The Transcription Profile Unveils the Cardioprotective Effect of Aspalathin against Lipid Toxicity in an In Vitro H9c2 Model

 ,
,  ,
,  ,
,
Abstract
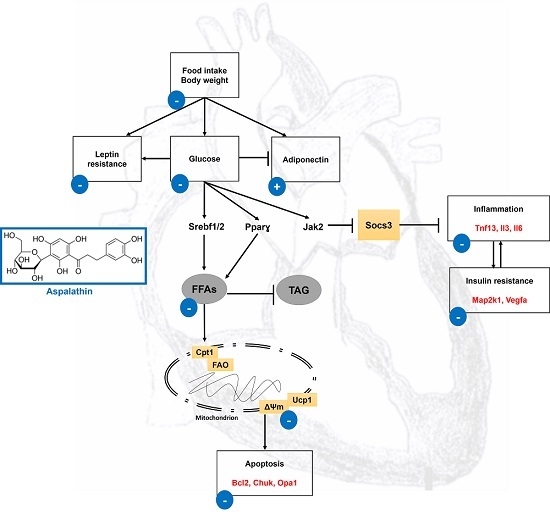
:
1. Introduction
2. Results and Discussion
2.1. Effect of Aspalathin on Lipid Profiles
2.2. In Vitro Effects of Aspalathin
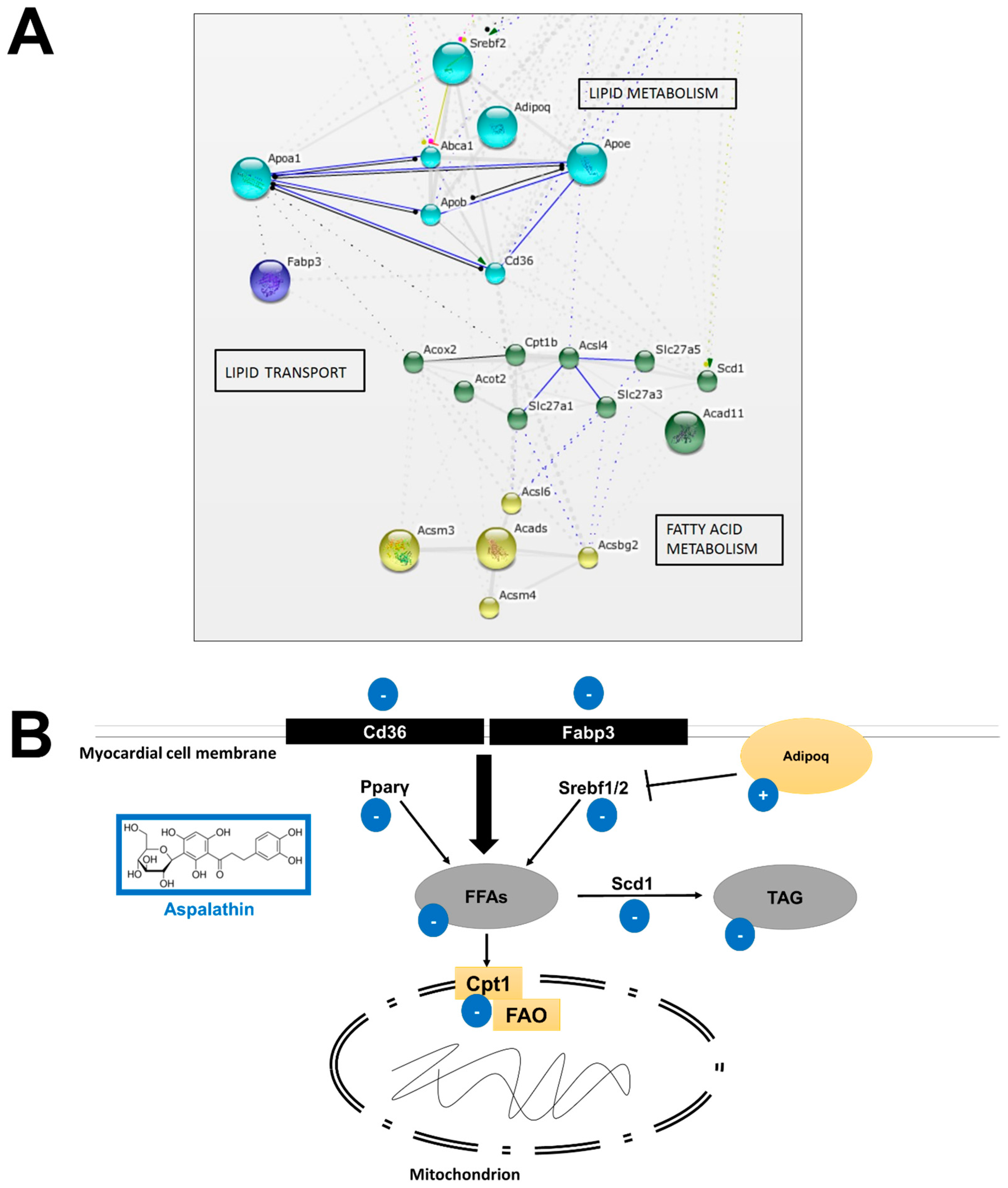
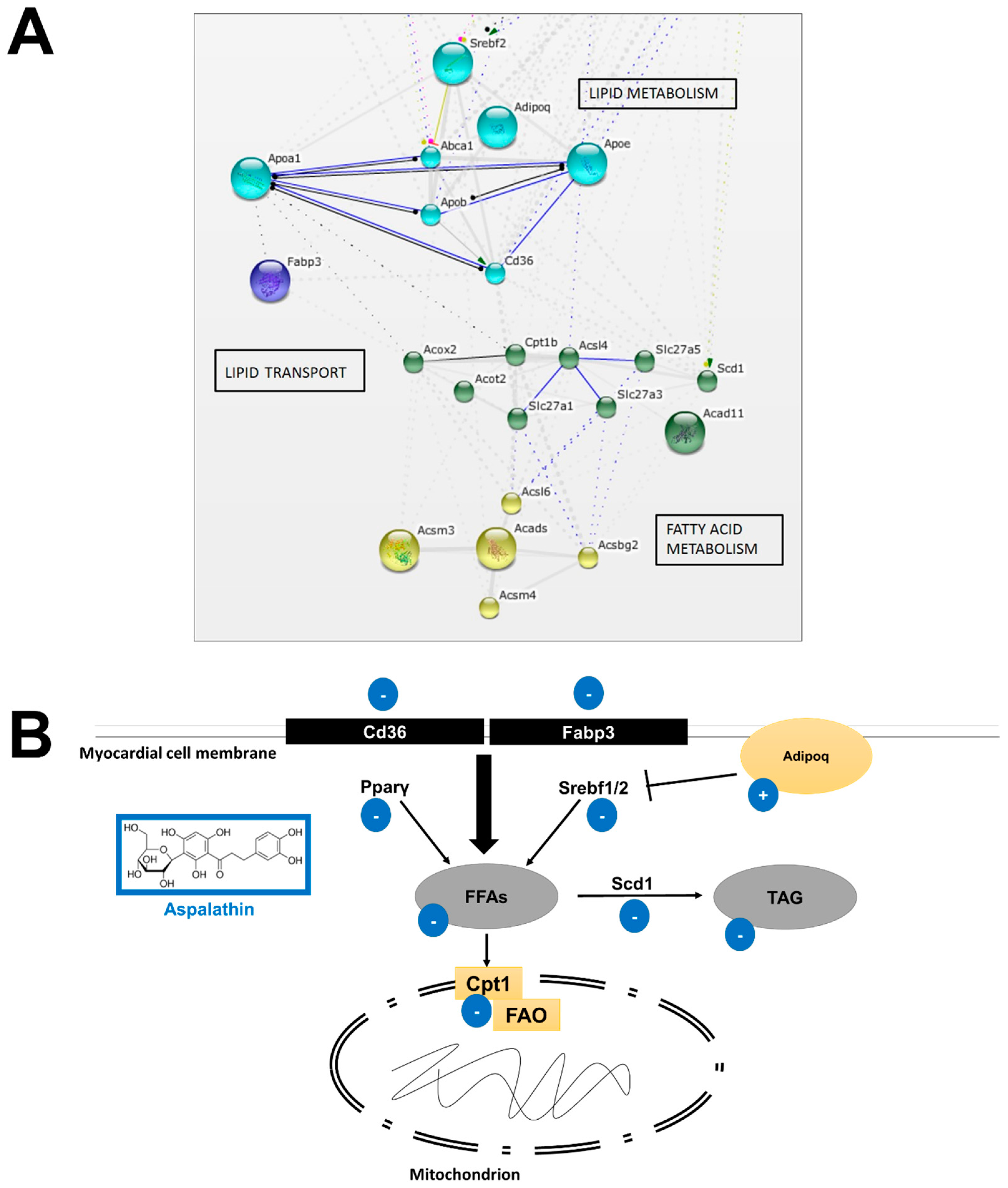
2.2.1. In Vitro Effect of Aspalathin on Fatty Acid and Lipid Metabolism
Increased β-oxidation
Increased Supply of Long-Chain Fatty Acids
Altered Lipid Metabolism and Increased Cholesterol Flux
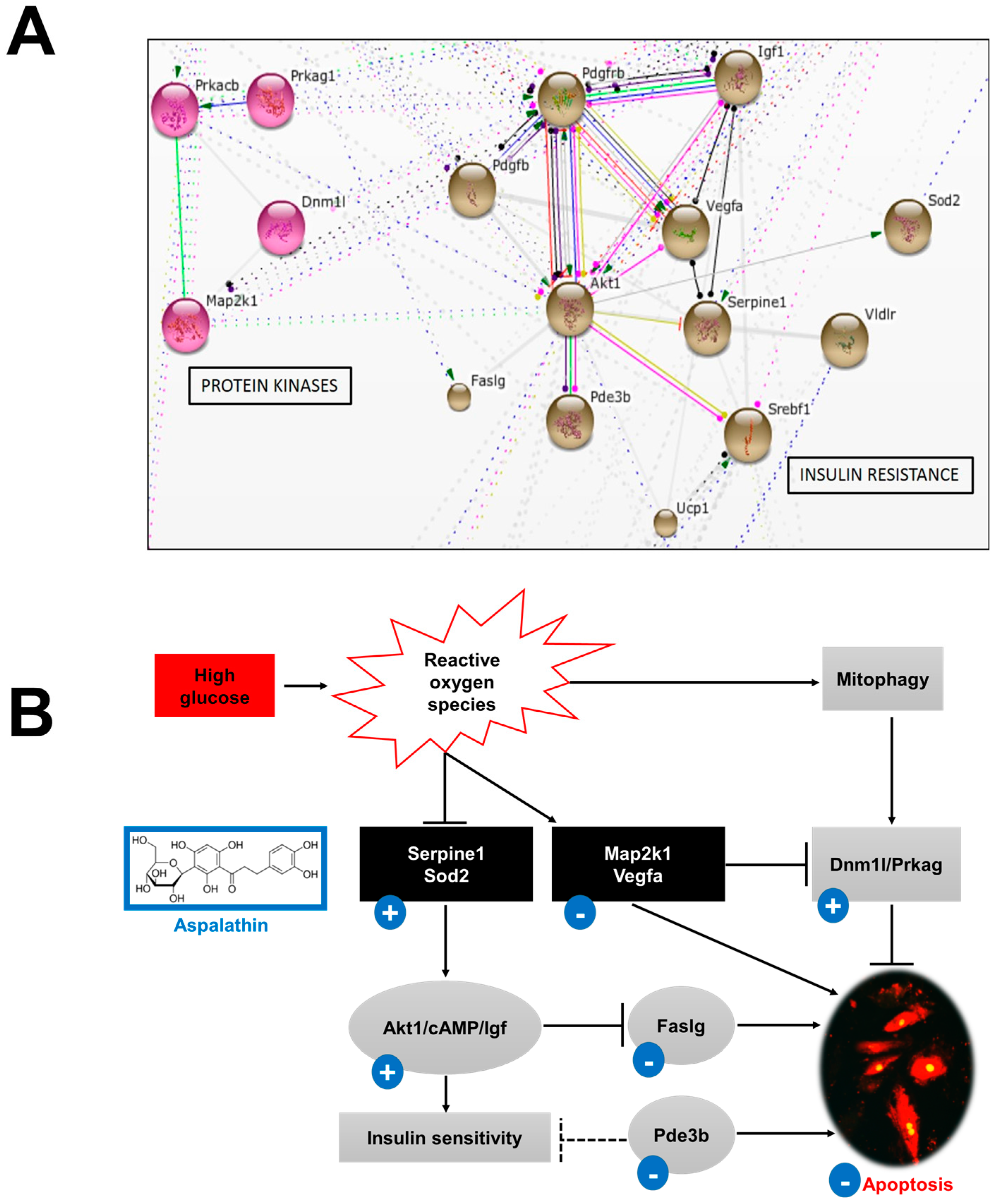
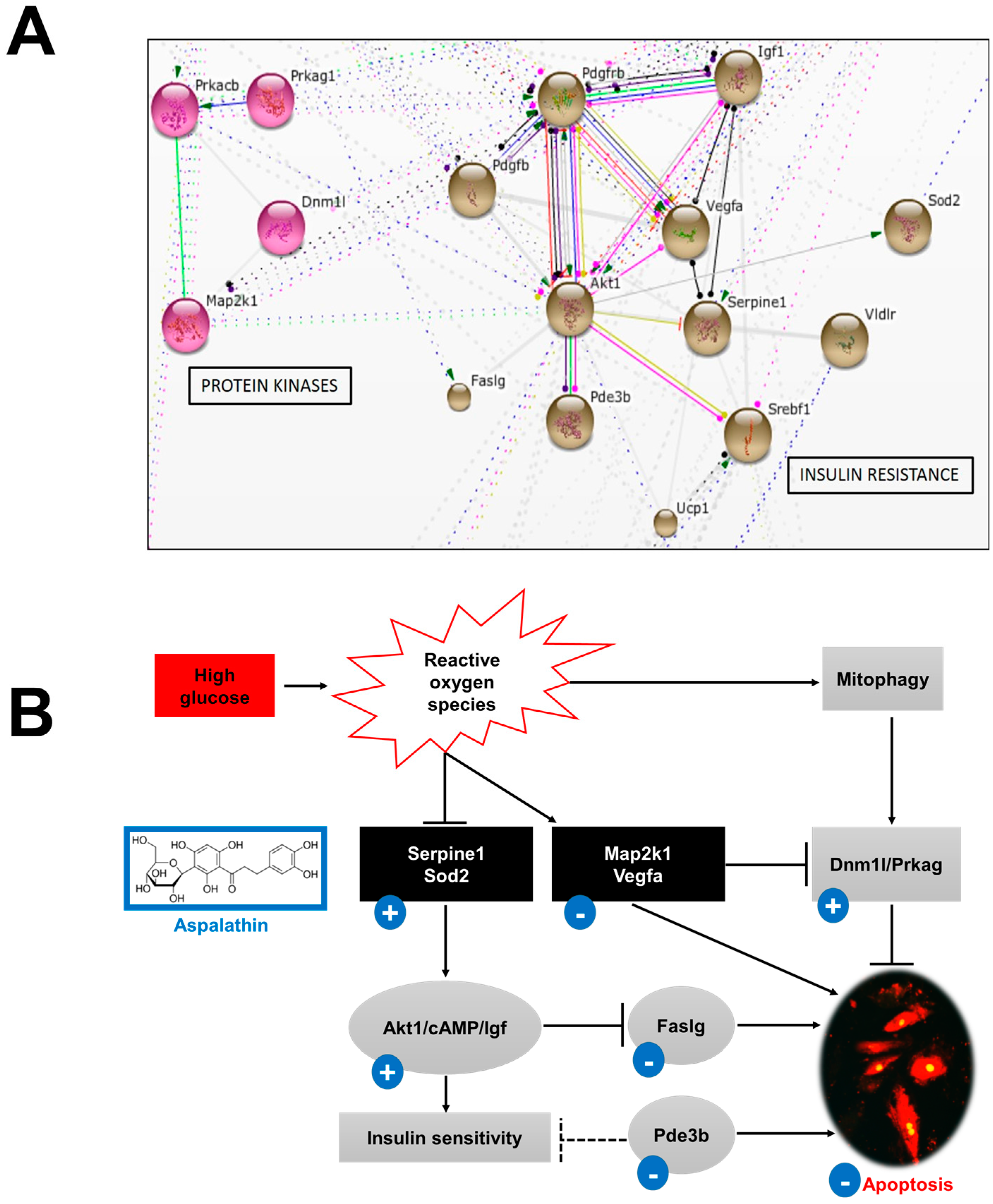
2.2.2. In Vitro Effect of Aspalathin on the Development of Insulin Resistance
Insulin Signaling and Effect on Myocardium
Protein Kinases and Mitochondrial Function
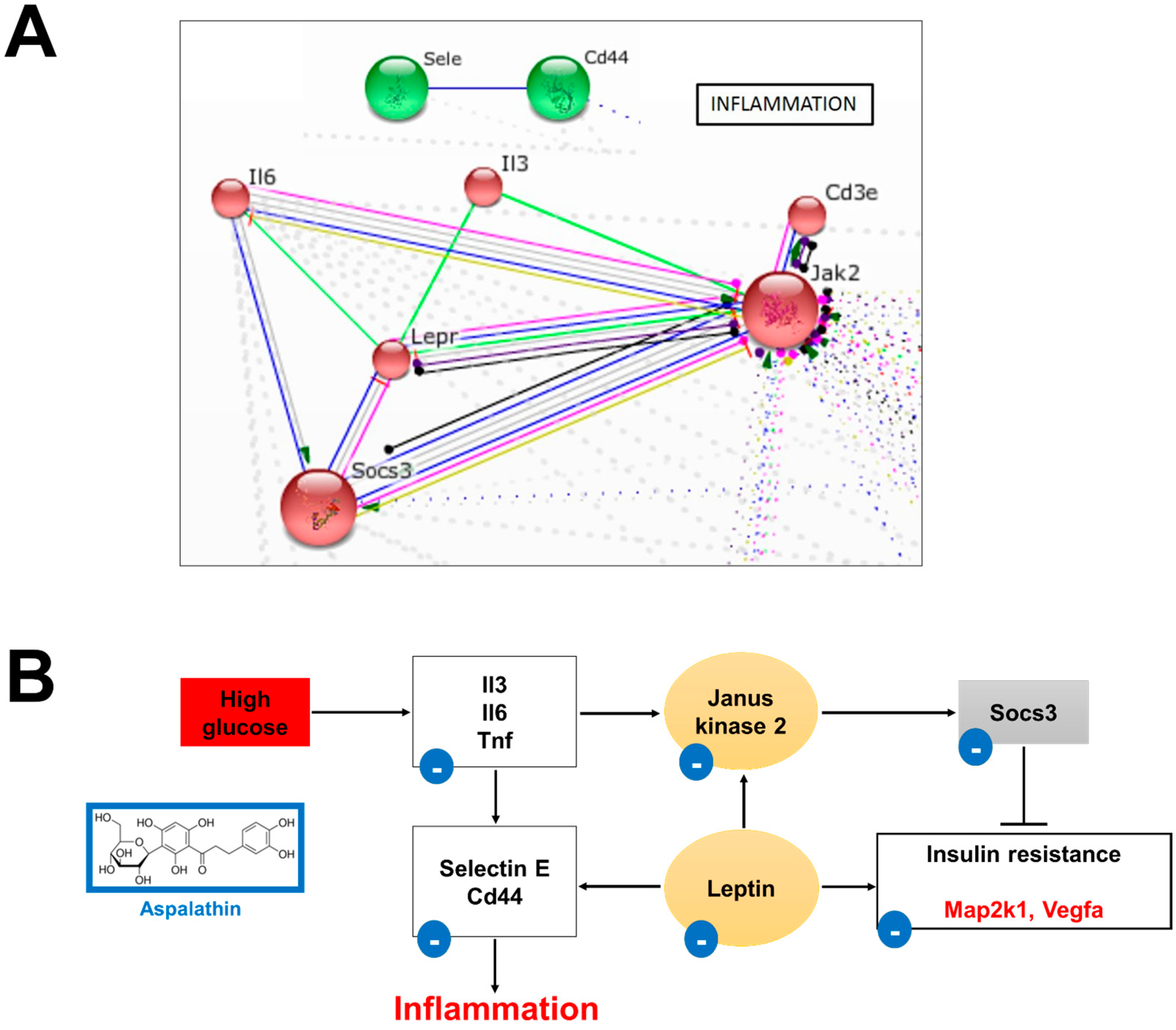
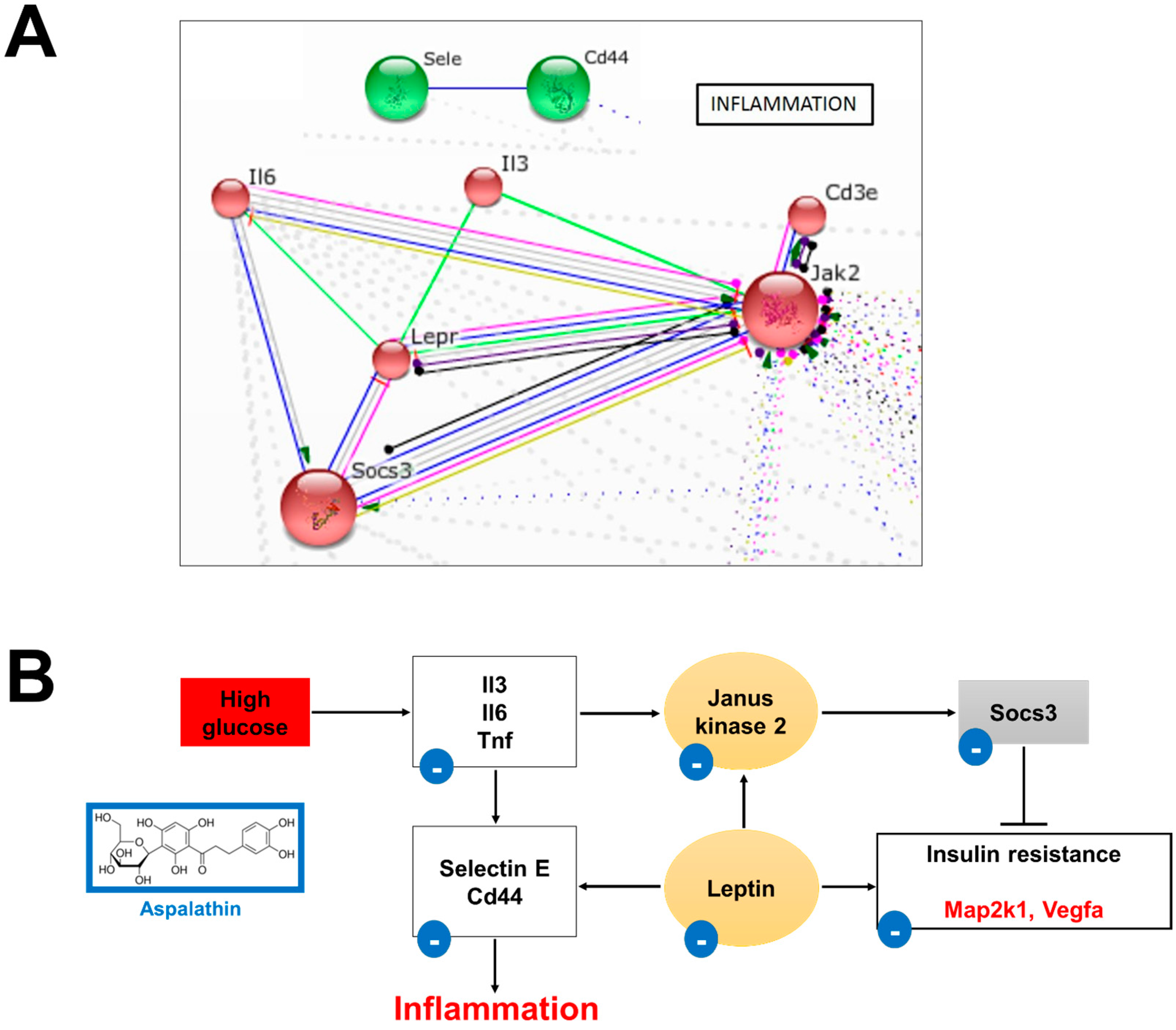
2.2.3. In Vitro Effect of Aspalathin on Inflammation
Leptin Signaling
Cytokine Signaling
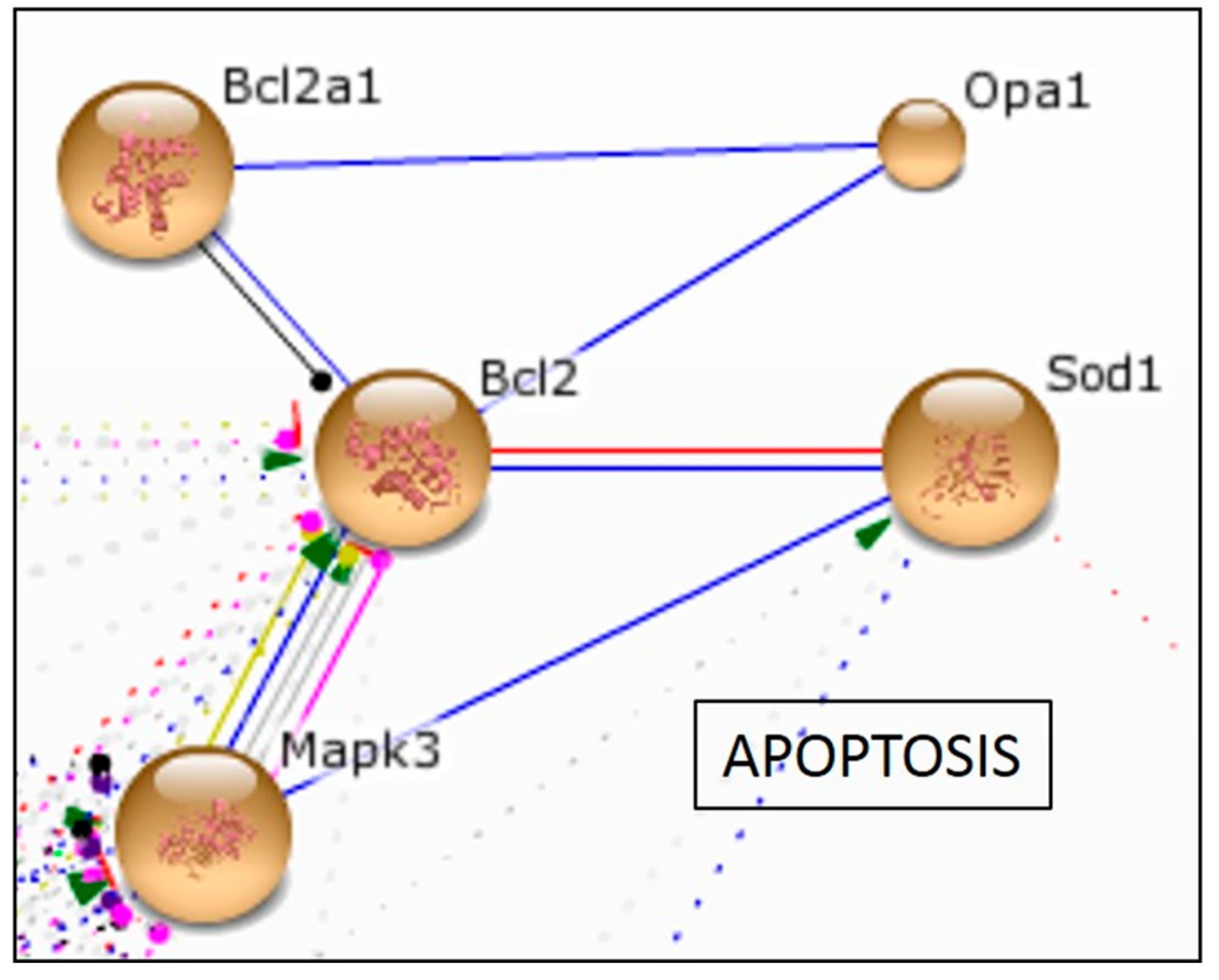
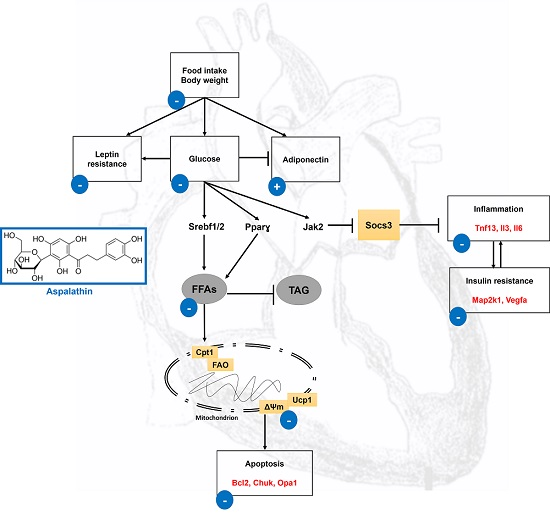
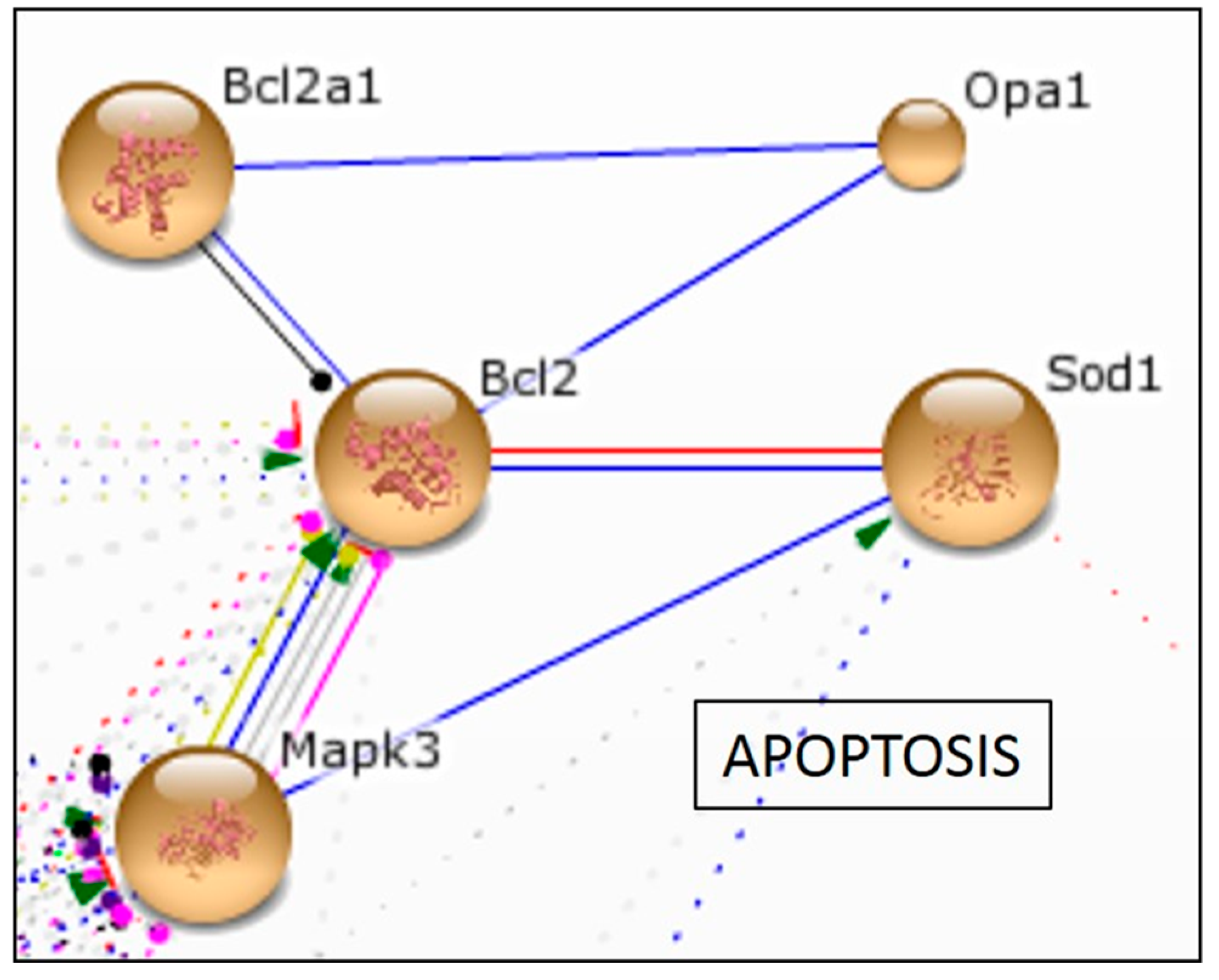
2.2.4. In Vitro Effect of Aspalathin on Apoptosis
2.3. Limitations to This Study
3. Materials and Methods
3.1. Reagents and Kits
3.2. Animal Ethics
3.3. Aspalathin Treatment of Mice
3.4. Effect of Aspalathin on HOMA-IR
3.5. Effect of Aspalathin on Lipid Profiles
3.6. Cell Culture and Treatment of H9c2 Cardiomyocytes with Aspalathin for Subsequent RT2 PCR Profiler Array Analysis
3.7. RT2-PCR Array Analysis of H9c2 Cells Treated with and without Aspalathin
3.8. Gene Interaction and Network Analysis
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gollucke, A.P.; Peres, R.C.; Odair, A.J.; Ribeiro, D.A. Polyphenols: A nutraceutical approach against diseases. Recent Pat. Food Nutr. Agric. 2013, 5, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, Z.; Mirmiran, P.; Azizi, F. Dietary polyphenols as potential nutraceuticals in management of diabetes: A review. J. Diabetes Metab. Disord. 2013, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Pantsi, W.G.; Marnewick, J.L.; Esterhuyse, A.J.; Rautenbach, F.; van Rooyen, J. Rooibos (Aspalathus linearis) offers cardiac protection against ischaemia/reperfusion in the isolated perfused rat heart. Phytomedicine 2011, 18, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Ulicná, O.; Vancová, O.; Bozek, P.; Cársky, J.; Sebeková, K.; Boor, P.; Nakano, M.; Greksák, M. Rooibos tea (Aspalathus linearis) partially prevents oxidative stress in streptozotocin-induced diabetic rats. Physiol. Res. 2006, 55, 157–164. [Google Scholar] [PubMed]
