
Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors



Abstract
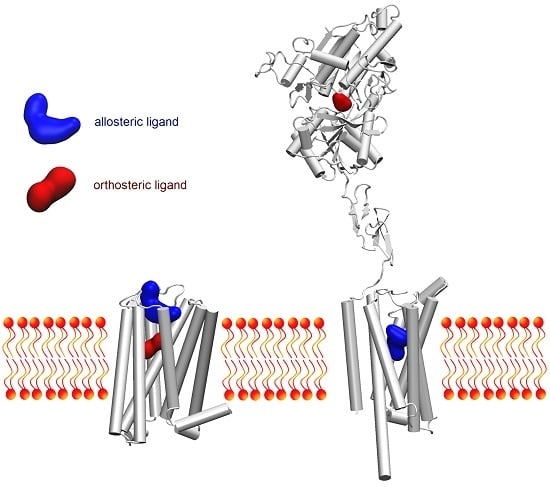
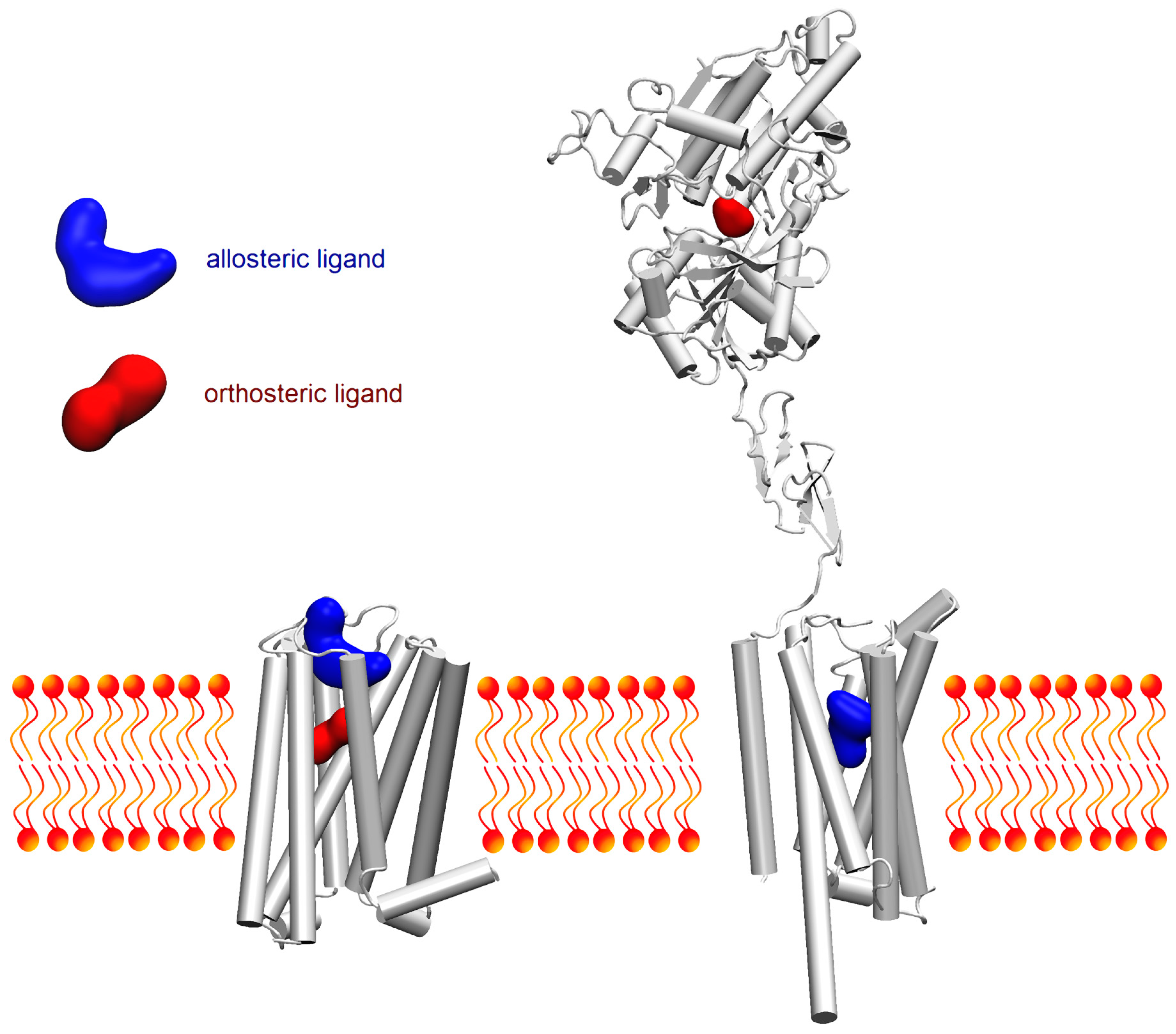
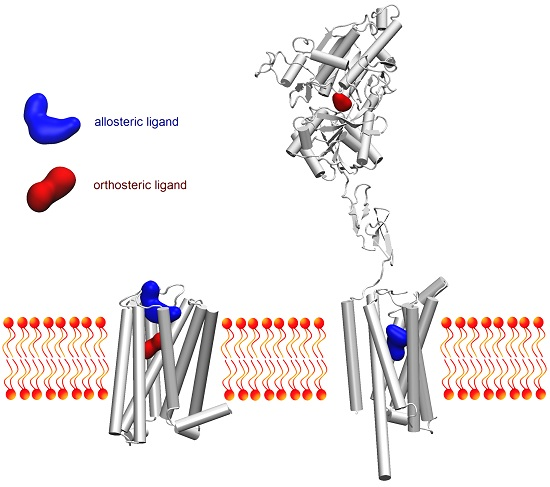
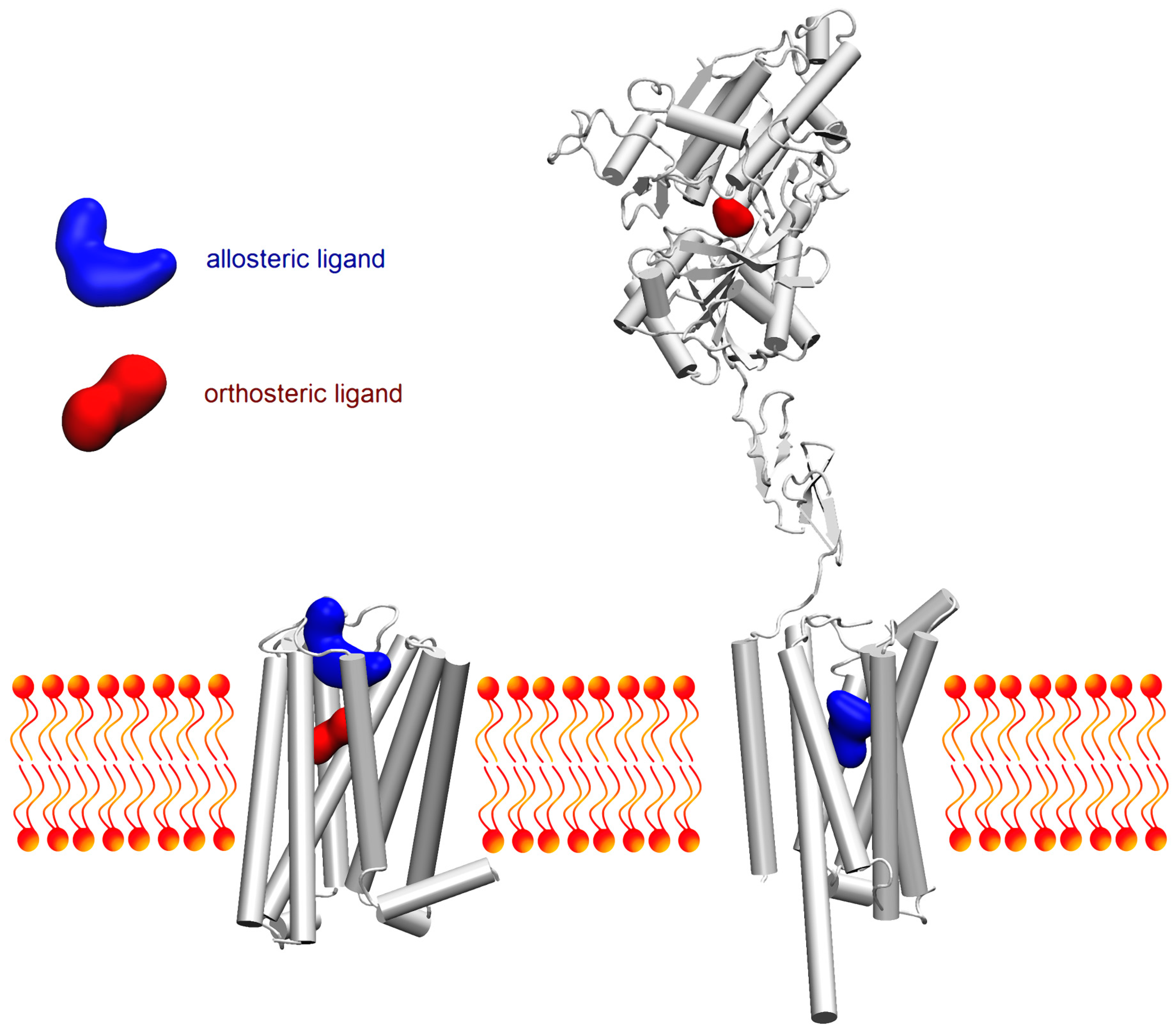
:
1. Introduction
2. An Overview of Docking of Small Molecules to GPCRs
Recently Introduced Docking Algorithms
3. Modeling of Water Molecules within the Binding Site
4. High-Throughput Docking as a Method of Virtual Screening
5. Docking by Molecular Dynamics Simulations and Their Modifications
6. Protein-Protein Docking as a Method to Model GPCR Dimers
7. Summary and Perspectives
Acknowledgments
Conflicts of Interest
References
- Dohlman, H.G. Thematic Minireview Series: New Directions in G Protein-coupled Receptor Pharmacology. J. Biol. Chem. 2015, 290, 19469–19470. [Google Scholar] [CrossRef]
- Gloriam, D.E.; Fredriksson, R.; Schiöth, H.B. The G protein-coupled receptor subset of the rat genome. BMC Genom. 2007, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Soppa, J. Two hypotheses—one answer. Sequence comparison does not support an evolutionary link between halobacterial retinal proteins including bacteriorhodopsin and eukaryotic G-protein-coupled receptors. FEBS Lett. 1994, 342, 7–11. [Google Scholar] [CrossRef]
- Römpler, H.; Stäubert, C.; Thor, D.; Schulz, A.; Hofreiter, M.; Schöneberg, T. G protein-coupled time travel: evolutionary aspects of GPCR research. Mol. Interv. 2007, 7, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, B.D.; Kramer, I.M.; Tatham, P.E.R. Chapter 4—GTP-binding Proteins and Signal Transduction. In Signal Transduction (Second Edition); Gomperts, B.D., Kramer, I.M., Tatham, P.E.R., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 81–129. [Google Scholar]
- Krauss, G. G Protein-Coupled Signal Transmission Pathways. In Biochemistry of Signal Transduction and Regulation; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 179–229. [Google Scholar]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Ferguson, S.S.G. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Mol. Endocrinol. 2015, 29, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, W.L.; Altenbach, C.; Hubbell, C.M.; Khorana, H.G. Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 2003, 63, 243–290. [Google Scholar] [PubMed]
- Farrens, D.L.; Altenbach, C.; Yang, K.; Hubbell, W.L.; Khorana, H.G. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996, 274, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Gether, U.; Lin, S.; Ghanouni, P.; Ballesteros, J.A.; Weinstein, H.; Kobilka, B.K. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J. 1997, 16, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Parnot, C.; Deupi, X.; Ratnala, V.R.P.; Swaminath, G.; Farrens, D.; Kobilka, B. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat. Chem. Biol. 2006, 2, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Altenbach, C.; Kusnetzow, A.K.; Ernst, O.P.; Hofmann, K.P.; Hubbell, W.L. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. USA 2008, 105, 7439–7444. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauss, N.; Choe, H.-W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, H.; Liu, J.; Li, F.; Li, X.; Shi, L.; Chen, J. Rational Selection of the 3D Structure of Biomacromolecules for Molecular Docking Studies on the Mechanism of Endocrine Disruptor Action. Chem. Res. Toxicol. 2016, 29, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Computer analysis of protein-protein interaction. J. Mol. Biol. 1978, 124, 323–342. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Selent, J.; Poso, A. Structure-based molecular modeling approaches to GPCR oligomerization. Methods Cell Biol. 2013, 117, 91–104. [Google Scholar] [PubMed]
