
Three Chalconoids and a Pterocarpene from the Roots of Tephrosia aequilata

 , , and
, , and
Abstract
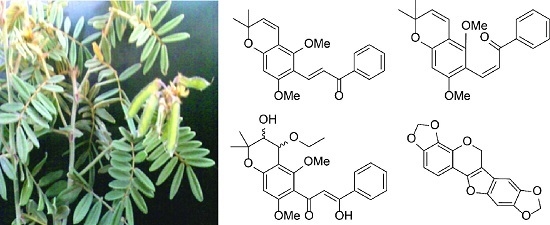
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Plasmodium Falciparum Culture
3.5. Plasmodium falciparum Growth Inhibition Assay
3.6. Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tarus, P.K.; Machocho, A.K.; Lang’at-Thoruwa, C.C.; Chhabra, S.C. Flavonoids from Tephrosia aequilata. Phytochemistry 2002, 60, 375–379. [Google Scholar] [CrossRef]
- Roy, M.; Mitra, S.R.; Bhattacharyya, A.; Adityachaudhury, N. Candidone, a flavanone from Tephrosia Candida. Phytochemistry 1986, 25, 961–962. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, T.; Gao, C.; Cao, W.; Huang, R. Natural products from the genus Tephrosia. Molecules 2014, 19, 1432–1458. [Google Scholar] [CrossRef] [PubMed]
- Kokwaro, J.O. Medicinal Plants of East Africa, 3rd ed.; University of Nairobi Press: Nairobi, Kenya, 2009; pp. 185–187. [Google Scholar]
- Batista, R.; Silva, A.J., Jr.; de Oliveira, A.B. Plant-derived antimalarial agents: New leads and efficient phytomedicines. Part II. Non-alkaloidal natural products. Molecules 2009, 14, 3037–3072. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Garibay, F.; Quijano, L.; Calderon, J.S.; Morales, S.; Rios, T. Prenylflavanols from Tephrosia quercetorum. Phytochemistry 1988, 27, 2971–2973. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Wang, Y.-S.; Lin, Y.-L.; Munakata, K.; Ohta, K. Obovatin, obovatin methyl ether and obovatachalcone, new piscicidal flavonoids from Tephrosia obovata. Agric. Biol. Chem. 1978, 42, 2431–2432. [Google Scholar] [CrossRef]
- Camele, G.; Monache, F.; Delle Monache, G.; Bettolo, G.M. Three new flavonoids from Tephrosia Praecans. Phytochemistry 1980, 19, 707–709. [Google Scholar] [CrossRef]
