Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation

 , ,
, ,  and
and 
Abstract
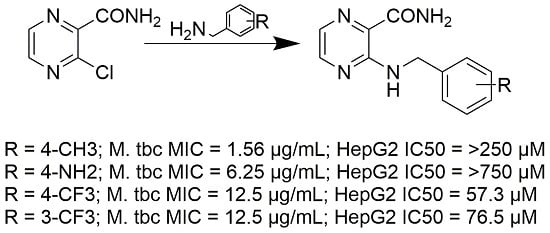
:
1. Introduction
2. Results and Discussion
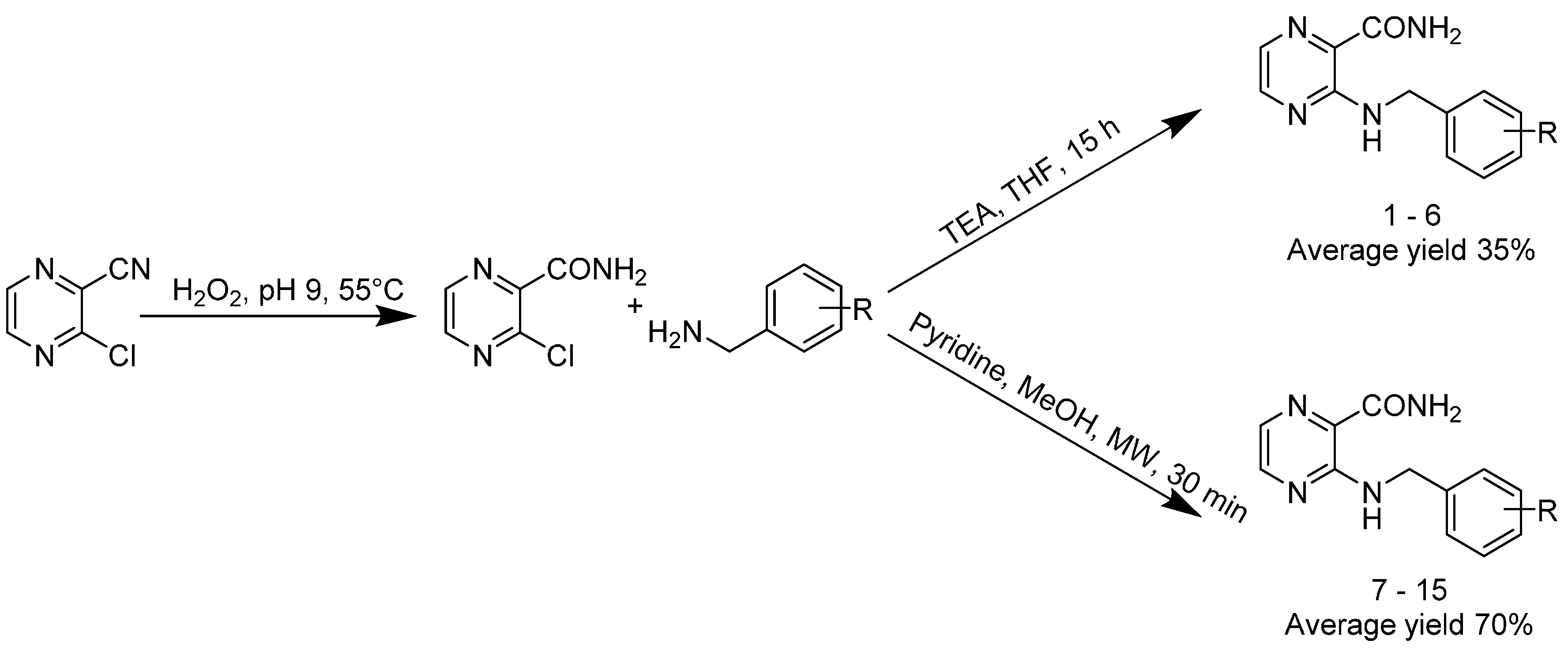
2.1. Chemistry
2.2. Biological Assays
2.2.1. Antimycobacterial Screening
2.2.2. Mycobacterium smegmatis Screening
2.2.3. Antibacterial and Antifungal Screening
2.2.4. Cytotoxicity Assays
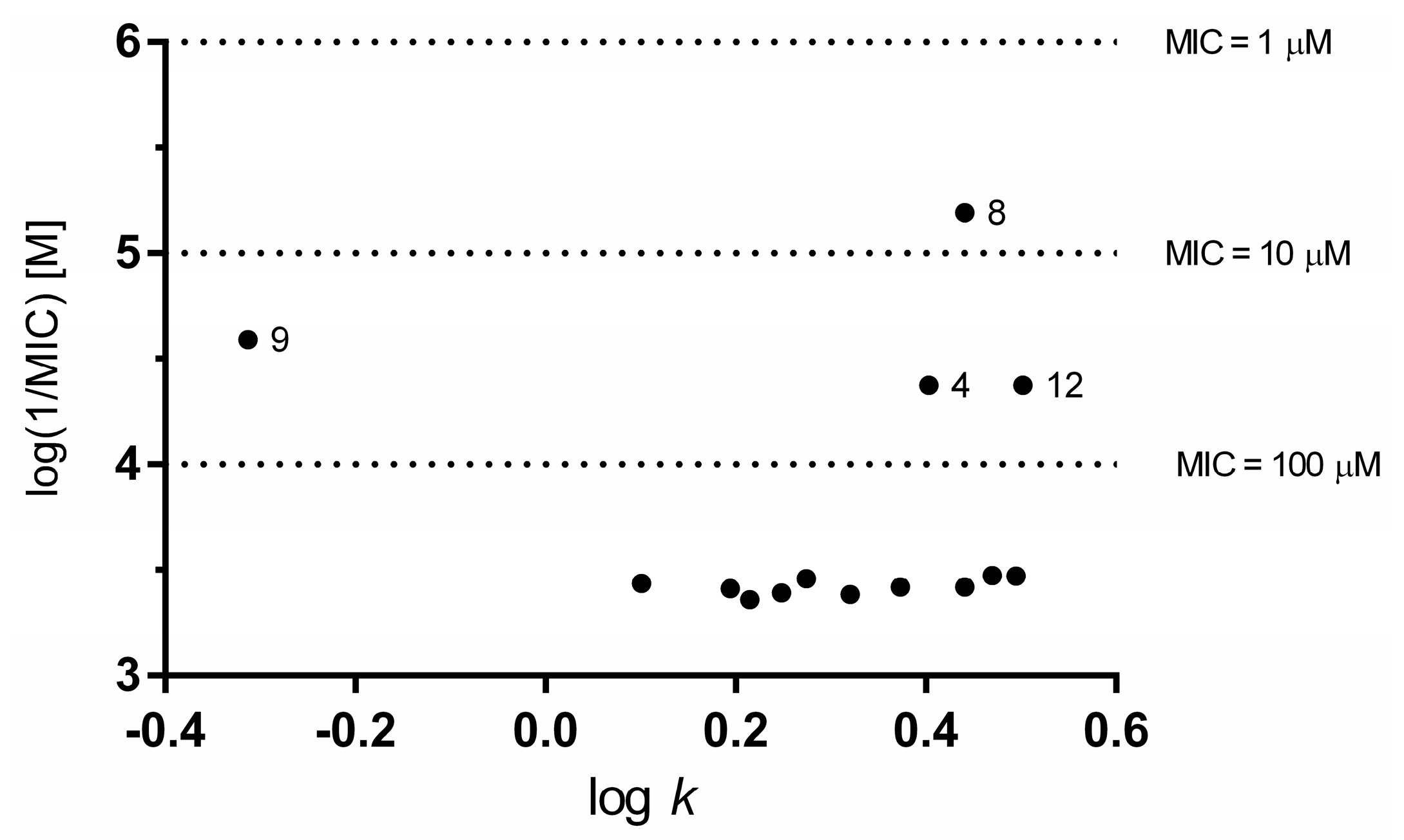
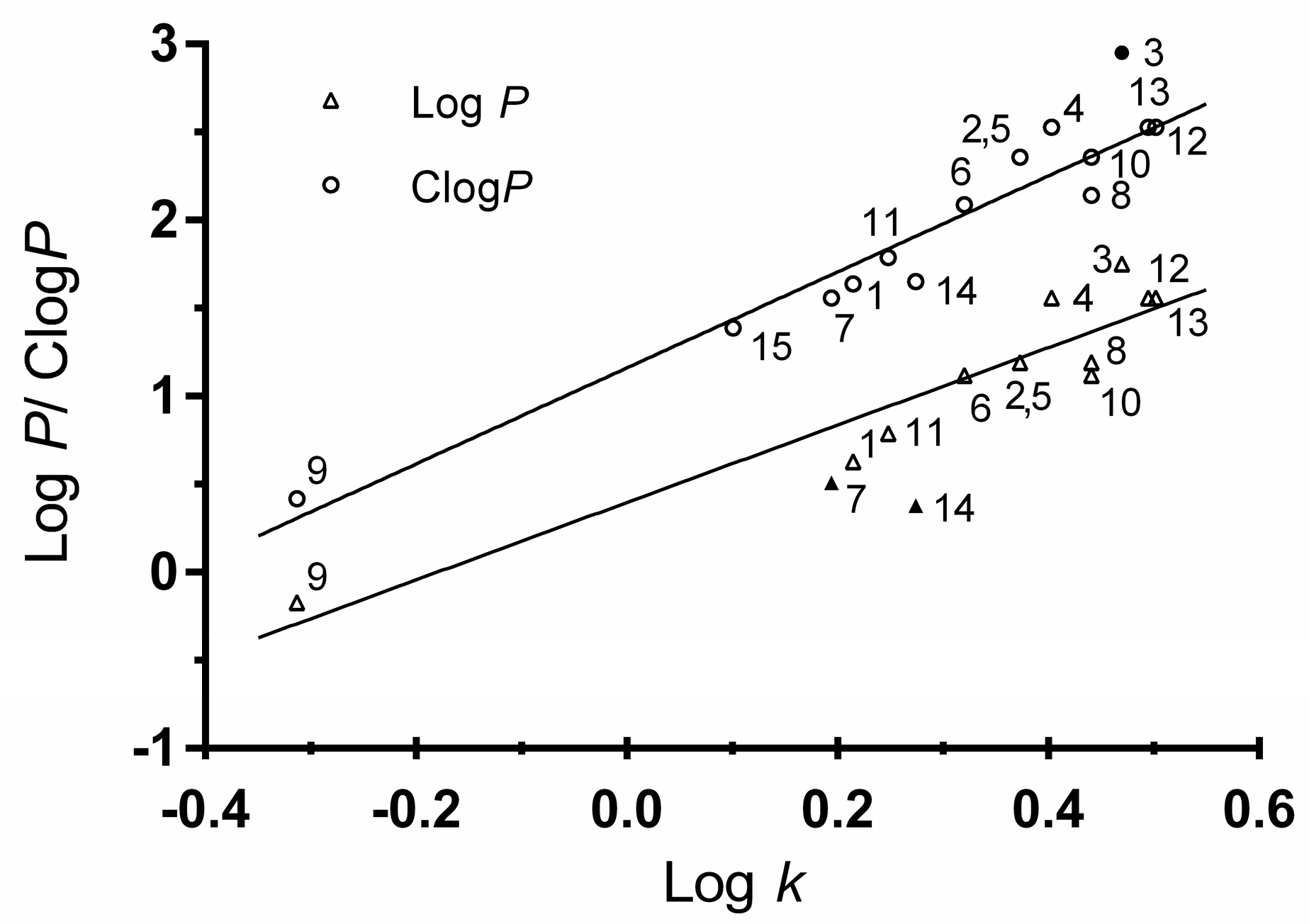
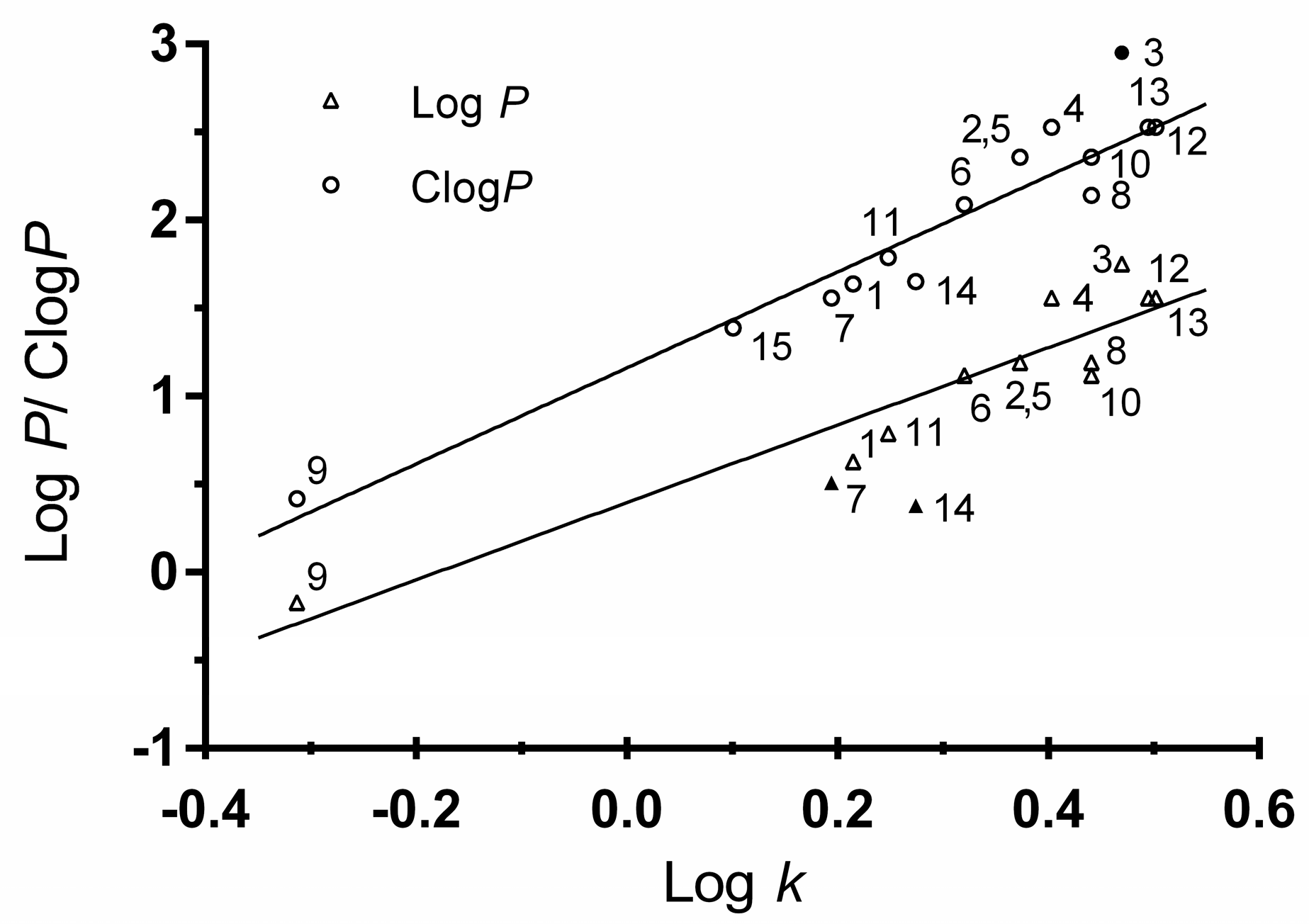
2.2.5. Lipophilicity Determination
R2 = 0.875, s = 0.194, F = 69.7, n = 12
R2 = 0.934, s = 0.159, F = 168.6, n = 14
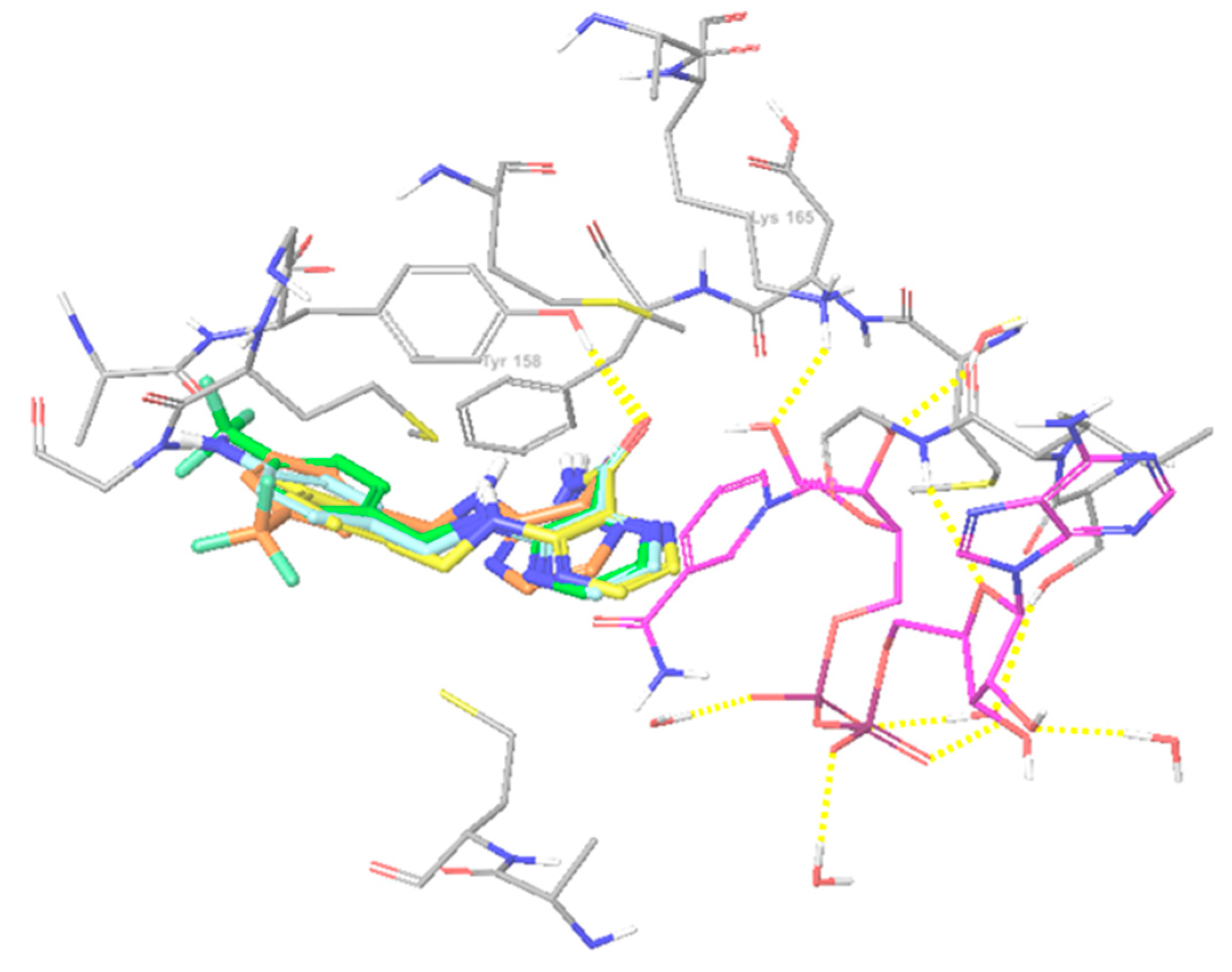
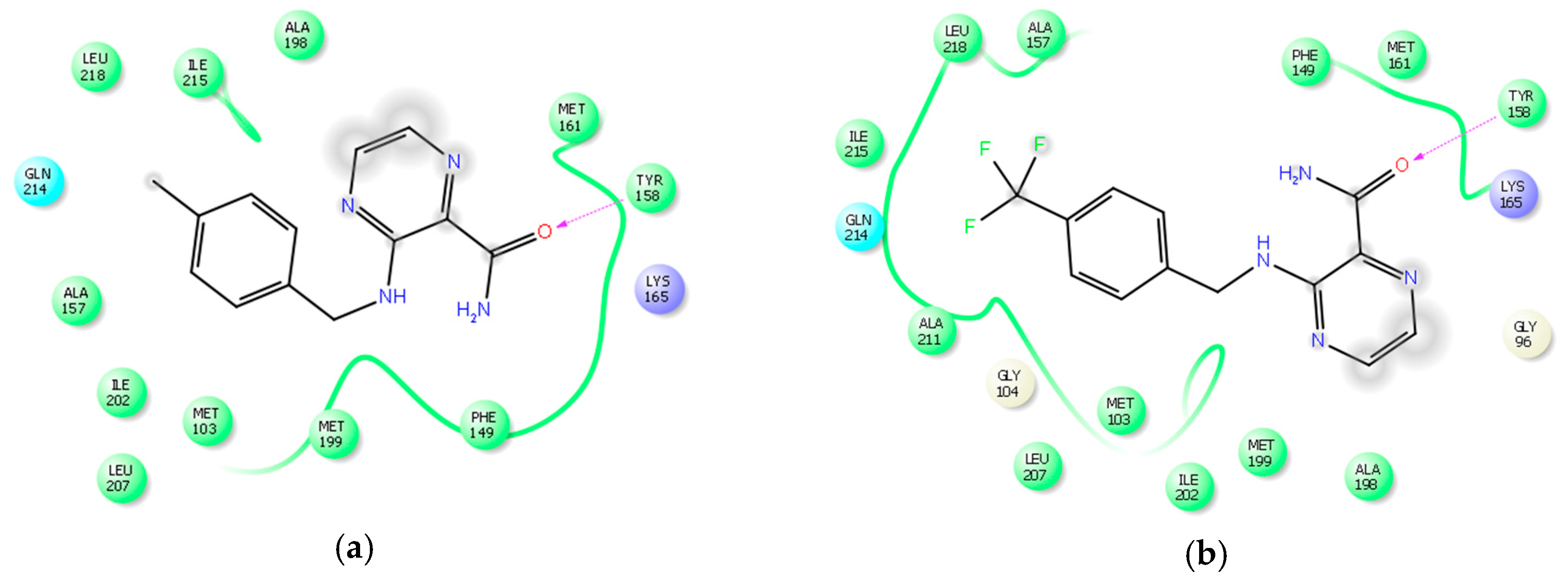
2.3. Computational Studies—Docking
3. Materials and Methods
3.1. General Methods
3.2. General Synthetic Procedure
3.3. Analytical Data of the Prepared Compounds
3.4. Biological Screening
3.4.1. Antimycobacterial Evaluation
3.4.2. Evaluation of Activity Against Mycobacterium smegmatis
3.4.3. Antibacterial and Antifungal Activity Evaluation
3.4.4. Cytotoxicity Determination
3.4.5. Lipophilicity Determination
3.5. Computational Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2016; WHO Press: Geneva, Switzerland, 2016; WHO/HTM/TB/2016.13; ISBN 978 92 4 156539 4. [Google Scholar]
- Palomino, J.C.; Martin, A. TMC207 becomes bedaquiline, a new anti-TB drug. Future Microbiol. 2013, 8, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar] [PubMed]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Peterson, N.D.; Rosen, B.C.; Dillon, N.A.; Baughn, A.D. Uncoupling environmental pH and intrabacterial acidification from pyrazinamide susceptibility in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Sayahi, H.; Zimhony, O.; Jacobs, W.R.; Shekthman, A.; Welch, J.T. Pyrazinamide, but not pyrazinoic acid, is a competitive inhibitor of NADPH binding to Mycobacterium tuberculosis fatty acid synthase I. Bioorg. Med. Chem. Lett. 2011, 21, 4804–4807. