persicinum. J. Nat. Prod. 2013, 76, 1432–1440. [Google Scholar]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, D.J. Walking in the woods with quantum chemistry-applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 2013, 30, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide to small molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Prot. 2014, 9, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Saielli, G.; Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Bagno, A. Addressing the stereochemistry of complex organic molecules by density functional theory-NMR: Vannusal B in retrospective. J. Am. Chem. Soc. 2011, 133, 6072–6077. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Tantillo, D.J. Prediction of the structure of nobilisitine using computed NMR chemical shifts. J. Nat. Prod. 2011, 74, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Spartan Software. Available online: https://www.wavefun.com./products/spartan.html (accessed on 21 February 2017).
- Sarotti, A.M.; Pellegrinet, S.C. A multi-standard approach for GIAO 13C-NMR calculations. J. Org. Chem. 2009, 74, 7254–7260. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Mukhina, O.A. Parametrization of 13C-1H nuclear spin-spin coupling constants. J. Org. Chem. 2015, 80, 10838–10848. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical assignments of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Cryle, M.J. Carbon-carbon bond cleavage by P450 systems. Met. Ions Life Sci. 2007, 3, 397–435. [Google Scholar]
Sample Availability: A sample of acremine P is available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).