PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
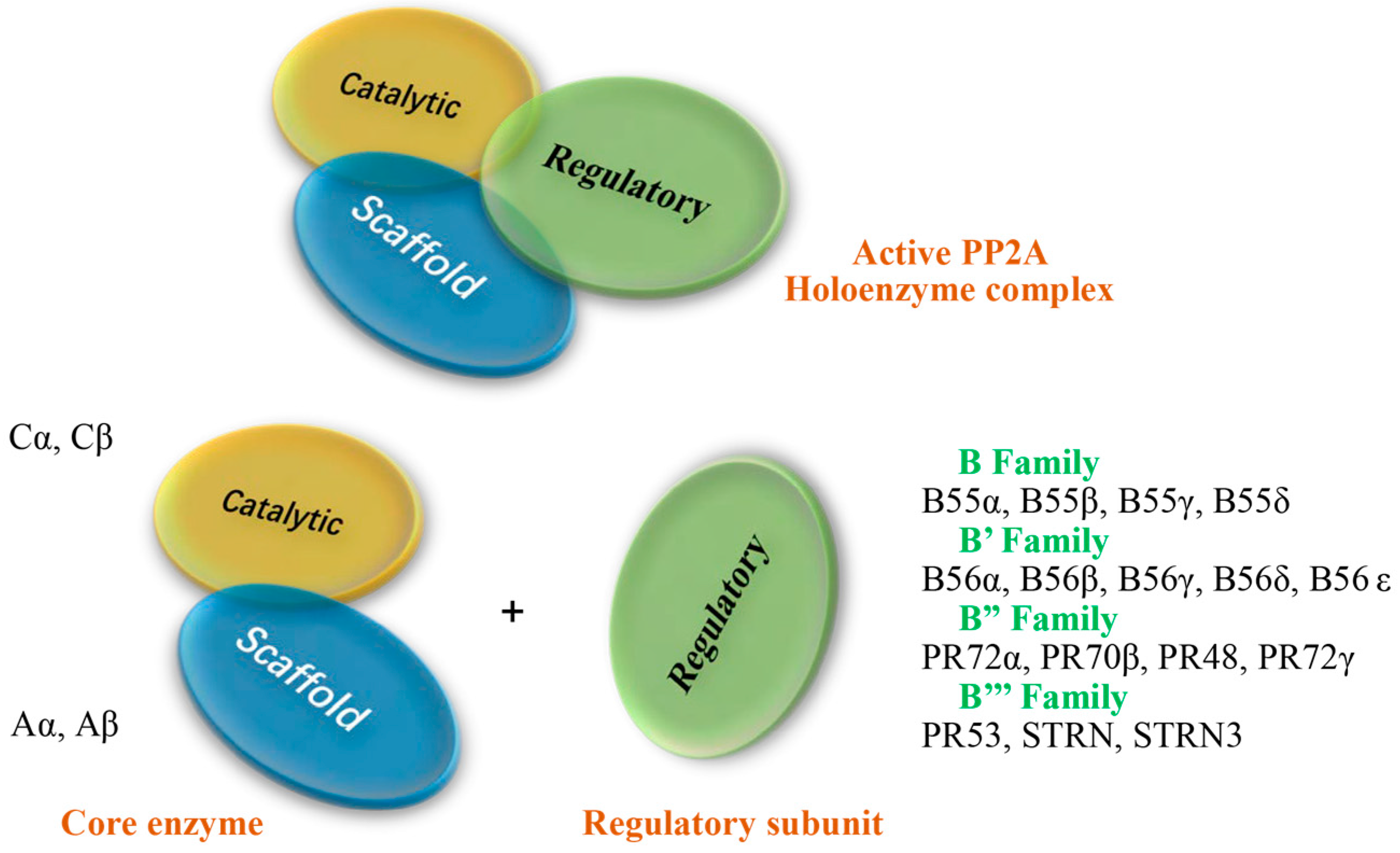
2. Subunit Proteins of PP2A
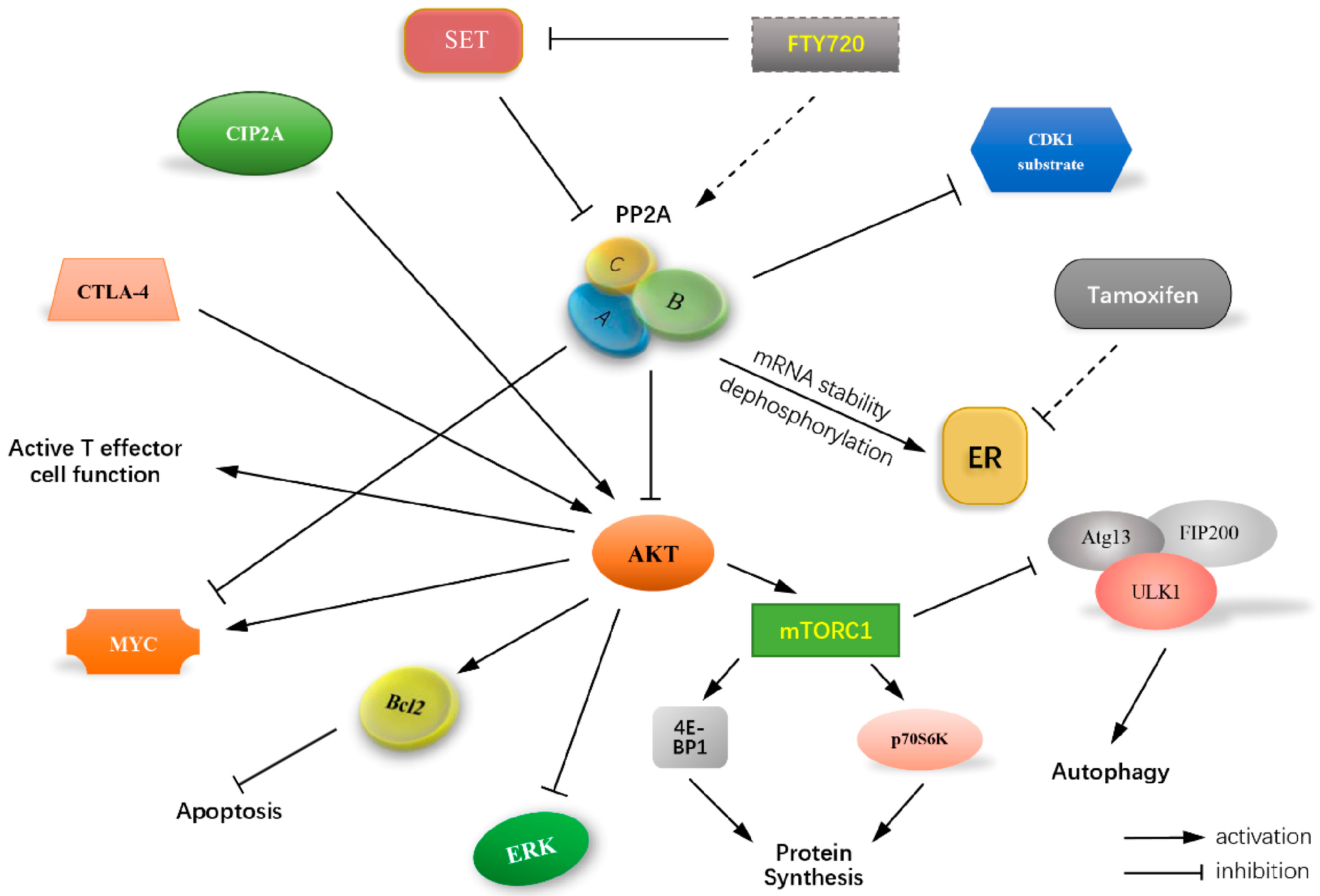
3. PP2A Regulating Cell Cycle Control in TNBCs
4. Relevance of PP2A Controlling DNA Damage Responses and PARP Inhibitors in Breast Cancer
5. Epidermal Growth Factor Receptor and PP2A Regulation
6. Benefits of PP2A Activity in Immunotherapy for Breast Cancer Patients
7. Effect of PP2A on the Cell Death Resistance Mechanisms
7.1. PP2A, an Undeniable Regulator of Apoptosis Escape
7.2. Role of PP2A in Autophagy Ambiguity
8. Significance of PP2A as Regulator of Estrogen Receptor
9. PP2A-Activity Regulating Agents/Drugs
10. PP2A as a Main Node in Treatment in TNBC Subtype
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, B.; Farahmand, L.; Eslami-S, Z.; Majidzadeh-A, K. NF-κB as the main node of resistance to receptor tyrosine kinase inhibitors in triple-negative breast cancer. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P. Biology and Management of Patients with Triple-Negative Breast Cancer. Oncologist 2016, 21, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Nakayama, T.; Naoi, Y.; Yamamoto, N.; Otani, Y.; Kim, S.J.; Shimazu, K.; Shimomura, A.; Maruyama, N.; Tamaki, Y.; et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012, 103, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Horak, C.E.; Pusztai, L.; Xing, G.; Trifan, O.C.; Saura, C.; Tseng, L.M.; Chan, S.; Welcher, R.; Liu, D. Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin. Cancer Res. 2013, 19, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Tseng, L.M.; Chan, S.; Chacko, R.T.; Campone, M.; Manikhas, A.; Nag, S.M.; Leichman, C.G.; Dasappa, L.; Fasching, P.A.; et al. Neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or paclitaxel in early stage breast cancer and evaluation of βIII-tubulin expression as a predictive marker. Oncologist 2013, 18, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Guestini, F.; McNamara, K.M.; Ishida, T.; Sasano, H. Triple negative breast cancer chemosensitivity and chemoresistance: Current advances in biomarkers indentification. Expert Opin. Ther. Targets 2016, 20, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Marmé, F.; Schneeweiss, A. Targeted Therapies in Triple-Negative Breast Cancer. Breast Care (Basel) 2015, 10, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Masoud, V.; Pagès, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Alberts, A.S.; Thorburn, A.M.; Shenolikar, S.; Mumby, M.C.; Feramisco, J.R. Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc. Natl. Acad. Sci. USA 1993, 90, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Glenn, G.M.; Eckhart, W. Mutation of a cysteine residue in polyomavirus middle T antigen abolishes interactions with protein phosphatase 2A, pp60c-src, and phosphatidylinositol-3 kinase, activation of c-fos expression, and cellular transformation. J. Virol. 1993, 67, 1945–1952. [Google Scholar] [PubMed]