- Dagne, E.; Yenesew, A.; Gray, A.I.; Waterman, P.G. Praecansone A: Evidence for the existence of 8,9-(E) and 8,9-(Z) isomers in extracts from Tephrosia pumila. Bull. Chem. Soc. Ethiopia 1990, 4, 141–145. [Google Scholar]
- Parmar, V.S.; Jain, R.; Gupta, S.R.; Boll, P.M.; Mikkelsen, J.M. Phytochemical investigation of Tephrosia candida: HPLC separation of tephrosin and 12a-hydroxyrotenone. J. Nat. Prod. 1988, 51, 185. [Google Scholar] [CrossRef]
- Khalid, S.A.; Waterman, P.G. 8-C-prenylflavonoids from the seed of Tephrosia bracteolata. Phytochemistry 1981, 20, 1719–1720. [Google Scholar] [CrossRef]
- Sabry, O.; El Sayed, A.M.; Ezzat, S.M.; Yousef, Z. Bioactive compounds from Acokanthera oblongifolia. World J. Pharm. Pharm. Sci. 2016, 28, 222–232. [Google Scholar]
- Peng, J.; Risinger, A.L.; Da, C.; Fest, G.A.; Kellogg, G.E.; Mooberry, S.L. Structure-activity relationships of retro-dihydrochalcones isolated from tacca sp. J. Nat. Prod. 2013, 76, 2189–2194. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, K.; Demizu, S.; Hiraga, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tamura, Y.; Okada, K.; Kinoshita, T. Two prenylated retrochalcones from Glycyrrhiza inflata. Phytochemistry 1992, 31, 3229–3232. [Google Scholar] [CrossRef]
- Saitoh, T.; Shibata, S. New type chalcones from licorice root. Tetrahedron Lett. 1975, 50, 4461–4462. [Google Scholar] [CrossRef]
- Ayabe, S.-I.; Furuya, T. Biosynthesis of a retrochalcone, echinatin: A feeding study with advanced precursors. Tetrahedron Lett. 1981, 22, 2097–2098. [Google Scholar] [CrossRef]
- Karé, M.; Koné, M.; Boulanger, A.; Niassy, B.; Lenouen, D.; Muckensturm, B.; Nongonierma, A. Isolation, identification et tests antibacteriens des chalcones et rotenoïdes de Tephrosia deflexa baker. J. Soc. Ouest-Afr. Chim. 2006, 22, 41. [Google Scholar]