- Marnewick, J.L.; Rautenbach, F.; Venter, I.; Neethling, H.; Blackhurst, D.M.; Wolmarans, P.; Macharia, M. Effects of rooibos (Aspalathus linearis) on oxidative stress and biochemical parameters in adults at risk for cardiovascular disease. J. Ethnopharmacol. 2011, 133, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Mazibuko, S.E.; Joubert, E.; Johnson, R.; Louw, J.; Opoku, A.R.; Muller, C.J.F. Aspalathin improves glucose and lipid metabolism in 3T3-L1 adipocytes exposed to palmitate. Mol. Nutr. Food Res. 2015, 59, 2199–2208. [Google Scholar] [CrossRef]
- Muller, C.J.F.; Joubert, E.; De Beer, D.; Sanderson, M.; Malherbe, C.J.; Fey, S.J.; Louw, J. Acute assessment of an aspalathin-enriched green rooibos (Aspalathus linearis) extract with hypoglycemic potential. Phytomedicine 2012, 20, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kawano, A.; Nakamura, H.; Hata, S.; Minakawa, M.; Miura, Y.; Yagasaki, K. Hypoglycemic effect of aspalathin, a rooibos tea component from Aspalathus linearis, in type 2 diabetic model db/db mice. Phytomedicine 2009, 16, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Muller, C.J.F.; Louw, J.; Joubert, E.; Salie, R.; Opoku, A.R.; Johnson, R. The cardioprotective effect of an aqueous extract of fermented rooibos (Aspalathus linearis) on cultured cardiomyocytes derived from diabetic rats. Phytomedicine 2014, 21, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Dludla, P.; Joubert, E.; February, F.; Mazibuko, S.; Ghoor, S.; Muller, C.; Louw, J. Aspalathin, a dihydrochalcone C-glucoside, protects H9c2 cardiomyocytes against high glucose-induced shifts in substrate preference and apoptosis. Mol. Nutr. Food Res. 2016, 60, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Smit, S.E. An Investigation into the Effects of Aspalathin on Myocardial Glucose Transport Using Cardiomyocytes from Control and Obesity-Induced Insulin Resistant Rats, and Terminally Differentiated H9c2 Cells. Master's