- Mayer-Jaekel, R.E.; Hemmings, B.A. Protein phosphatase 2A—A ‘ménage à trois’. Trends Cell Biol. 1994, 4, 287–291. [Google Scholar] [CrossRef]
- Kamibayashi, C.; Estes, R.; Lickteig, R.L.; Yang, S.I.; Craft, C.; Mumby, M.C. Comparison of heterotrimeric protein phosphatase 2A containing different B subunits. J. Biol. Chem. 1994, 269, 20139–20148. [Google Scholar] [PubMed]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef] [PubMed]
- Côme, C.; Laine, A.; Chanrion, M.; Edgren, H.; Mattila, E.; Liu, X.; Jonkers, J.; Ivaska, J.; Isola, J.; Darbon, J.M.; et al. CIP2A is associated with human breast cancer aggressivity. Clin. Cancer Res. 2009, 15, 5092–5100. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Chu, P.Y.; Tung, S.L.; Liu, C.Y.; Tsai, Y.F.; Lin, Y.S.; Wang, W.L.; Wang, Y.L.; Lien, P.J.; Chao, T.C.; et al. Overexpression of phosphoprotein phosphatase 2A predicts worse prognosis in patients with breast cancer: A 15-year follow-up. Hum. Pathol. 2017, 66, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Feng, T.T.; Guo, Y.; Yu, X.; Huang, Q.; Zhang, L.; Tang, W.; Liu, Y. Expression of cancerous inhibitor of protein phosphatase 2A in human triple negative breast cancer correlates with tumor survival, invasion and autophagy. Oncol. Lett. 2016, 12, 5370–5376. [Google Scholar] [PubMed]
- Hu, X.; Garcia, C.; Fazli, L.; Gleave, M.; Vitek, M.P.; Jansen, M.; Christensen, D.; Mulholland, D.J. Inhibition of Pten deficient Castration Resistant Prostate Cancer by Targeting of the SET—PP2A Signaling axis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Hung, M.H.; Wang, D.S.; Chu, P.Y.; Su, J.C.; Teng, T.H.; Huang, C.T.; Chao, T.T.; Wang, C.Y.; Shiau, C.W.; et al. Tamoxifen induces apoptosis through cancerous inhibitor of protein phosphatase 2A-dependent phospho-Akt inactivation in estrogen receptor-negative human breast cancer cells. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Cristóbal, I.; Torrejón, B.; Martínez-Useros, J.; Madoz-Gurpide, J.; Rojo, F.; García-Foncillas, J. PP2A regulates signaling through hormonal receptors in breast cancer with important therapeutic implications. Biochim. Biophys. Acta 2017, 1868, 435–438. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Virshup, D.M. Identification of a new family of protein phosphatase 2A regulatory subunits. J. Biol. Chem. 1995, 270, 26123–26128. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.R.; Hofsteenge, J.; Hemmings, B.A. Molecular cloning of cDNAs encoding two isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry 1987, 26, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Adams-Pearson, C.; Maurer, F.; Müller, P.; Goris, J.; Merlevede, W.; Hofsteenge, J.; Stone, S.R. alpha- and beta-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry 1990, 29, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Grech, G.; Baldacchino, S.; Saliba, C.; Grixti, M.P.; Gauci, R.; Petroni, V.; Fenech, A.G.; Scerri, C. Deregulation of the protein phosphatase 2A, PP2A in cancer: Complexity and therapeutic options. Tumour Biol. 2016, 37, 11691–11700. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.E.; Hendrix, P.; Cron, P.; Matthies, R.; Stone, S.R.; Goris, J.; Merlevede, W.; Hofsteenge, J.; Hemmings, B.A. Structure of the 55-kDa regulatory subunit of protein phosphatase 2A: Evidence for a neuronal-specific isoform. Biochemistry 1991, 30, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, P.; Mayer-Jackel, R.E.; Cron, P.; Goris, J.; Hofsteenge, J.; Merlevede, W.; Hemmings, B.A. Structure and expression of a 72-kDa regulatory subunit of protein phosphatase 2A. Evidence for different size forms produced by alternative splicing. J. Biol. Chem. 1993, 268, 15267–15276. [Google Scholar] [PubMed]
- Lambrecht, C.; Haesen, D.; Sents, W.; Ivanova, E.; Janssens, V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol. Biol. 2013, 1053, 283–305. [Google Scholar] [PubMed]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Sun, D.; Jiang, W.; Klotz-Noack, K.; Vashisht, A.A.; Wohlschlegel, J.; Widschwendter, M.; Spruck, C. PP2A-B55β antagonizes cyclin E1 proteolysis and promotes its dysregulation in cancer. Cancer Res. 2014, 74, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Keyomarsi, K.; Tucker, S.L.; Buchholz, T.A.; Callister, M.; Ding, Y.; Hortobagyi, G.N.; Bedrosian, I.; Knickerbocker, C.; Toyofuku, W.; Lowe, M.; et al. Cyclin E and survival in patients with breast cancer. N. Engl. J. Med. 2002, 347, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Brunet, J.S.; Stefansson, I.M.; Straume, O.; Chappuis, P.O.; Bégin, L.R.; Hamel, N.; Goffin, J.R.; Wong, N.; Trudel, M.; et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004, 64, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Eichhorn, P.J.; Cortés, J.; Prudkin, L.; Aura, C.; Jiménez, J.; Chandarlapaty, S.; Serra, V.; Prat, A.; Ibrahim, Y.H.; et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. USA 2011, 108, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Gao, H.; Lin, S.Y.; Goss, J.A.; Brunicardi, F.C.; Li, K. siRNA-based targeting of cyclin E overexpression inhibits breast cancer cell growth and suppresses tumor development in breast cancer mouse model. PLoS ONE 2010, 5, e12860. [Google Scholar] [CrossRef] [PubMed]
- Funabiki, H.; Wynne, D.J. Making an effective switch at the kinetochore by phosphorylation and dephosphorylation. Chromosoma 2013, 122, 135–158. [Google Scholar] [CrossRef] [PubMed]