- Ciancetta, A.; Sabbadin, D.; Federico, S.; Spalluto, G.; Moro, S. Advances in Computational Techniques to Study GPCR-Ligand Recognition. Trends Pharmacol. Sci. 2015, 36, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. Advances and Challenges in Protein-Ligand Docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Vuong, Q.V.; Nguyen, T.T.; Li, M.S. A New Method for Navigating Optimal Direction for Pulling Ligand from Binding Pocket: Application to Ranking Binding Affinity by Steered Molecular Dynamics. J. Chem. Inf. Model. 2015, 55, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Katritch, V.; Participants of GPCR Dock 2013; Stevens, R.C.; Abagyan, R. Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: Meeting new challenges. Structure 2014, 22, 1120–1139. [Google Scholar]
- Carlson, H.A.; Smith, R.D.; Damm-Ganamet, K.L.; Stuckey, J.A.; Ahmed, A.; Convery, M.A.; Somers, D.O.; Kranz, M.; Elkins, P.A.; Cui, G.; et al. CSAR 2014: A Benchmark Exercise Using Unpublished Data from Pharma. J. Chem. Inf. Model. 2016, 56, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Gathiaka, S.; Liu, S.; Chiu, M.; Yang, H.; Stuckey, J.A.; Kang, Y.N.; Delproposto, J.; Kubish, G.; Dunbar, J.B.; Carlson, H.A.; et al. D3R grand challenge 2015: Evaluation of protein-ligand pose and affinity predictions. J. Comput. Aided Mol. Des. 2016, 30, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Tidgewell, K.; Groer, C.E.; Harding, W.W.; Lozama, A.; Schmidt, M.; Marquam, A.; Hiemstra, J.; Partilla, J.S.; Dersch, C.M.; Rothman, R.B.; et al. Herkinorin analogues with differential beta-arrestin-2 interactions. J. Med. Chem. 2008, 51, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Perez-Aguilar, J.M.; Shan, J.; LeVine, M.V.; Khelashvili, G.; Weinstein, H. A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J. Am. Chem. Soc. 2014, 136, 16044–16054. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Interplay between Two Allosteric Sites and Their Influence on Agonist Binding in Human μ Opioid Receptor. J. Chem. Inf. Model. 2016, 56, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What Can Crystal Structures of Aminergic Receptors Tell Us about Designing Subtype-Selective Ligands? Pharmacol. Rev. 2015, 67, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Receptor Molecular Biology; Academic Press: San Diego, CA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular Docking Screening Using Agonist-Bound GPCR Structures: Probing the A 2A Adenosine Receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Levitt, D.G.; Banaszak, L.J. POCKET: A Computer Graphics Method for Identifying and Displaying Protein Cavities and Their Surrounding Amino Acids. J. Mol. Graph. 1992, 10, 229–234. [Google Scholar] [CrossRef]
- Laskowski, R.A. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph. 1995, 13, 323–330. [Google Scholar] [CrossRef]
- Brady, G.P.; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M.; Feinstein, W.P. eFindSite: Improved prediction of ligand binding sites in protein models using meta-threading, machine learning and auxiliary ligands. J. Comput. Aided Mol. Des. 2013, 27, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Skolnick, J. APoc: Large-scale identification of similar protein pockets. Bioinformatics 2013, 29, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Rutkowska, E.; Bartuzi, D.; Targowska-Duda, K.M.; Matosiuk, D.; Selent, J. Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 2016, 132, 359–399. [Google Scholar] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Jaśkowska, J.; Bojarski, A.J.; Duszyńska, B.; Bucki, A.; Kołaczkowski, M. Evaluation of 1-arylpiperazine derivative of hydroxybenzamides as 5-HT1A and 5-HT7 serotonin receptor ligands: An experimental and molecular modeling approach. J. Heterocycl. Chem. 2011, 48, 192–198. [Google Scholar] [CrossRef]
- Sturlese, M.; Bellanda, M.; Moro, S. NMR-Assisted Molecular Docking Methodologies. Mol. Inform. 2015, 34, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Michino, M.; Abola, E.; GPCR Dock 2008 participants; Brooks, C.L.; Dixon, J.S.; Moult, J.; Stevens, R.C. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat. Rev. Drug Discov. 2009, 8, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Rueda, M.; Katritch, V.; Stevens, R.C.; Abagyan, R.; GPCR Dock 2010 participants. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19, 1108–1126. [Google Scholar]