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.L.; Zhang, X.L.; Jiang, X.; Yuan, H.M.; Lee, J.S.; Barry, C.E.; Wang, H.H.; Zhang, W.H.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chen, J.; Feng, J.; Cui, P.; Zhang, S.; Weng, X.; Zhang, W.; Zhang, Y. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2014, 3, e58. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Dolezal, M.; Svobodova, M.; Vejsova, M.; Kunes, J.; Kucera, R.; Jilek, P. Synthesis and antimycobacterial properties of N-substituted-6-amino-5-cyanopyrazine-2-carboxamides. Bioorg. Med. Chem. 2011, 19, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Jampilek, J.; Dobrovolny, L.; Svobodova, M.; Kunes, J.; Dolezal, M. Synthesis and antimycobacterial evaluation of N-substituted 3-aminopyrazine-2,5-dicarbonitriles. Bioorg. Med. Chem. Lett. 2012, 22, 1598–1601. [Google Scholar] [CrossRef] [PubMed]
- Servusova, B.; Paterova, P.; Mandikova, J.; Kubicek, V.; Kucera, R.; Kunes, J.; Dolezal, M.; Zitko, J. Alkylamino derivatives of pyrazinamide: Synthesis and antimycobacterial evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusova, B.; Janoutova, A.; Paterova, P.; Mandikova, J.; Garaj, V.; Vejsova, M.; Marek, J.; Dolezal, M. Synthesis and antimycobacterial evaluation of 5-alkylamino-N-phenylpyrazine-2-carboxamides. Bioorg. Med. Chem. 2015, 23, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Servusova-Vanaskova, B.; Jandourek, O.; Paterova, P.; Kordulakova, J.; Plevakova, M.; Kubicek, V.; Kucera, R.; Garaj, V.; Naesens, L.; Kunes, J.; et al. Alkylamino derivatives of N-benzylpyrazine-2-carboxamide: synthesis and antimycobacterial evaluation. MedChemComm 2015, 6, 1311–1317. [Google Scholar] [CrossRef]
- Servusova, B.; Eibinova, D.; Dolezal, M.; Kubicek, V.; Paterova, P.; Pesko, M.; Kralova, K. Substituted N-benzylpyrazine-2-carboxamides: Synthesis and biological evaluation. Molecules 2012, 17, 13183–13198. [Google Scholar] [CrossRef] [PubMed]