- Cadot, B.; Brunetti, M.; Coppari, S.; Fedeli, S.; de Rinaldis, E.; Dello Russo, C.; Gallinari, P.; De Francesco, R.; Steinkühler, C.; Filocamo, G. Loss of histone deacetylase 4 causes segregation defects during mitosis of p53-deficient human tumor cells. Cancer Res. 2009, 69, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.A.; Maldonado, M.; Kapoor, T.M. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 2011, 13, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Zhang, G.; Larsen, M.S.; Lischetti, T.; Streicher, W.; Kragh Nielsen, T.; Bjørn, S.P.; Nilsson, J. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J. Cell Sci. 2013, 126, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Porter, I.M.; Schleicher, K.; Porter, M.; Swedlow, J.R. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Virshup, D.M.; Lee, S.H. B56-PP2A regulates motor dynamics for mitotic chromosome alignment. J. Cell Sci. 2014, 127, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, W.; Vallardi, G.; Teixeira, A.; Kops, G.J.; Saurin, A.T. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat. Cell Biol. 2014, 16, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Grallert, A.; Boke, E.; Hagting, A.; Hodgson, B.; Connolly, Y.; Griffiths, J.R.; Smith, D.L.; Pines, J.; Hagan, I.M. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 2015, 517, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Beullens, M.; Huang, J.; De Munter, S.; Lesage, B.; Bollen, M. Cdk1 orders mitotic events through coordination of a chromosome-associated phosphatase switch. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ying, H.; Liu, R.; Li, L.; Bergholz, J.; Xiao, Z.X. Pin1 inhibits PP2A-mediated Rb dephosphorylation in regulation of cell cycle and S-phase DNA damage. Cell Death Dis. 2015, 6, e1640. [Google Scholar] [CrossRef] [PubMed]
- Kolupaeva, V.; Laplantine, E.; Basilico, C. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS ONE 2008, 3, e3447. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.A.; Makris, A.; Wu, M.F.; Rimawi, M.; Froehlich, A.; Dave, B.; Hilsenbeck, S.G.; Chamness, G.C.; Lewis, M.T.; Dobrolecki, L.E.; et al. DNA repair signature is associated with anthracycline response in triple negative breast cancer patients. Breast Cancer Res. Treat. 2010, 123, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Müller, B.M.; Loibl, S.; Budczies, J.; Hanusch, C.; Darb-Esfahani, S.; Hilfrich, J.; Weiss, E.; Huober, J.; Blohmer, J.U.; et al. Cytoplasmic poly(adenosine diphosphate-ribose) polymerase expression is predictive and prognostic in patients with breast cancer treated with neoadjuvant chemotherapy. J. Clin. Oncol. 2011, 29, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Zwaenepoel, K.; Goris, J.; Erneux, C.; Parker, P.J.; Janssens, V. Protein phosphatase 2A PR130/B′′alpha1 subunit binds to the SH2 domain-containing inositol polyphosphate 5-phosphatase 2 and prevents epidermal growth factor (EGF)-induced EGF receptor degradation sustaining EGF-mediated signaling. FASEB J. 2010, 24, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Zwaenepoel, K.; Rossé, C.; Petit, M.M.; Goris, J.; Parker, P.J. PP2A binds to the LIM domains of lipoma-preferred partner through its PR130/B′′ subunit to regulate cell adhesion and migration. J. Cell Sci. 2016, 129, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Parsels, L.A.; Karnak, D.; Davis, M.A.; Parsels, J.D.; Marsh, A.C.