- Totrov, M.; Abagyan, R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins 1997, 29 (Suppl. 1), 215–220. [Google Scholar] [CrossRef]
- Piotto, S.; Biasi, L.D.; Fino, R.; Parisi, R.; Sessa, L.; Concilio, S. Yada: A novel tool for molecular docking calculations. J. Comput. Aided Mol. Des. 2016, 30, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. DockBench as docking selector tool: The lesson learned from D3R Grand Challenge 2015. J. Comput. Aided Mol. Des. 2016, 30, 773–789. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Farelli, J.D.; Brown, S.D.; Liu, C.; Huang, H.; Korczynska, M.; Al-Obaidi, N.F.; Babbitt, P.C.; Almo, S.C.; Allen, K.N.; et al. Covalent Docking Predicts Substrates for Haloalkanoate Dehalogenase Superfamily Phosphatases. Biochemistry (Mosc.) 2015, 54, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Bikbulatov, R.V.; Mocanu, V.; Dicheva, N.; Parker, C.E.; Wetsel, W.C.; Mosier, P.D.; Westkaemper, R.B.; Allen, J.A.; Zjawiony, J.K.; et al. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry (Mosc.) 2009, 48, 6898–6908. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shimon, A.; Niv, M.Y. AnchorDock: Blind and Flexible Anchor-Driven Peptide Docking. Structure 2015, 23, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Xu, X.; Zou, X. Fully Blind Docking at the Atomic Level for Protein-Peptide Complex Structure Prediction. Structure 2016, 24, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein–peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.E.; Lightstone, F.C. Accounting for water molecules in drug design. Expert Opin. Drug Discov. 2011, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Cavasotto, C.N. Open challenges in structure-based virtual screening: Receptor modeling, target flexibility consideration and active site water molecules description. Arch. Biochem. Biophys. 2015, 583, 105–119. [Google Scholar] [CrossRef] [PubMed]
- De Beer, S.B.A.; Vermeulen, N.P.E.; Oostenbrink, C. The role of water molecules in computational drug design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Amadasi, A.; Spyrakis, F.; Cozzini, P.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Mapping the energetics of water-protein and water-ligand interactions with the “natural” HINT forcefield: Predictive tools for characterizing the roles of water in biomolecules. J. Mol. Biol. 2006, 358, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Taylor, R. SuperStar: A knowledge-based approach for identifying interaction sites in proteins. J. Mol. Biol. 1999, 289, 1093–1108. [Google Scholar] [CrossRef]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Prediction of the water content in protein binding sites. J. Phys. Chem. B 2009, 113, 13337–13346. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Li, Y.; Xiong, B.; Jiang, H.; Shen, J. Water PMF for predicting the properties of water molecules in protein binding site. J. Comput. Chem. 2013, 34, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Cho, A.E. Incorporating QM and solvation into docking for applications to GPCR targets. Phys. Chem. Chem. Phys. 2016, 18, 28281–28289. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Beuming, T.; Sherman, W.; van Vlijmen, H.W.T.; IJzerman, A.P. Selecting an optimal number of binding site waters to improve virtual screening enrichments against the adenosine A2A receptor. J. Chem. Inf. Model. 2014, 54, 1737–1746. [Google Scholar] [CrossRef]
- Yang, Y.; Lightstone, F.C.; Wong, S.E. Approaches to efficiently estimate solvation and explicit water energetics in ligand binding: The use of WaterMap. Expert Opin. Drug Discov. 2013, 8, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Ghoshdastider, U.; Trzaskowski, B.; Latek, D.; Debinski, A.; Pulawski, W.; Wu, R.; Gerke, V.; Filipek, S. The role of water in activation mechanism of human N-formyl peptide receptor 1 (FPR1) based on molecular dynamics simulations. PLoS ONE 2012, 7, e47114. [Google Scholar] [CrossRef] [PubMed]