- Servusova, B.; Vobickova, J.; Paterova, P.; Kubicek, V.; Kunes, J.; Dolezal, M.; Zitko, J. Synthesis and antimycobacterial evaluation of N-substituted 5-chloropyrazine-2-carboxamides. Bioorg. Med. Chem. Lett. 2013, 23, 3589–3591. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Paterova, P.; Kubicek, V.; Mandikova, J.; Trejtnar, F.; Kunes, J.; Dolezal, M. Synthesis and antimycobacterial evaluation of pyrazinamide derivatives with benzylamino substitution. Bioorg. Med. Chem. Lett. 2013, 23, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Jandourek, O.; Dolezal, M.; Paterova, P.; Kubicek, V.; Pesko, M.; Kunes, J.; Coffey, A.; Guo, J.; Kralova, K. N-Substituted 5-amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-assisted synthesis and biological properties. Molecules 2014, 19, 651–671. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Pub.: Matthews, NC, USA, 2002. [Google Scholar]
- Jandourek, O.; Dolezal, M.; Klementova, M.; Kralova, K.; Pesko, M. Microwave assisted synthesis of new pyrazinamide analogues and their biological evaluation. In Proceedings of the 16th International Electronic Conference on Synthetic Organic Chemistry, 29 November 2012; MDPI: Basel, Switzerland, 2012; 16, p. 12. [Google Scholar]
- Kralova, K.; Pesko, M.; Paterova, P.; Kunes, J.; Tauchman, M.; Eibinova, D.; Carillo, C.; Zitko, J.; Dolezal, M. Substituted N-benzylpyrazine-2-carboxamides, Their Synthesis, Hydro-lipophilic Properties and Evaluation of Their Antimycobacterial and Photosynthesis-inhibiting Activities. In Proceedings of the 15th International Electronic Conference on Synthetic Organic Chemistry, 1–30 November 2011; MDPI: Basel, Switzerland, 2011; 15, p. 6. [Google Scholar]
- Kuo, M.R.; Morbidoni, M.R.; Alland, D.; Sneddon, S.F.; Gourlie, B.B.; Staveski, M.M.; Leonard, M.; Gregory, J.S.; Janjigian, A.D.; Yee, C.; et al. Targeting tuberculosis and malaria through inhibition of enoyl reductase compound activity and structural data. J. Biol. Chem. 2003, 278, 20851–20859. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, M.A.; Mansour, R.A.; Atatreh, N.; Bryce, R.A. Analysis of Enoyl-Acyl Carrier Protein Reductase Structure and Interactions Yields an Efficient Virtual Screening Approach and Suggests a Potential Allosteric Site. Chem. Biol. Drug Des. 2016, 87, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Bi, J.; Cai, Q.; Liao, X.; Li, W.; Guo, C.; Zhang, Q.; Lin, T.; Zhao, Y.; et al. Structural basis for targeting the ribosomal protein S1 of Mycobacterium tuberculosis by pyrazinamide. Mol. Microbiol. 2015, 95, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Petrella, S.; Gelus-Ziental, N.; Maudry, A.; Laurans, C.; Boudjelloul, R.; Sougakoff, W. Crystal structure of the pyrazinamidase of Mycobacterium tuberculosis: insights into natural and acquired resistance to pyrazinamide. PLoS ONE 2011, 6, e15785. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Kaur, J.; Grover, A. Molecular principles behind pyrazinamide resistance due to mutations in panD gene in Mycobacterium tuberculosis. Gene 2016, 581, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, L.; Connell, S.R.; Enderle, M.; Mills, D.J.; Vonck, J.; Grininger, M. Structure and conformational variability of the Mycobacterium tuberculosis fatty acid synthase multienzyme complex. Structure 2013, 21, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Dlabal, K.; Palat, K.; Lycka, A.; Odlerova, Z. Synthesis and 1H and 13C NMR spectra of sulfur derivatives of pyrazine derived from amidation product of 2-chloropyrazine and 6-chloro-2-pyrazinecarbonitrile. Tuberculostatic activity. Collect. Czech Chem. Commun. 1990, 55, 2493–2500. [Google Scholar] [CrossRef]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Satinsky, D.; Raich, I. Novel regioselective preparation of 5-chloropyrazine-2-carbonitrile from pyrazine-2-carboxamide and coupling study of substituted phenylsulfanylpyrazine-2-carboxylic acid derivatives. Curr. Org. Chem. 2005, 9, 49–60. [Google Scholar] [CrossRef]
- Jandourek, O.; Dolezal, M.; Kunes, J. Microwave-Assisted Synthesis of Pyrazinamide Derivatives: The Coupling Reaction of 3-Chloropyrazine-2-Carboxamide and Ring-Substituted Anilines. Curr. Org. Synth. 2015, 12, 189–196. [Google Scholar] [CrossRef]