- Colegate, S.M.; Din, L.B.; Ghisalberti, E.L.; Latiff, A. Tepanone, a retrochalcone from Ellipeia cuneifolia. Phytochemistry 1992, 31, 2123–2126. [Google Scholar] [CrossRef]
- Waldeck, D.H. Photoisomerization dynamics of stilbenes. Chem. Rev. 1991, 91, 415–436. [Google Scholar] [CrossRef]
- Dinda, B. Essentials of Pericyclic and Photochemical Reactions; Springer: Cham, Switzerland, 2017; pp. 215–218. [Google Scholar]
- Cis isomers can be more stable than trans. Chem. Eng. News 1963, 41, 38–40.
- Viehe, H.G. 1-Mono- und 1.4-dihalogen-1.3-butadiene mit bevorzugter cis-Struktur. Angew. Chem. 1963, 75, 793–794. [Google Scholar] [CrossRef]
- Horspool, W.M. Enone Rearrangements and Cycloadditions: Photoreactions of Cyclohexadienes, Quionones, Tropones, etc. In Photochemistry; The Chemical Society: London, UK, 1972; Volume 3, pp. 430–434. [Google Scholar]
- Parsons, I.C.; Gary, A.I.; Hartley, T.G.; Waterman, P.G. Acetophenones and coumarins from stem bark and leaves of Melzcope stzpztata. Phytochemistry 1994, 37, 565–570. [Google Scholar] [CrossRef]
- Oberholzer, M.E.; Rall, G.J.H.; Roux, D.G. New natural rotenoid and eyterocarpanoid analogues from Neorautanenia amboensis. Phytochemistry 1976, 15, 1283–1284. [Google Scholar] [CrossRef]
- Yenesew, A.; Derese, S.; Irungu, B.; Midiwo, J.O.; Waters, N.C.; Liyala, P.; Akala, H.; Heydenreich, M.; Peter, M.G. Flavonoids and isoflavonoids with antiplasmodial activities from the root bark of Erythrina abyssinica. Planta Med. 2003, 69, 658–661. [Google Scholar] [PubMed]
- Marco, M.; Deyou, T.; Gruhonjic, A.; Holleran, J.P.; Duffy, S.; Heydenreich, M.; Fitzpatrick, P.A.; Landberg, G.; Koch, A.; Derese, S.; et al. Pterocarpans and isoflavones from the root bark of Millettia micans and of Millettia dura. Adv. Drug Discov. Dev. 2016, 1–8. [Google Scholar]