- Higgs, C.; Beuming, T.; Sherman, W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.B.; Repasky, M.P.; Greenwood, J.R.; Tubert-Brohman, I.; Jerome, S.; Annabhimoju, R.; Boyles, N.A.; Schmitz, C.D.; Abel, R.; Farid, R.; et al. WScore: A Flexible and Accurate Treatment of Explicit Water Molecules in Ligand-Receptor Docking. J. Med. Chem. 2016, 59, 4364–4384. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Zhang, W.; Hobrath, J.V. Recent advances in structure-based virtual screening of G-protein coupled receptors. AAPS J. 2009, 11, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kobilka, B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Sherman, W. Current assessment of docking into GPCR crystal structures and homology models: Successes, challenges, and guidelines. J. Chem. Inf. Model. 2012, 52, 3263–3277. [Google Scholar] [CrossRef] [PubMed]
- Senderowitz, H.; Marantz, Y. G Protein-Coupled Receptors: Target-based in silico screening. Curr. Pharm. Des. 2009, 15, 4049–4068. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M.; Liu, Z. Structure-based design in the GPCR target space. Curr. Med. Chem. 2012, 19, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. From three-dimensional GPCR structure to rational ligand discovery. Adv. Exp. Med. Biol. 2014, 796, 129–157. [Google Scholar] [PubMed]
- Sanders, M.P.A.; Roumen, L.; van der Horst, E.; Lane, J.R.; Vischer, H.F.; van Offenbeek, J.; de Vries, H.; Verhoeven, S.; Chow, K.Y.; Verkaar, F.; et al. A prospective cross-screening study on G-protein-coupled receptors: Lessons learned in virtual compound library design. J. Med. Chem. 2012, 55, 5311–5325. [Google Scholar] [CrossRef] [PubMed]
- Kołaczkowski, M.; Bucki, A.; Feder, M.; Pawłowski, M. Ligand-optimized homology models of D₁ and D₂ dopamine receptors: Application for virtual screening. J. Chem. Inf. Model. 2013, 53, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Silva, A.G.; Loza, M.I.; Kolb, P.; Castro, M.; Poso, A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. ChemMedChem 2016, 11, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Gandhimathi, A.; Sowdhamini, R. Molecular modelling of human 5-hydroxytryptamine receptor (5-HT2A) and virtual screening studies towards the identification of agonist and antagonist molecules. J. Biomol. Struct. Dyn. 2016, 34, 952–970. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Coleman, R.G.; Setola, V.; Irwin, J.J.; Fan, H.; Schlessinger, A.; Sali, A.; Roth, B.L.; Shoichet, B.K. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 2011, 7, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, K.; Li, X.-D.; Zhang, D.-L.; Xu, F. Discovery of novel antagonists of human neurotensin receptor 1 on the basis of ligand and protein structure. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 84, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Daga, P.R.; Polgar, W.E.; Zaveri, N.T. Structure-based virtual screening of the nociceptin receptor: Hybrid docking and shape-based approaches for improved hit identification. J. Chem. Inf. Model. 2014, 54, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.R.; Bortolato, A.; Tehan, B.; Mason, J.S. GPCR-Bench: A Benchmarking Set and Practitioners’ Guide for G Protein-Coupled Receptor Docking. J. Chem. Inf. Model. 2016, 56, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, M.-B.; Chen, Z.-J.; Chen, H.; Lin, J.-P.; Yang, L.-R. Fragment-based drug discovery and molecular docking in drug design. Curr. Pharm. Biotechnol. 2015, 16, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Phan, K.; Gao, Z.-G.; Gakh, A.A.; Xu, F.; Deflorian, F.; Abagyan, R.; Stevens, R.C.; Jacobson, K.A.; Katritch, V. Optimization of adenosine 5’-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. J. Med. Chem. 2012, 55, 4297–4308. [Google Scholar] [CrossRef] [PubMed]