Thesis, Stellenbosch University, Stellenbosch, South Africa, March 2016. Available online: http://scholar.sun.ac.za/handle/10019.1/98490 (accessed on 1 November 2016). [Google Scholar]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.L.; Jiang, X.C.; Tall, A.R. Increased high density lipoprotein (HDL), defective hepatic catabolism of ApoA-I and ApoA-II, and decreased ApoA-I mRNA in ob/ob mice. Possible role of leptin in stimulation of HDL turnover. J. Biol. Chem. 1999, 274, 4140–4146. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overload heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocytes expression of PPARγ leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Rajapurohitam, V.; Gan, X.T.; Kirshenbaum, L.A.; Karmazyn, M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ. Res. 2003, 93, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, A.K.; Sack, M.N.; Mjøs, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Manzella, D.; Montano, N.; Gambardella, A.; Varricchio, M. Plasma leptin concentrations and cardiac autonomic nervous system in healthy subjects with different body weights. J. Clin. Endocrinol. Metab. 1999, 85, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Khan, R.S.; Trent, C.M.; Jiang, H.; Son, N.; Blaner, W.S.; Homma, S.; Schulze, P.C.; Goldberg, I.J. PPARγ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ. Heart Fail. 2013, 6, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Muller, C.J.F.; Joubert, E.; Louw, J.; Gabuza, K.B.; Huisamen, B.; Essop, M.F.; Johnson, R. Phenylpyruvic acid-2-O-β-d-glucoside attenuates high glucose-induced apoptosis in H9c2 cardiomyocytes. Planta Med. 2016, 82, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Portoghese, M.; Ferraraccio, F.; Siniscalchi, M.; Babieri, M.; Di Filippo, C.; D’Amico, M.; Rossi, F.; Paolisso, G. Thiazolidinediones may contribute to the intramyocardial lipid accumulation in diabetic myocardium: Effects on cardiac function. Heart 2009, 95, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Tamang, H.K.; Timilsina, U.; Singh, K.P.; Shrestha, S.; Raman, R.K.; Panta, P.; Karna, P.; Khadka, L.; Dahal, C. Apo B/Apo A-I ratio is statistically a better predictor of cardiovascular disease (CVD) than conventional lipid profile: A study from Kathmandu valley, Nepal. J. Clin. Diagn. Res. 2014, 8, 34–36. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F. Apolipoproteins as markers and managers of coronary risk. QJM 2006, 99, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Sundell, J.; Huupponen, R.; Raitakari, O.T.; Nuutila, P.; Knuuti, J. High serum leptin is associated with attenuated coronary vasoreactivity. Obes. Res. 2003, 11, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Zdychová, J.; Komers, R. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol. Res. 2005, 54, 1–16. [Google Scholar] [PubMed]
- Bockus, L.B.; Humphries, K.M. cAMP-dependent protein kinase (PKA) signaling is impaired in the diabetic heart. J. Biol. Chem. 2015, 290, 29250–29258. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.E.; Menahan, L.A.; Chaudhuri, S.N.; Shipp, J.C. Effect of experimental diabetes and glucagon on cAMP-dependent protein kinase in rat liver. Diabetologia 1977, 13, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. PDE3A regulates basal myocardial contractility through Interacting with SERCA2a-signaling complexes in mouse heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hoerter, J.; Gonzalez-Barroso, M.D.; Couplan, E.; Mateo, P.; Gelly, C.; Cassard-Doulcier, A.M.; Diolez, P.; Bouillaud, F. Mitochondrial uncoupling protein 1 expressed in the heart of transgenic mice protects against ischemic-reperfusion damage. Circulation 2004, 110, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Vettor, R.; Fabris, R.; Serra, R.; Lombardi, A.M.; Tonello, C.; Granzotto, M.; Marzolo, M.O.; Carruba, M.O.; Ricquier, D.; Federspil, G.; et al. Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 838–847. [Google Scholar] [PubMed]
- Barreto, P.; Okura, V.K.; Neshich, I.A.; Maia Ide, G.; Arruda, P. Overexpression of UCP1 in tobacco induces mitochondrial biogenesis and amplifies a broad stress response. BMC Plant Biol. 2014, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Ghirlanda-Keller, C.; Schmid, C. Stimulation of glucose transport in osteoblastic cells by parathyroid hormone and insulin-like growth factor I. Mol. Cell Biochem. 2011, 348, 33–42. [Google Scholar] [CrossRef]