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Sun, Y.; et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin. Cancer Res. 2013, 19, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Kalev, P.; Simicek, M.; Vazquez, I.; Munck, S.; Chen, L.; Soin, T.; Danda, N.; Chen, W.; Sablina, A. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res. 2012, 72, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, S3–S8. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Terrasi, M.; Rizzo, S.; Sivestris, N.; Ficorella, C.; Cajozzo, M.; Di Gaudio, F.; Gulotta, G.; Siragusa, S.; Gebbia, N.; et al. EGFR genomic alterations in cancer: Prognostic and predictive values. Front. Biosci. (Elite Ed.) 2011, 3, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.K.; Chamness, G.C.; Elledge, R.M. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.F.; Wong, C.; Wu, J.M. Anti-EGFR Therapy: Mechanism and Advances in Clinical Efficacy in Breast Cancer. J. Oncol. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Jang, M.H.; Lee, S.J.; Bae, Y.K. Mutations of the Epidermal Growth Factor Receptor Gene in Triple-Negative Breast Cancer. J. Breast Cancer 2017, 20, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Nogi, H.; Kobayashi, T.; Suzuki, M.; Tabei, I.; Kawase, K.; Toriumi, Y.; Fukushima, H.; Uchida, K. EGFR as paradoxical predictor of chemosensitivity and outcome among triple-negative breast cancer. Oncol. Rep. 2009, 21, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.Y.; Chang, M.H.; Choi, Y.L.; Lee, J.E.; Nam, S.J.; Yang, J.H.; Park, Y.H.; Ahn, J.S.; Im, Y.H. Potential candidate biomarkers for heterogeneity in triple-negative breast cancer (TNBC). Cancer Chemother. Pharmacol. 2011, 68, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhu, L.; Li, Y.; Ji, J.; Li, J.; Yuan, F.; Wang, D.; Chen, W.; Huang, O.; Chen, X.; et al. Overexpression of epithelial growth factor receptor (EGFR) predicts better response to neo-adjuvant chemotherapy in patients with triple-negative breast cancer. J. Transl. Med. 2012, 10 (Suppl. 1). [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Howard, E.W.; Parris, A.B.; Guo, Z.; Zhao, Q.; Ma, Z.; Xing, Y.; Liu, B.; Edgerton, S.M.; Thor, A.D.; et al. Activation of cancerous inhibitor of PP2A (CIP2A) contributes to lapatinib resistance through induction of CIP2A-Akt feedback loop in ErbB2-positive breast cancer cells. Oncotarget 2017, 8, 58847–58864. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Zhang, D.; Chang, C.F.; Koay, E.S. Silencing of the PP2A catalytic subunit causes HER-2/neu positive breast cancer cells to undergo apoptosis. Exp. Cell Res. 2010, 316, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, E.; Kelly, C.M.; McDaid, H.M. Therapeutic Advances and New Directions for Triple-Negative Breast Cancer. Breast Care (Basel) 2017, 12, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Mohammed, S.; Al-Omair, A.; Qattan, A.; Lehe, C.; Al-Qudaihi, G.; Elkum, N.; Alshabanah, M.; Amer, S.B.; Tulbah, A.; et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: Correlation with important high-risk prognostic factors. Neoplasia 2006, 8, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Glont, S.E.; Blows, F.M.; Provenzano, E.; Dawson, S.J.; Liu, B.; Hiller, L.; Dunn, J.; Poole, C.J.; Bowden, S.; et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann. Oncol. 2015, 26, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Chau, T.A.; Madrenas, J. Structure-Function analysis of the CTLA-4 interaction with PP2A. BMC Immunol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Baroja, M.L.; Vijayakrishnan, L.; Bettelli, E.; Darlington, P.J.; Chau, T.A.; Ling, V.; Collins, M.; Carreno, B.M.; Madrenas, J.; Kuchroo, V.K. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 2002, 168, 5070–5078. [Google Scholar] [CrossRef] [PubMed]
- Lienlaf, M.; Perez-Villarroel, P.; Knox, T.; Pabon, M.; Sahakian, E.; Powers, J.; Woan, K.V.; Lee, C.; Cheng, F.; Deng, S.; et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol. Oncol. 2016, 10, 735–750. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, V.R.; Kurinna, S.M.; Karanjeet, K.B.; Schuster, T.F.; Martelli, A.M.; McCubrey, J.A.; Ruvolo, P.P. PKR regulates B56(alpha)-mediated BCL2 phosphatase activity in acute lymphoblastic leukemia-derived REH cells. J. Biol. Chem. 2008, 283, 35474–35485. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M.; Barrow, C.A.; Davis, A.J.; Mumby, M.C. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. USA 2002, 99, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Clark, W.; Mumby, M.; Gao, F.; May, W.S. A functional role for the B56 alpha-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J. Biol. Chem. 2002, 277, 22847–22852. [Google Scholar] [CrossRef] [PubMed]
- Patturajan, M.; Nomoto, S.; Sommer, M.; Fomenkov, A.; Hibi, K.; Zangen, R.; Poliak, N.; Califano, J.; Trink, B.; Ratovitski, E.; et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell 2002, 1, 369–379. [Google Scholar] [CrossRef]
- Letourneux, C.; Rocher, G.; Porteu, F. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 2006, 25, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Silverstein, A.M.; Shum, H.; Martinez, B.; Mumby, M.C. A functional genomics analysis of the B56 isoforms of Drosophila protein phosphatase 2A. Mol. Cell. Proteom. 