- Sirci, F.; Istyastono, E.P.; Vischer, H.F.; Kooistra, A.J.; Nijmeijer, S.; Kuijer, M.; Wijtmans, M.; Mannhold, R.; Leurs, R.; de Esch, I.J.P.; et al. Virtual fragment screening: Discovery of histamine H3 receptor ligands using ligand-based and protein-based molecular fingerprints. J. Chem. Inf. Model. 2012, 52, 3308–3324. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.; Stoddart, L.A.; Hill, S.J.; Carlsson, J. Fragment-Based Discovery of Subtype-Selective Adenosine Receptor Ligands from Homology Models. J. Med. Chem. 2015, 58, 9578–9590. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Kooistra, A.J.; Vischer, H.F.; Katritch, V.; Kuijer, M.; Shiroishi, M.; Iwata, S.; Shimamura, T.; Stevens, R.C.; de Esch, I.J.P.; et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J. Med. Chem. 2011, 54, 8195–8206. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; IJzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A(2A) adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Schmidt, É.; Horti, F.; Keserű, G.M. Virtual fragment screening on GPCRs: A case study on dopamine D3 and histamine H4 receptors. Eur. J. Med. Chem. 2014, 77, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Espigares, I.; Kaczor, A.A.; Selent, J. In silico Exploration of the Conformational Universe of GPCRs. Mol. Inform. 2016, 35, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Tarcsay, A.; Paragi, G.; Vass, M.; Jójárt, B.; Bogár, F.; Keserű, G.M. The impact of molecular dynamics sampling on the performance of virtual screening against GPCRs. J. Chem. Inf. Model. 2013, 53, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Kohlhoff, K.J.; Shukla, D.; Lawrenz, M.; Bowman, G.R.; Konerding, D.E.; Belov, D.; Altman, R.B.; Pande, V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2013, 6, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Vaidehi, N. Computational mapping of the conformational transitions in agonist selective pathways of a G-protein coupled receptor. J. Am. Chem. Soc. 2010, 132, 5205–5214. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Ranganathan, A.; Carlsson, J. Strategies for improved modeling of GPCR-drug complexes: Blind predictions of serotonin receptors bound to ergotamine. J. Chem. Inf. Model. 2014, 54, 2004–2021. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.R.; Ahn, S.; Sassano, M.F.; Kleist, A.; Zhu, X.; Strachan, R.; Roth, B.L.; Lefkowitz, R.J.; Shoichet, B.K. Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem. Biol. 2013, 8, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Bernat, V.; Brox, R.; Tschammer, N.; Kolb, P. Identifying modulators of CXC receptors 3 and 4 with tailored selectivity using multi-target docking. ACS Chem. Biol. 2015, 10, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Brea, J.; Loza, M.I.; Carlsson, J. Structure-based discovery of selective serotonin 5-HT(1B) receptor ligands. Structure 2014, 22, 1140–1151. [Google Scholar]
- Miao, Y.; Goldfeld, D.A.; Moo, E.V.; Sexton, P.M.; Christopoulos, A.; McCammon, J.A.; Valant, C. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E5675–E5684. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Cho, N.-C.; Min, S.-J.; Cho, Y.S.; Park, K.D.; Seo, S.H.; No, K.T.; Pae, A.N. Novel Scaffold Identification of mGlu1 Receptor Negative Allosteric Modulators Using a Hierarchical Virtual Screening Approach. Chem. Biol. Drug Des. 2016, 87, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-P.; Karpiak, J.; Kroeze, W.K.; Zhu, H.; Chen, X.; Moy, S.S.; Saddoris, K.A.; Nikolova, V.D.; Farrell, M.S.; Wang, S.; et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, A.; Pérez-Nueno, V.I.; Lentini, G.; Ritchie, D.W. Recent trends and future prospects in computational GPCR drug discovery: From virtual screening to polypharmacology. Curr. Top. Med. Chem. 2013, 13, 1069–1097. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Kooistra, A.J.; Ritschel, T.; Leurs, R.; de Esch, I.J.; de Graaf, C. Molecular interaction fingerprint approaches for GPCR drug discovery. Curr. Opin. Pharmacol. 2016, 30, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Hertig, S.; Latorraca, N.R.; Dror, R.O. Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations. PLoS Comput. Biol. 2016, 12, e1004746. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Nevins, N.; Lakdawala, A.S.; Bahar, I. Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. [Google Scholar] [CrossRef] [PubMed]