- Kajstura, J.; Fiordaliso, F.; Andreoli, A.M.; Li, B.; Chimenti, S.; Medow, M.S.; Limana, F.; Nadal-Ginard, B.; Leri, A.; Anversa, P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001, 50, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Lacana, E.; D’Adamio, L. Regulation of Fas ligand expression and cell death by apoptosis-linked gene 4. Nat. Med. 1999, 5, 542–547. [Google Scholar] [PubMed]
- Xue, W.; Cai, L.; Tan, Y.; Thistlethwaite, P.; Kang, Y.J.; Li, X.; Feng, W. Cardiac-specific overexpression of HIF-1α prevents deterioration of glycolytic pathway and cardiac remodeling in streptozotocin-induced diabetic mice. Am. J. Pathol. 2010, 177, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Bai, W.; Lanting, L.; Gonzales, N.; Nadler, J. Effects of high glucose on vascular endothelial growth factor expression in vascular smooth muscle cells. Am. J. Physiol. 1997, 273, H2224–H2231. [Google Scholar] [PubMed]
- Xu, Z.; Castellino, F.J.; Ploplis, V.A. Plasminogen activator inhibitor-1 (PAI-1) is cardioprotective in mice by maintaining microvascular integrity and cardiac architecture. Blood 2010, 115, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Arabacilar, P.; Marber, M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front. Pharmacol. 2015, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Kageyama, Y.; Iijima, M.; Sesaki, H. PARK2/Parkin becomes critical when DNM1L/Drp1 is absent. Autophagy 2015, 119, 573–574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 149, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.P.; Chen, M.S.; Wang, Y.Z.; Yi, Q.; Lin, S.B.; Chen, A.F.; Luo, J.D. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004, 110, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Barouch, L.A. Leptin signaling and obesity: Cardiovascular consequences. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Cuff, C.A.; Kothapalli, D.; Azonobi, I.; Chun, S.; Zhang, Y.; Belkin, R.; Yeh, C.; Secreto, A.; Assoian, R.K.; Rader, D.J.; et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J. Clin. Investig. 2001, 108, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Chapman, S.M.; Brown, A.A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of P- and E-Selectins in atherosclerosis. J. Clin. Investig. 