- Duffy, S.; Avery, V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.C.; Bhattacharya, A.; Bhasin, V.K. Phytochemical licochalcone a enhances antimalarial activity of artemisinin in vitro. Acta Top. 2009, 109, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Muiva, L.M.; Yenesew, A.; Derese, S.; Heydenreich, M.; Peter, M.G.; Akala, H.M.; Eyase, F.; Waters, N.C.; Mutai, C.; Keriko, J.M.; et al. Antiplasmodial β-hydroxydihydrochalcone from seedpods of Tephrosia elata. Phytochem. Lett. 2009, 2, 99–102. [Google Scholar] [CrossRef]
- Yenesew, A.; Dagne, E.; Waterman, P.G. Flavonoids from the seed pods of Tephrosia Pumila. Phytochemistry 1989, 28, 1291–1292. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
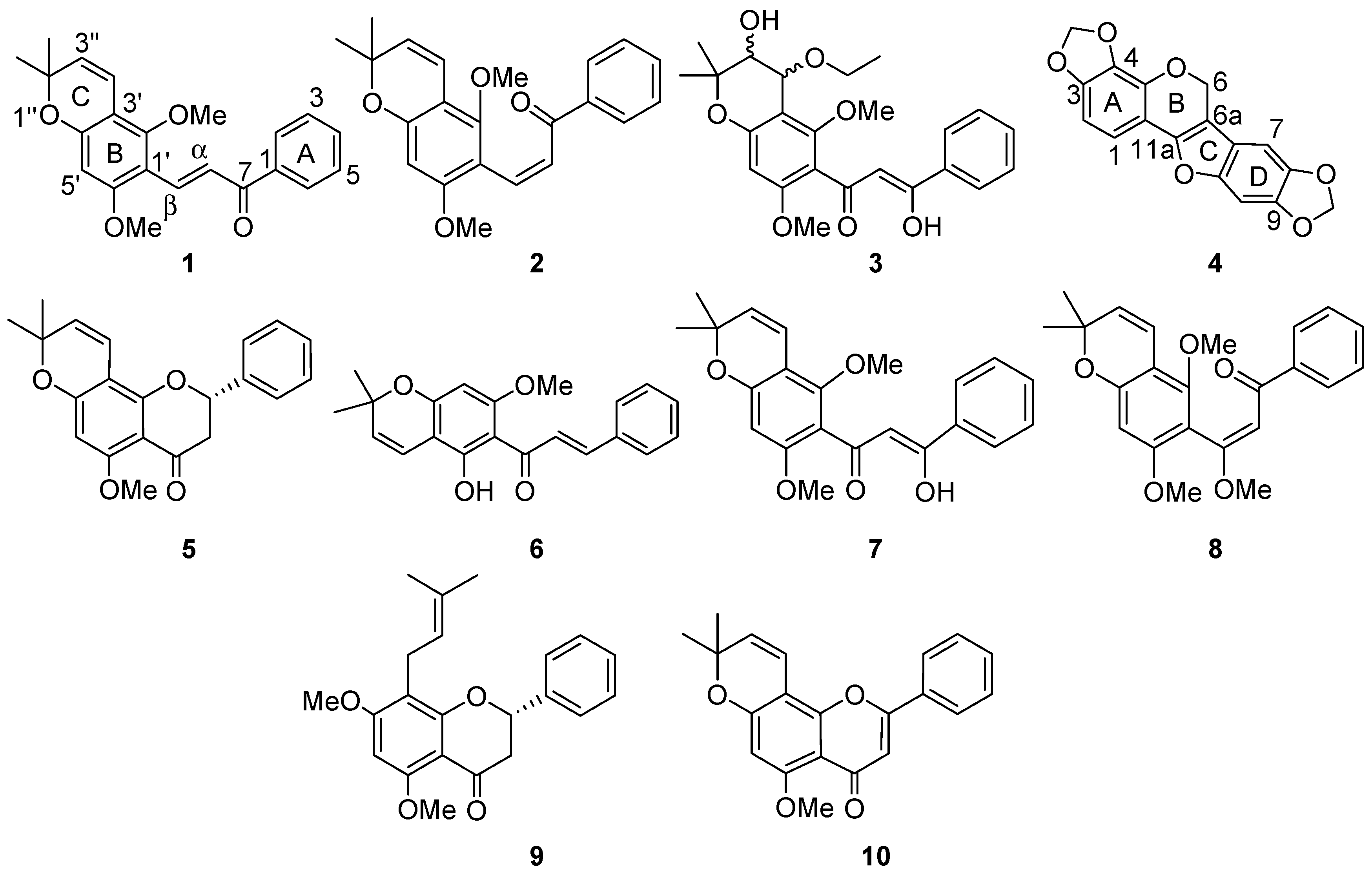

{kind=link}
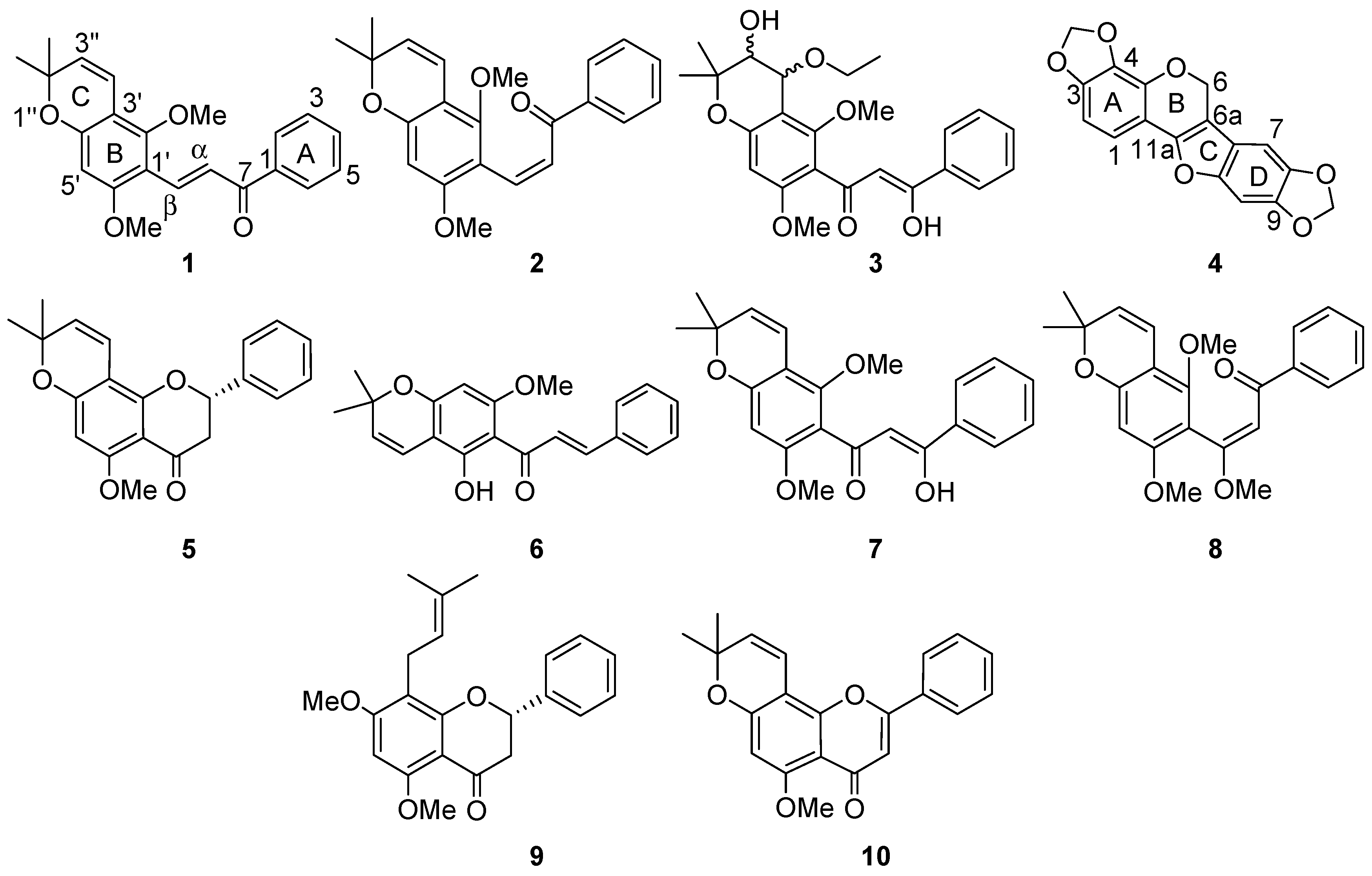{kind=link}
| Position | 1 | 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| δC | δH, m, (J in Hz) | HMBC | NOE | δC | δH, m, (J in Hz) | HMBC | NOE | |
| 1 | 139.0 | - | 137.6 | - | ||||
| 2/6 | 128.7 | 8.01 dd (7.7, 1.4) | C-3/5, C-4, C-7 | 128.7 | 7. 86 dd (6.9,1.4) | C-3/5, C-4, C-7 | H-α, H-3/5 | |
| 3/5 | 128.0 | 7.47 dd (7.7, 7.7) | C-1, C-2/6 | 128.0 | 7.34 dd (7.4,6.9) | C-1, C-2/6, | ||
| 4 | 132.2 | 7.53 tt (7.7, 1.4) | C-2/6, C-3/5 | 132.1 | 7.43 tt (7.4,1.4) | C-2/6, C-3/5 | ||
| 7 | 192.0 | 194.4 | ||||||
| α | 122.8 | 7.96 d (16.0) | C-7, C-1′ | 127.3 | 6.57 d (12.6) | C-1′, C-7 | H-β | |
| β | 136.1 | 8.15 d (16.0) | C-α, C-7, C-6′, C-2′ | OMe-6′ | 130.0 | 6.94 d (12.6) | C-1′, C-2′, C-6′, C-7 | H-α |
| 1′ | 110.5 | - | 111.4 | - | ||||
| 2′ | 161.2 | - | 155.0 | - | ||||
| 3′ | 108.2 | - | 107.7 | - | ||||
| 4′ | 157.0 | - | 155.2 | - | ||||