- Ebeid, F.M.; Gheit, A.K.A.; Ezzo, E.M.; Ali, L.I. Decomposition of Triethylamine over Acid Catalysts. J. Chin. Chem. Soc.-Taip. 1982, 29, 125–129. [Google Scholar] [CrossRef]
- Dickinson, J.M.; Mitchison, D.A. Observations in vitro on the suitability of pyrazinamide for intermittent chemotherapy of tuberculosis. Tubercle 1970, 51, 389–396. [Google Scholar] [CrossRef]
- McDermott, W.; Tomsett, R. Activation of pyrazinamide and nicotinamide in acidic environments in vitro. Am. Rev. Tuberc. 1954, 70, 748–754. [Google Scholar] [PubMed]
- Heifets, L.; Lindholm-Levy, P. Pyrazinamide sterilizing activity in vitro against semidormant Mycobacterium tuberculosis bacterial populations. Am. Rev. Respir. Dis. 1992, 145, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Butler, W.R.; Kilburn, J.O. Improved method for testing susceptibility of Mycobacterium tuberculosis to pyrazinamide. J. Clin. Microbiol. 1982, 16, 1106–1109. [Google Scholar] [PubMed]
- Portaels, F.; Pattyn, S.R. Growth of mycobacteria in relation to the pH of the medium. Ann. Microbiol. 1982, 133, 213–221. [Google Scholar]
- Sula, J. WHO Co-operative studies on a simple culture technique for the isolation of mycobacteria: 1. Preparation, lyophilization and reconstitution of a simple semi-synthetic concentrated liquid medium; culture technique; growth pattern of different mycobacteria. Bull. Wld. Hlth. Org. 1963, 29, 589–606. [Google Scholar]
- Sula, J.; Sundaresan, T.K. WHO Co-operative studies on a simple culture technique for the isolation of mycobacteria: 2. Comparison of the efficacy of lyophilized liquid medium with that of Löwenstein-Jensen (LJ) medium. Bull. Wld. Hlth. Org. 1963, 29, 607–625. [Google Scholar]
- Alegre, O.S. Estudio comparativo de las posibilidades de los medios de Löwenstein-Jensen y liofilizado reconsititudo de Sula para el aislamiento del Myocbaterium tuberculosis. Bol. Oficina Sanit. Panam. 1967, 63, 13–16. [Google Scholar] [PubMed]