1998, 102, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Kochan, J.P.; Campfield, L.A.; Burn, P.; Smeyne, R.J. Splice variants of the OB receptor gene are differentially expressed in brain and peripheral tissues of mice. J. Recept. Signal Transduct. Res. 1999, 19, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Achilonu, M.C.; Kendrekar, P.S.; Joubert, E.; Ferreira, D.; Bonnet, S.L.; van der Westhuizen, J.H. Concise and scalable synthesis of aspalathin, a powerful plasma sugar-lowering natural product. J. Nat. Prod. 2014, 77, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; Lin, A.J.; Ververis, K.; Chang, L.; Tang, M.M.; Okabe, J.; El-Osta, A. Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging 2010, 2, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Profile Parameter | Leprdb/+_UC | Leprdb/db_UC | Leprdb/db_MET | Leprdb/db_ASP_LD | Leprdb/db_ASP_HD |
|---|---|---|---|---|---|
| Body weight (g) | 27.1±0.33 | 37.9 ± 0.82 *** | 38.0 ± 0.61 *** | 37.7 ± 0.74 *** | 35.0 ± 0.99 ***,# |
| Total cholesterol (mmol/L) | 2.4 ± 0.09 | 3.5 ± 0.18 *** | 2.9 ± 0.18 *,# | 3.2 ± 0.18 ** | 2.7 ± 0.24 # |
| LDL (mmol/L) | 0.1 ± 0.06 | 0.4 ± 0.07 * | 0.2 ± 0.04 # | 0.2 ± 0.05 | 0.1 ± 0.04 ## |
| HDL (mmol/L) | 1.7 ± 0.06 | 2.5 ± 0.10 *** | 2.2 ± 0.13 ** | 2.3 ± 0.13 ** | 2.3 ± 0.14 ** |
| Triglycerides (mmol/L) | 0.9 ± 0.04 | 3.2 ± 0.38 *** | 2.3 ± 0.46 * | 2.6 ± 0.19 *** | 2.3 ± 0.28 ** |
| Insulin (ng/mL) | 0.4 ± 0.09 | 1.9 ± 1.02 | 1.1 ± 0.22 | 1.5 ± 0.34 | 0.7 ± 0.10 |
| FPG (mmol/L) | 5.6 ± 0.29 | 21.2 ± 1.34 *** | 18.1 ± 1.68 *** | 22.2 ± 1.50 *** | 22.0 ± 1.41 *** |
| HOMA-IR | 0.1 ± 0.01 | 0.6 ± 0.28 | 0.3 ± 0.05 | 0.4 ± 0.07 | 0.2 ± 0.03 |
| Gene Name | Gene Symbol | Gene Fold Regulation | |
|---|---|---|---|
| High Glucose (33 mM) | Aspalathin (1 µM) | ||
| Lipid metabolism | |||
| ATP-binding cassette, subfamily A (ABC1), member 1 | Abca1 | 2.0776 | −1.1921 |
| Acyl-Coenzyme A dehydrogenase, C-2 to C-3 short chain | Acads | −1.6611 | −2.425 |
| Acyl-CoA thioesterase 2 | Acot2 | 2.8239 | −1.2178 |
| Acyl-Coenzyme A oxidase 2, branched chain | Acox2 | 2.0959 | 1.8895 |
| Acyl-CoA synthetase bubblegum family member 2 | Acsbg2 | 4.3205 | −1.5973 |
| Acyl-CoA synthetase long-chain family member 4 | Acsl4 | 5.6547 | 1.7361 |
| Acyl-CoA synthetase long-chain family member 6 | Acsl6 | 2.465 | −1.5973 |
| Acyl-CoA synthetase medium-chain family member 3 | Acsm3 | 14.2353 | 5.697 |