2007, 6, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.C.; Nica, A.F.; Kurinna, S.M.; Jiffar, T.; Mumby, M.; Ruvolo, P.P. Mitochondrial protein phosphatase 2A regulates cell death induced by simulated ischemia in kidney NRK-52E cells. Cell Cycle 2007, 6, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wallace, L.; Harper, S.Q.; Yang, J. PP2A:B56ε, a substrate of caspase-3, regulates p53-dependent and p53-independent apoptosis during development. J. Biol. Chem. 2010, 285, 34493–34502. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Jono, H.; Miller, C.L.; Cai, Y.; Lim, S.; Liu, X.; Gao, P.; Abe, J.; Li, J.D.; Yan, C. Ca2+/calmodulin-stimulated PDE1 regulates the beta-catenin/TCF signaling through PP2A B56 gamma subunit in proliferating vascular smooth muscle cells. FEBS J. 2010, 277, 5026–5039. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, E.; Maenaka, S.; Shimada, E.; Nishimura, Y.; Sakurai, H. Dynamic regulation of extracellular signal-regulated kinase (ERK) by protein phosphatase 2A regulatory subunit B56γ1 in nuclei induces cell migration. PLoS ONE 2013, 8, e63729. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Deng, X.; Ito, T.; Carr, B.K.; May, W.S. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J. Biol. Chem. 1999, 274, 20296–20300. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 2003, 47, 383–392. [Google Scholar] [CrossRef]
- Kim, T.; Han, W.; Kim, M.K.; Lee, J.W.; Kim, J.; Ahn, S.K.; Lee, H.B.; Moon, H.G.; Lee, K.H.; Kim, T.Y.; et al. Predictive Significance of p53, Ki-67, and Bcl-2 Expression for Pathologic Complete Response after Neoadjuvant Chemotherapy for Triple-Negative Breast Cancer. J. Breast Cancer 2015, 18, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Perry, C.; Dickinson, P.; Ball, G.; Moseley, P.; Madhusudan, S.; Ellis, I.O.; Chan, S.Y. Bcl2 is an independent prognostic marker of triple negative breast cancer (TNBC) and predicts response to anthracycline combination (ATC) chemotherapy (CT) in adjuvant and neoadjuvant settings. Ann. Oncol. 2013, 24, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Arnold, H.K.; Sears, R.C. A tumor suppressor role for PP2A-B56alpha through negative regulation of c-Myc and other key oncoproteins. Cancer Metastasis Rev. 2008, 27, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gao, F.; Flagg, T.; May, W.S., Jr. Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc. Natl. Acad. Sci. USA 2004, 101, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gao, F.; Flagg, T.; Anderson, J.; May, W.S. Bcl2’s flexible loop domain regulates p53 binding and survival. Mol. Cell. Biol. 2006, 26, 4421–4434. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Kurzrock, R.; Shankar, S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol. Cancer Ther. 2010, 9, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Zang, F.; Wei, X.; Leng, X.; Yu, M.; Sun, B. C-FLIP(L) contributes to TRAIL resistance in HER2-positive breast cancer. Biochem. Biophys. Res. Commun. 