- Zitko, J.; Servusova, B.; Paterova, P.; Mandikova, J.; Kubicek, V.; Kucera, R.; Hrabcova, V.; Kunes, J.; Soukup, O.; Dolezal, M. Synthesis, antimycobacterial activity and in vitro cytotoxicity of 5-chloro-N-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807–14825. [Google Scholar] [CrossRef] [PubMed]
- Bielenica, A.; Stefańska, J.; Stępień, K.; Napiórkowska, A.; Augustynowicz-Kopeć, E.; Sanna, G.; Madeddu, S.; Boi, S.; Giliberti, G.; Wrzosek, M.; et al. Synthesis, cytotoxicity and antimicrobial activity of thiourea derivatives incorporating 3-(trifluoromethyl) phenyl moiety. Eur. J. Med. Chem. 2015, 101, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Kratky, M.; Vinsova, J.; Novotna, E.; Mandikova, J.; Trejtnar, F.; Stolarikova, J. Antibacterial activity of salicylanilide 4-(trifluoromethyl)-benzoates. Molecules 2013, 18, 3674–3688. [Google Scholar] [CrossRef] [PubMed]
- Keir, W.F.; MacLennan, A.H.; Wood, H.C. Amidinoacetamides in the synthesis of pyrazines and pteridines. J. Chem. Soc. Perk. T. 1 1977, 11, 1321–1325. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [PubMed]
- Jones, R.N.; Barry, A.L. Optimal dilution susceptibility testing conditions, recommendations for MIC interpretation, and quality control guidelines for the ampicillin-sulbactam combination. J. Clin. Microbiol. 1987, 25, 1920–1925. [Google Scholar] [PubMed]
- National Committee for Clinical Laboratory Standards. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Proposed Standard M 27-P; National Committee for Clinical Laboratory Standards: Villanova, PA, USA, 1992. [Google Scholar]
- Sample Availability: Samples of the compounds 1–15 are available from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | R | MW | LogP/ClogP | Logk | Minimum Inhibitory Concentrations | |||
|---|---|---|---|---|---|---|---|---|
| M. tbc H37Rv (µg/mL) | M. tbc H37Rv (µM) | M. smegmatis (µg/mL) | Bacteria (µM) | |||||
| 1 | H | 228.25 | 0.63/1.64 | 0.215 | >100 | n.a. | ≥500 | >500 |
| 2 | 3-Cl | 262.69 | 1.19/2.36 | 0.373 | >100 | n.a. | ≥500 | >500 |
| 3 | 3,4-Cl | 298.14 | 1.75/2.95 | 0.470 | >100 | n.a. | ≥500 | >500 |