| Acyl-CoA synthetase medium-chain family member 4 | Acsm4 | 6.1301 | 3.1806 |
| Adiponectin, C1Q and collagen domain containing | Adipoq | −6.0324 | 3.2588 |
| Apolipoprotein A-I | Apoa1 | 4.2006 | 2.9178 |
| Apolipoprotein B | Apob | 7.7651 | −3.0228 |
| Apolipoprotein E | Apoe | 4.4207 | −1.0317 |
| CD36 antigen | Cd36 | 3.7512 | 1.7387 |
| Carnitine palmitoyltransferase 1b, muscle | Cpt1b | 3.3366 | 2.0449 |
| Fatty acid binding protein 3, muscle and heart | Fabp3 | 2.7003 | −1.5135 |
| Lysophospholipase 1 | Lypla1 | 3.5198 | 1.6047 |
| Peroxisome proliferator activated receptor gamma | Pparγ | 8.3847 | −1.6135 |
| Stearoyl-Coenzyme A desaturase 1 | Scd1 | 5.5982 | 1.0879 |
| Solute carrier family 25, member 30 | Slc25a30 | 2.6123 | 1.6634 |
| Solute carrier family 27 (fatty acid transporter), member 1 | Slc27a1 | 1.0514 | −2.2919 |
| Solute carrier family 27 (fatty acid transporter), member 3 | Slc27a3 | 6.323 | 2.2207 |
| Solute carrier family 27 (fatty acid transporter), member 5 | Slc27a5 | 2.0184 | −1.5973 |
| Sterol regulatory element binding transcription factor 1 | Srebf1 | 3.4573 | −4.544 |
| Sterol regulatory element binding factor 2 | Srebf2 | 2.0208 | −26.826 |
| Very low density lipoprotein receptor | Vldlr | 2.0148 | 1.3077 |
| Insulin resistance | |||
| V-akt murine thymoma viral oncogene homolog 1 | Akt1 | −2.2173 | 1.2477 |
| Dynamin 1-like | Dnm1l | 1.4367 | 2.8522 |
| Fas ligand (TNF superfamily, member 6) | Faslg | 4.8622 | −1.4856 |
| Insulin-like growth factor 1 | Igf1 | 1.3447 | 2.4477 |
| Mitogen-activated protein kinase kinase 1 | Map2k1 | 2.3446 | −2.1822 |
| Phosphodiesterase 3B, cGMP-inhibited | Pde3b | 2.1481 | 1.5951 |
| Protein kinase, cAMP dependent, catalytic, β | Prkacb | 1.687 | 2.4466 |
| Protein kinase, AMP-activated, γ 1 non-catalytic subunit | Prkag1 | −2.6381 | 1.3516 |
| Serpin peptidase inhibitor, clade B (ovalbumin), member 2 | Serpinb2 | −2.7789 | 1.8746 |
| Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | Serpine1 | 46.1814 | −1.4005 |
| Superoxide dismutase 1, soluble | Sod1 | 2.8805 | 1.0032 |
| Superoxide dismutase 2, mitochondrial | Sod2 | −1.5522 | 3.1195 |
| Uncoupling protein 1 (mitochondrial, proton carrier) | Ucp1 | 58.6622 | −1.7821 |
| Vascular endothelial growth factor A | Vegfa | 2.0015 | 1.2082 |