| 5′ | 96.4 | 6.25 s | C-1′, C-2′, C-3′, C-4′ | OMe-6′ | 96.0 | 6.01 s | C-1′, C-3′, C-4′, C-6′ | OMe-6′ |
| 6′ | 157.7 | - | 157.6 | - | ||||
| 2′′ | 77.0 | - | 76.6 | - | ||||
| 3′′ | 128.4 | 5.55 d (9.9) | C-2′′, C-3′, 2′′-Me2 | 2′′-Me2 | 127.3 | 5.44 d (10.0) | C-2′′, C-3′, 2′′-Me2 | H-4′′ |
| 4′′ | 116.5 | 6.55 d (9.9) | C-2′, C-3′, C-4′, C-2′′ | OMe-2′ | 116.8 | 6.41 d (10.0) | C-′ C-3′, C-4′, C-2′′ | H-3′′, OMe-6′ |
| 2′′-Me2 | 28.1 | 1.44 s | C-2′′, C-3′′ | 27.9 | 1.37 s | C-2′′, C-3′′ | ||
| OMe-2′ | 62.3 | 3.77 s | C-2′ | H-3′ | 54.9 | 3.47 s | C-2′ | |
| OMe-6′ | 55.9 | 3.88 s | C-6′ | H-4′′, H-α, H-β | 61.8 | 3.67 s | C-6′ | H-α, H-β |
| Position | δC | δH, m, J in Hz | HMBC | NOE |
|---|---|---|---|---|
| 1 | 135.0 | - | ||
| 2/6 | 127.0 | 7. 97 m | C-2/6, C4, C-7 | H-8 |
| 3/5 | 128.6 | 7.52 m | C-2/6, C-1 | |
| 4 | 132.2 | 7.59 m | C-1, C-3/5, C-2/6 | |
| 7 | 182.2 | |||
| 8α | 100.6 | 6.57 s | C-1, C-1′, C-7, C-9, | |
| 9β | 188.1 | |||
| 1′ | 114.6 | |||
| 2′ | 160.2 | |||
| 3′ | 107.2 | |||
| 4′ | 155.8 | |||
| 5′ | 95.9 | 6.27 s | C-1′,C-3′, C-4′, C-6′, C-9, C-4′′ | OMe-6′ |
| 6′ | 158.7 | |||
| 2′′ | 77.5 | |||
| 3′′ | 70.3 | 3.86 d (2.8) | C-4′′ | 2′′-Me2 |
| 4′′ | 72.8 | 4.40 d (2.8) | C-2′, C-3′, C-4′, C-2′′, C-3′′, C-2′′′ | |
| OCH2CH3 | 64.8 | 3.75 m | OCH2CH3, C-4′′ | |
| OCH2CH3 | 15.3 | 1.25 t (7.0, 14.0) | OCH2CH3 | |
| 2′′-Me2 | 24.8 23.3 | 1.47 s 1.49 s | C-2′′, C-3′′ | |
| OMe-2′ | 62.6 | 3.87 s | ||
| OMe-6′ | 55.9 | 3.82 s | ||
| OH-9 | 16.37 |
| Position | δC | δH, m, (J in Hz) | HMBC |
|---|---|---|---|
| 1 | 113.3 | 6.98 d (8.0) | C-3, C-4a, C-11a |
| 2 | 101.8 | 6.50 d (8.0) | C-3, C-4, C-11b, |
| 3 | 149.5 | ||
| 4 | 134.5 | ||
| 4a | 137.0 | ||
| 6 | 65.8 | 5.54 s | C-4a, C-6a, C-6b, C-11a, C-11b (w) |