| 4 | 3-CF3 | 297.14 | 1.56/2.53 | 0.403 | 12.5 | 42 | 250 | 31.25 SA a |
| 5 | 4-Cl | 262.69 | 1.19/2.36 | 0.373 | >100 | n.a. | 250 | 250 SA a |
| 6 | 2-CH3 | 242.28 | 1.12/2.09 | 0.320 | >100 | n.a. | ≥500 | >500 |
| 7 | 4-OCH3 | 258.25 | 0.51/1.56 | 0.194 | >100 | n.a. | ≥500 | >500 |
| 8 | 4-CH3 | 242.28 | 1.12/2.14 | 0.441 | 1.56 | 6 | ≥500 | >500 |
| 9 | 4-NH2 | 243.26 | −0.17/0.42 | −0.313 | 6.25 | 26 | 250 | >500 |
| 10 | 2-Cl | 262.69 | 1.19/2.36 | 0.441 | >100 | n.a. | ≥500 | >500 |
| 11 | 2-F | 246.24 | 0.79/1.79 | 0.248 | >100 | n.a. | ≥500 | 125 EF b |
| 12 | 4-CF3 | 296.25 | 1.56/2.53 | 0.502 | 12.5 | 42 | ≥500 | 125 EF b |
| 13 | 2-CF3 | 296.25 | 1.56/2.53 | 0.495 | >100 | n.a. | ≥500 | 125 EF b |
| 14 | 2,4-OCH3 | 288.30 | 0.38/1.65 | 0.274 | >100 | n.a. | ≥500 | 62.5 EF b |
| 15 | 3-NO2 | 273.25 | n.d./1.39 | 0.101 | >100 | n.a. | ≥500 | >500 |
| INH | - | 137.14 | - | - | 0.39 | 3 | 7.81–15.63 | - |
| PZA | - | 123.12 | - | - | 12.5 | 102 | ≥500 | - |
| RFM | - | 822.94 | - | - | n.d. | n.d. | 0.78–1.56 | - |
| CPX | - | 331.37 | - | - | n.d. | n.d. | 0.10–0.20 | - |
 | MIC against M. tbc H37Rv | |||||
|---|---|---|---|---|---|---|
| No. | R | R2 | R5 | R6 | µg/mL | µM |
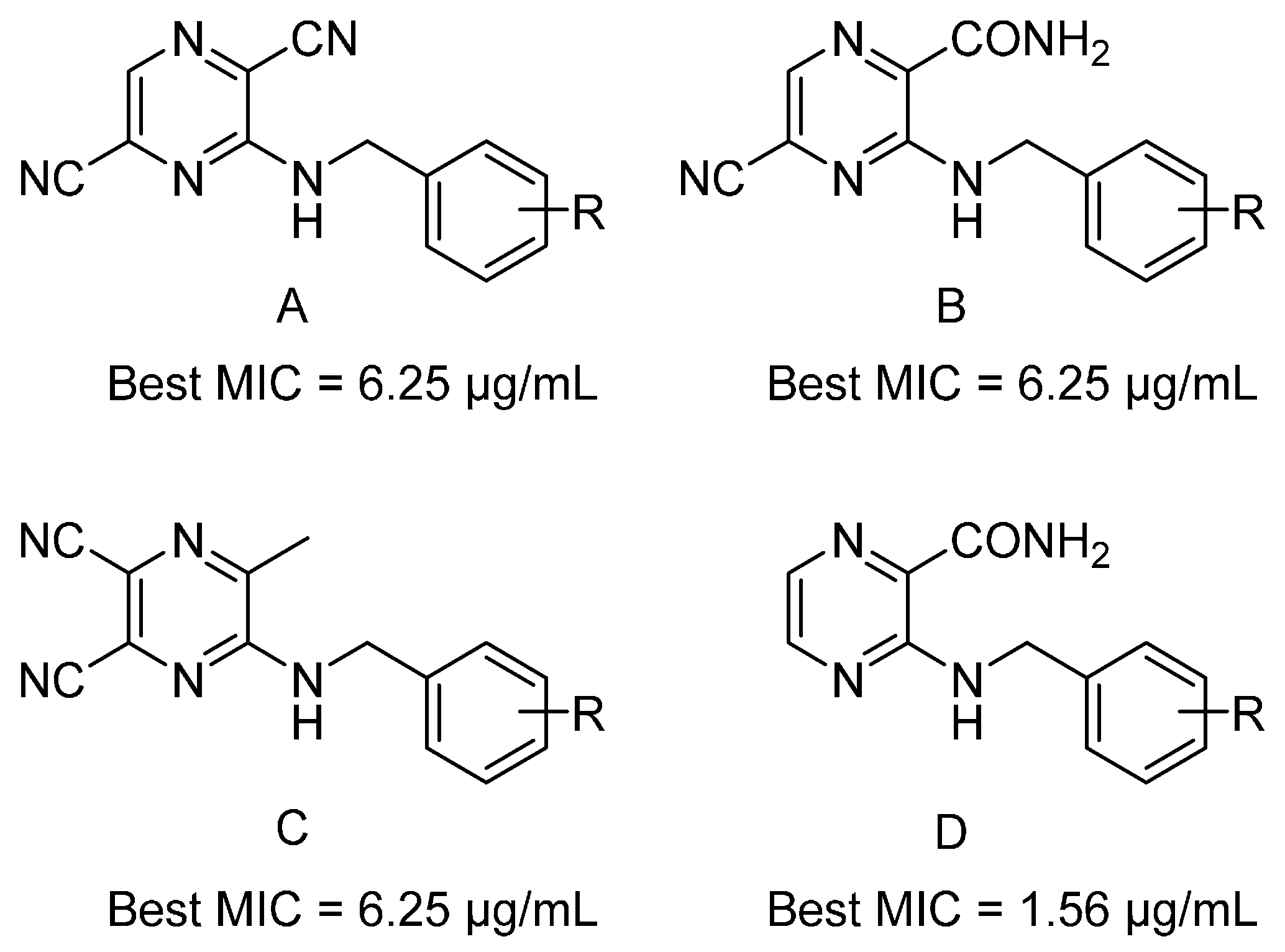
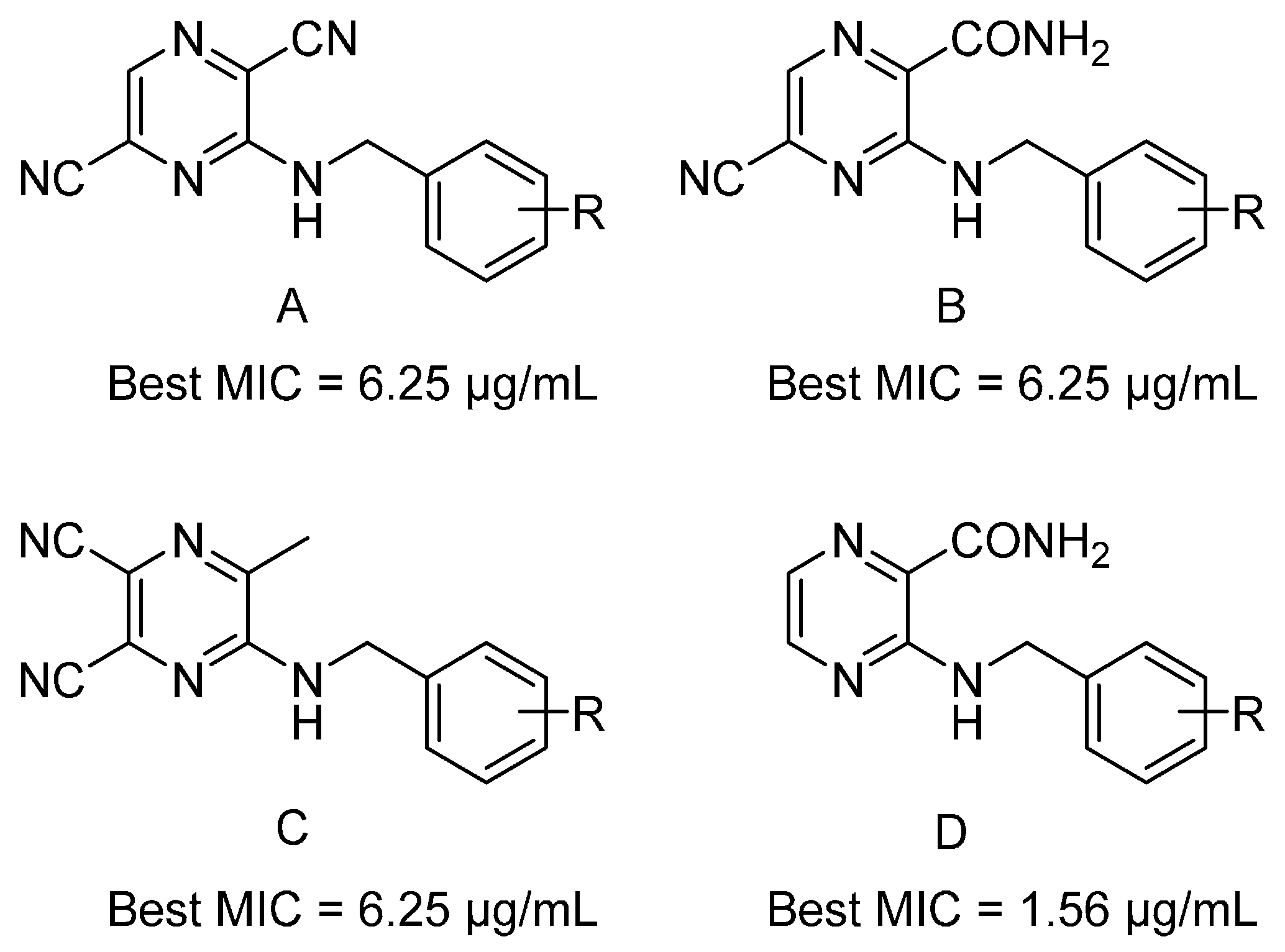
| 1 | H | CONH2 | H | H | >100 | - |
| 1-A | H | CONH2 | CN | H | >100 | - |
| 1-B | H | CN | CN | H | 12.5 | 78 |
| 1-C | H | CH3 | CN | CN | 25 | 100 |
| 4 | 3-CF3 | CONH2 | H | H | 12.5 | 42 |
| 4-A | 3-CF3 | CONH2 | CN | H | 25 | 78 |
| 4-B | 3-CF3 | CN | CN | H | >100 | - |
| 4-C | 3-CF3 | CH3 | CN | CN | 12.5 | 39 |
| 8 | 4-CH3 | CONH2 | H | H | 1.56 | 6 |
| 8-A | 4-CH3 | CONH2 | CN | H | 12.5 | 47 |
| 8-B | 4-CH3 | CN | CN | H | 6.25 | 25 |
| 8-C | 4-CH3 | CH3 | CN | CN | 25 | 95 |
| 9 | 4-NH2 | CONH2 | H | H | 6.25 | 26 |
| 9-A | 4-NH2 | CONH2 | CN | H | 12.5 | 47 |
| 9-B | 4-NH2 | CN | CN | H | 25 | 100 |
| 9-C | 4-NH2 | CH3 | CN | CN | 25 | 95 |
| INH | - | - | - | - | 0.39 | 3 |
| PZA | - | - | - | - | 12.5 | 102 |