- Ivetac, A.; McCammon, J.A. Mapping the druggable allosteric space of G-protein coupled receptors: A fragment-based molecular dynamics approach. Chem. Biol. Drug Des. 2010, 76, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Śledź, P.; Lang, S.; Stubbs, C.J.; Spring, D.R.; Abell, C.; Best, R.B. Using ligand-mapping simulations to design a ligand selectively targeting a cryptic surface pocket of polo-like kinase 1. Angew. Chem. 2012, 51, 10078–10081. [Google Scholar] [CrossRef] [PubMed]
- Buch, I.; Giorgino, T.; De Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Kim, E.T.; Eastwood, M.P.; Dror, R.O.; Seeliger, M.A.; Shaw, D.E. How does a drug molecule find its target binding site? J. Am. Chem. Soc. 2011, 133, 9181–9183. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Filizola, M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Curr. Opin. Struct. Biol. 2011, 21, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Deneroff, M.M.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C.; Batson, B.; Bowers, K.J.; Chao, J.C.; et al. Anton, A Special-Purpose Machine for Molecular Dynamics Simulation. Commun. ACM 2008, 51. [Google Scholar] [CrossRef]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry (Mosc.) 2009, 48, 10020–10029. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Provasi, D.; Filizola, M. How Oliceridine (TRV-130) Binds and Stabilizes a μ-Opioid Receptor Conformational State that Selectively Triggers G Protein-Signaling Pathways. Biochemistry (Mosc.) 2016. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Miao, Y.; McCammon, J.A. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015, 48, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, X.; Sengupta, D.; Chattopadhyay, A. Cholesterol-dependent Conformational Plasticity in GPCR Dimers. Sci. Rep. 2016, 6, 31858. [Google Scholar] [CrossRef] [PubMed]
- Pluhackova, K.; Gahbauer, S.; Kranz, F.; Wassenaar, T.A.; Böckmann, R.A. Dynamic Cholesterol-Conditioned Dimerization of the G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016, 12, e1005169. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Activation and allosteric modulation of human μ opioid receptor in molecular dynamics. J. Chem. Inf. Model. 2015, 55, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kastner, K.W.; Izaguirre, J.A. Accelerated molecular dynamics simulations of the octopamine receptor using GPUs: Discovery of an alternate agonist-binding position. Proteins 2016, 84, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Milanos, L.; Saleh, N.; Kling, R.C.; Kaindl, J.; Tschammer, N.; Clark, T. Identification of Two Distinct Sites for Antagonist and Biased Agonist Binding to the Human Chemokine Receptor CXCR3. Angew. Chem. 2016, 55, 15277–15281. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, N.; Suenaga, A.; Taiji, M. Evaluation of protein–ligand affinity prediction using steered molecular dynamics simulations. J. Biomol. Struct. Dyn. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Berteotti, A.; Ronsisvalle, S.; Rocchia, W.; Cavalli, A. Steered Molecular Dynamics Simulations for Studying Protein–Ligand Interaction in Cyclin-Dependent Kinase 5. J. Chem. Inf. Model. 2014, 54, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.-P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Salahpour, A.; Angers, S.; Bouvier, M. Functional significance of oligomerization of G-protein-coupled receptors. Trends Endocrinol. Metab. 2000, 11, 163–168. [Google Scholar] [CrossRef]