| 6a | 119.0 | ||
| 6b | 107.3 | ||
| 7 | 93.8 | 7.02 s | C-6a, C-8, C-9, C-10a |
| 8 | 144.9 | ||
| 9 | 146.1 | ||
| 10 | 97.3 | 6.76 s | C-6b, C-7 (w), C-8, C-9, C-10a |
| 10a | 150.3 | ||
| 11a | 147.0 | ||
| 11b | 112.5 | ||
| 3,4-OCH2O | 101.7 | 6.00 s | C-3, C-4 |
| 8,9-OCH2O | 101.8 | 5.97 s | C-8, C-9 |
| Samples | IC50, μM |
|---|---|
| Aequichalcone A (1) | 9.20 ± 1.42 |
| Aequichalcone B (2) | 9.75 ± 0.81 |
| Aequichalcone C (3) | 2.48 ± 0.22 |
| 3,4:8,9-Dimethylenedioxypterocarpene (4) | > 40 |
| Obovatachalcone (6) | 4.23 ± 1.11 |
| Praecansone B (7) | 4.14 ± 0.26 |
| Praecansone A (8) | 6.45 ± 0.48 |
| Isopongaflavone (10) | 8.19 ± 1.48 |
| Chloroquine | 0.0047 |
| Artesunate | 0.00067 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atilaw, Y.; Duffy, S.; Heydenreich, M.; Muiva-Mutisya, L.; Avery, V.M.; Erdélyi, M.; Yenesew, A. Three Chalconoids and a Pterocarpene from the Roots of Tephrosia aequilata. Molecules 2017, 22, 318. https://doi.org/10.3390/molecules22020318
Atilaw Y, Duffy S, Heydenreich M, Muiva-Mutisya L, Avery VM, Erdélyi M, Yenesew A. Three Chalconoids and a Pterocarpene from the Roots of Tephrosia aequilata. Molecules. 2017; 22(2):318. https://doi.org/10.3390/molecules22020318
Chicago/Turabian StyleAtilaw, Yoseph, Sandra Duffy, Matthias Heydenreich, Lois Muiva-Mutisya, Vicky M. Avery, Máté Erdélyi, and Abiy Yenesew. 2017. "Three Chalconoids and a Pterocarpene from the Roots of Tephrosia aequilata" Molecules 22, no. 2: 318. https://doi.org/10.3390/molecules22020318
APA StyleAtilaw, Y., Duffy, S., Heydenreich, M., Muiva-Mutisya, L., Avery, V. M., Erdélyi, M., & Yenesew, A. (2017). Three Chalconoids and a Pterocarpene from the Roots of Tephrosia aequilata. Molecules, 22(2), 318. https://doi.org/10.3390/molecules22020318