| No. | R | Minimum inhibitory concentrations [µM] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M. tbc 9449/2007 | M. tbc 234/2005 | M. tbc PRAHA 1 | M. tbc PRAHA 4 | M. tbc PRAHA 131 | |||||||
| 14 days | 21 days | 14 days | 21 days | 14 days | 21 days | 14 days | 21 days | 14 days | 21 days | ||
| 8 | 4-CH3 | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 |
| 9 | 4-NH2 | 62.5 | 62.5 | 62.5 | 62.5 | 62.5 | 62.5 | 62.5 | 62.5 | 62.5 | 125 |
| INH | - | 62.5 | 62.5 | 15.63 | 15.63 | 15.63 | 15.63 | 15.63 | 31.25 | 15.63 | 31.25 |
| RFM | - | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 | >15.63 |
| Compound | IC50 (µM) | SI (IC50/MIC) |
|---|---|---|
| 4 | 76.5 | 1.8 |
| 8 | >250 | >41.7 |
| 9 | >750 | >28.8 |
| 12 | 57.3 | 1.4 |
| Compound | Affinity (kcal/mol) | MIC M. tbc H37Rv (µM) |
|---|---|---|
| 4TZK Ligand | −9.82 | - |
| 4 | −8.47 | 42 |
| 8 | −7.40 | 6 |
| 9 | −7.82 | 26 |
| 12 | −8.49 | 42 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandourek, O.; Tauchman, M.; Paterova, P.; Konecna, K.; Navratilova, L.; Kubicek, V.; Holas, O.; Zitko, J.; Dolezal, M. Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation. Molecules 2017, 22, 223. https://doi.org/10.3390/molecules22020223
Jandourek O, Tauchman M, Paterova P, Konecna K, Navratilova L, Kubicek V, Holas O, Zitko J, Dolezal M. Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation. Molecules. 2017; 22(2):223. https://doi.org/10.3390/molecules22020223
Chicago/Turabian StyleJandourek, Ondrej, Marek Tauchman, Pavla Paterova, Klara Konecna, Lucie Navratilova, Vladimir Kubicek, Ondrej Holas, Jan Zitko, and Martin Dolezal. 2017. "Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation" Molecules 22, no. 2: 223. https://doi.org/10.3390/molecules22020223
APA StyleJandourek, O., Tauchman, M., Paterova, P., Konecna, K., Navratilova, L., Kubicek, V., Holas, O., Zitko, J., & Dolezal, M. (2017). Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation. Molecules, 22(2), 223. https://doi.org/10.3390/molecules22020223