- Milligan, G. G protein-coupled receptor hetero-dimerization: Contribution to pharmacology and function. Br. J. Pharmacol. 2009, 158, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Selent, J.; Kaczor, A.A. Oligomerization of G protein-coupled receptors: Computational methods. Curr. Med. Chem. 2011, 18, 4588–4605. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Selent, J. Oligomerization of G protein-coupled receptors: Biochemical and biophysical methods. Curr. Med. Chem. 2011, 18, 4606–4634. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Makarska-Bialokoz, M.; Selent, J.; de la Fuente, R.A.; Martí-Solano, M.; Castro, M. Application of BRET for studying G protein-coupled receptors. Mini Rev. Med. Chem. 2014, 14, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Guixà-González, R.; Ramírez-Anguita, J.M.; Kaczor, A.A.; Selent, J. Simulating G protein-coupled receptors in native-like membranes: From monomers to oligomers. Methods Cell Biol. 2013, 117, 63–90. [Google Scholar] [PubMed]
- Lukasiewicz, S.; Polit, A.; Kędracka-Krok, S.; Wędzony, K.; Maćkowiak, M.; Dziedzicka-Wasylewska, M. Hetero-dimerization of serotonin 5-HT(2A) and dopamine D(2) receptors. Biochim. Biophys. Acta 2010, 1803, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Thor, D.; Zhou, Y.; Liu, T.; Wang, Y.; McMillin, S.M.; Mistry, R.; Challiss, R.A.J.; Costanzi, S.; Wess, J. Structural aspects of M₃ muscarinic acetylcholine receptor dimer formation and activation. FASEB J. 2012, 26, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Selent, J.; Sanz, F.; Pastor, M. Modeling Complexes of Transmembrane Proteins: Systematic Analysis of Protein-Protein Docking Tools. Mol. Inform. 2013, 32, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Biyani, N.; Baskaran, K.; Capitani, G. An analysis of oligomerization interfaces in transmembrane proteins. BMC Struct. Biol. 2013, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.-L.; Russo, O.; Giner, M.; Rivail, L.; Berthouze, M.; Ongeri, S.; Maigret, B.; Fischmeister, R.; Lezoualc’h, F.; Sicsic, S.; et al. Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J. Med. Chem. 2005, 48, 6220–6228. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Mauri, M.; Capra, V.; Raimondi, F.; Guzzi, F.; Ambrosio, M.; Rovati, G.E.; Parenti, M. Light on the structure of thromboxane A2 receptor heterodimers. Cell. Mol. Life Sci. 2011, 68, 3109–3120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Jacobson, K.A. Computational prediction of homodimerization of the A3 adenosine receptor. J. Mol. Graph. Model. 2006, 25, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; de Benedetti, P.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kai, M.; Jin, L.; Wang, R. Computational study of the heterodimerization between mu and delta receptors. J. Comput. Aided Mol. Des. 2009, 23, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Guadix, A.E.; Costantino, G. Molecular dynamics simulation of the heterodimeric mGluR2/5HT(2A) complex. An atomistic resolution study of a potential new target in psychiatric conditions. J. Chem. Inf. Model. 2009, 49, 1602–1616. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Guixà-González, R.; Carrió, P.; Poso, A.; Dove, S.; Pastor, M.; Selent, J. Multi-Component Protein—Protein Docking Based Protocol with External Scoring for Modeling Dimers of G Protein-Coupled Receptors. Mol. Inform. 2015, 34, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Guixà-González, R.; Carrió, P.; Obiol-Pardo, C.; Pastor, M.; Selent, J. Fractal dimension as a measure of surface roughness of G protein-coupled receptors: Implications for structure and function. J. Mol. Model. 2012, 18, 4465–4475. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Jörg, M.; Capuano, B. The dopamine D2 receptor dimer and its interaction with homobivalent antagonists: Homology modeling, docking and molecular dynamics. J. Mol. Model. 2016, 22, 203. [Google Scholar] [CrossRef] [PubMed]