| Inflammation | |||
| CD3 antigen, epsilon polypeptide | Cd3e | 5.4019 | 1.3557 |
| CD44 molecule | Cd44 | 2.3883 | 1.322 |
| Interleukin 3 | Il3 | 2.3815 | −1.1606 |
| Interleukin 6 | Il6 | 2.7362 | 1.9106 |
| Janus kinase 2 | Jak2 | 3.9723 | 1.721 |
| Leptin receptor | Lepr | 7.2781 | −1.6135 |
| Selectin E | Sele | 13.8787 | −1.5233 |
| Suppressor of cytokine signalling 3 | Socs3 | 4.5848 | −2.4959 |
| Tumor necrosis factor receptor superfamily, member 1b | Tnfrsf1b | 1.0099 | −3.114 |
| Tumor necrosis factor (ligand) superfamily, member 13 | Tnfsf13 | 4.7142 | 1.2849 |
| Tumor necrosis factor (ligand) superfamily, member 13b | Tnfsf13b | 2.0522 | 1.8583 |
| Apoptosis | |||
| Bcl-2 binding component 3 | Bbc3 | −1.3703 | −3.1586 |
| B-cell CLL/lymphoma 2 | Bcl2 | −2.6947 | 1.8312 |
| B-cell leukemia/lymphoma 2 related protein A1d | Bcl2a1 | −7.13 | 1.8085 |
| Conserved helix-loop-helix ubiquitous kinase | Chuk | 4.2959 | 2.1502 |
| Mitogen-activated protein kinase 3 | Mapk3 | 1.2583 | −3.0871 |
| Optic atrophy 1 homolog (human) | Opa1 | 1.3949 | 2.0255 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.; Dludla, P.V.; Muller, C.J.F.; Huisamen, B.; Essop, M.F.; Louw, J. The Transcription Profile Unveils the Cardioprotective Effect of Aspalathin against Lipid Toxicity in an In Vitro H9c2 Model. Molecules 2017, 22, 219. https://doi.org/10.3390/molecules22020219
Johnson R, Dludla PV, Muller CJF, Huisamen B, Essop MF, Louw J. The Transcription Profile Unveils the Cardioprotective Effect of Aspalathin against Lipid Toxicity in an In Vitro H9c2 Model. Molecules. 2017; 22(2):219. https://doi.org/10.3390/molecules22020219
Chicago/Turabian StyleJohnson, Rabia, Phiwayinkosi V. Dludla, Christo J. F. Muller, Barbara Huisamen, M. Faadiel Essop, and Johan Louw. 2017. "The Transcription Profile Unveils the Cardioprotective Effect of Aspalathin against Lipid Toxicity in an In Vitro H9c2 Model" Molecules 22, no. 2: 219. https://doi.org/10.3390/molecules22020219
APA StyleJohnson, R., Dludla, P. V., Muller, C. J. F., Huisamen, B., Essop, M. F., & Louw, J. (2017). The Transcription Profile Unveils the Cardioprotective Effect of Aspalathin against Lipid Toxicity in an In Vitro H9c2 Model. Molecules, 22(2), 219. https://doi.org/10.3390/molecules22020219