- Jorg, M.; Kaczor, A.A.; Mak, F.S.; Lee, K.C.K.; Poso, A.; Miller, N.D.; Scammells, P.J.; Capuano, B. Investigation of novel ropinirole analogues: Synthesis, pharmacological evaluation and computational analysis of dopamine D2 receptor functionalized congeners and homobivalent ligands. MedChemComm 2014, 5, 891–898. [Google Scholar] [CrossRef]
- Altuntaş, S.; Bozkus, Z.; Fraguela, B.B. GPU Accelerated Molecular Docking Simulation with Genetic Algorithms. In Applications of Evolutionary Computation; Squillero, G., Burelli, P., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2016; pp. 134–146. [Google Scholar]

{kind=link}
{kind=link}
| Software | Description |
|---|---|
| GRID | Developed in 1985 by Goodford. It aims to identify favorable interaction sites for probe molecules and water is one of available probes. GRID energy is computed by summing Lennard-Jones, electrostatic and hydrogen bonding interactions. |
| HINT | Developed by Kellogg and co-workers. Combination of Hydrophobic Interactions (HINT) and the geometric descriptor rank into a statistically robust method to detect water molecules for consideration in protein-ligand docking and structure-based drug discovery techniques. |
| Superstar | SuperStar is capable of combining IsoStar propensity maps in order to calculate hotspots in protein binding sites. |
| JAWM | Developed by Michel and co-workers. Just Add Water Molecules (JAWM) procedure applies a double decoupling technique to compare the energetic cost of removing a water molecule from the bulk and from a binding site. |
| WaterMap | WaterMap (Schrödinger) is based on explicit solvent molecular dynamics simulations followed by statistical thermodynamic analyses of water clusters based on inhomogeneous solvation theory. |
| Water PMF | Developed by Zheng and co-workers. Water PMF applies the potential of mean force on 3946 nonredundant high resolution crystal structures. |
| Water Flap | Fingerprint for Ligands and Proteins (FLAP), based on the GRID Molecular Interaction Fields enables the user to carry out molecular docking automatically calculating the probability of crystallographic or predicted water molecules to be retained upon ligand binding to the protein target. |
| Gold | In GOLD water molecules are allowed to spin and toggle on and off. Toggling a water molecule on introduces an entropic penalty to the scoring function which needs to be offset by forming hydrogen bonds to the protein and the ligand. If the hydrogen bonds formed by the water molecules does not offset the entropic penalty introduced by turning the water molecule on then the water molecule will be deselected for (turned off) during the genetic algorithm run. |
| DOCK | Flexible-receptor docking method considering displaced water and retained water states with variable water position. |
| FlexX | Algorithmic approach, called the particle concept, for integrating the placement of single water molecules in the docking algorithm of FLEXX. |
| AutoDock | Hydration force field accounting for the entropic and enthalpic contributions of discrete waters to ligand binding. |
| Glide | Statistics about the number of hydrogen bonds formed by polar and apolar groups. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules 2017, 22, 340. https://doi.org/10.3390/molecules22020340
Bartuzi D, Kaczor AA, Targowska-Duda KM, Matosiuk D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules. 2017; 22(2):340. https://doi.org/10.3390/molecules22020340
Chicago/Turabian StyleBartuzi, Damian, Agnieszka A. Kaczor, Katarzyna M. Targowska-Duda, and Dariusz Matosiuk. 2017. "Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors" Molecules 22, no. 2: 340. https://doi.org/10.3390/molecules22020340
APA StyleBartuzi, D., Kaczor, A. A., Targowska-Duda, K. M., & Matosiuk, D. (2017). Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules, 22(2), 340. https://doi.org/10.3390/molecules22020340