2014, 450, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, C.; Kong, X.; Li, X.; Kong, X.; Wang, Y.; Ding, X.; Yang, Q. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS ONE 2014, 9, e99067. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Davis, S.R.; Pumphrey, J.G.; Bao, J.; Nau, M.M.; Meltzer, P.S.; Lipkowitz, S. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 2009, 113, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Creyghton, M.P.; Wilhelmsen, K.; van Dam, H.; Bernards, R. A RNA interference screen identifies the protein phosphatase 2A subunit PR55gamma as a stress-sensitive inhibitor of c-SRC. PLoS Genet. 2007, 3, e218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, Z.; Zhou, J.Y.; Zhuang, Z.; Wang, E.; Boerner, J.; Wu, G.S. Regulation of the Src-PP2A interaction in tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J. Biol. Chem. 2013, 288, 33263–33271. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Notte, A.; Leclere, L.; Michiels, C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 2011, 82, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Rikiishi, H. Novel Insights into the Interplay between Apoptosis and Autophagy. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Usui, T.; Ohama, T.; Sato, K. Regulation of Beclin 1 Protein Phosphorylation and Autophagy by Protein Phosphatase 2A (PP2A) and Death-associated Protein Kinase 3 (DAPK3). J. Biol. Chem. 2016, 291, 10858–10866. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.Z.; Yuan, Z.Y.; Li, M.; Xi, S.Y.; Fu, J.; He, J. Accumulation of p62 is associated with poor prognosis in patients with triple-negative breast cancer. OncoTargets Ther. 2013, 6, 883–888. [Google Scholar]
- Magnaudeix, A.; Wilson, C.M.; Page, G.; Bauvy, C.; Codogno, P.; Lévêque, P.; Labrousse, F.; Corre-Delage, M.; Yardin, C.; Terro, F. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol. Aging 2013, 34, 770–790. [Google Scholar] [CrossRef] [PubMed]
- Keen, J.C.; Zhou, Q.; Park, B.H.; Pettit, C.; Mack, K.M.; Blair, B.; Brenner, K.; Davidson, N.E. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. J. Biol. Chem. 2005, 280, 29519–29524. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sidell, N. Anti-estrogenic effects of conjugated linoleic acid through modulation of estrogen receptor phosphorylation. Breast Cancer Res. Treat. 2005, 94, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rincón, R.; Cristóbal, I.; Zazo, S.; Arpí, O.; Menéndez, S.; Manso, R.; Lluch, A.; Eroles, P.; Rovira, A.; Albanell, J.; et al. PP2A inhibition determines poor outcome and doxorubicin resistance in early breast cancer and its activation shows promising therapeutic effects. Oncotarget 2015, 6, 4299–4314. [Google Scholar] [CrossRef] [PubMed]
- Baldacchino, S.; Saliba, C.; Petroni, V.; Fenech, A.G.; Borg, N.; Grech, G. Deregulation of the phosphatase, PP2A is a common event in breast cancer, predicting sensitivity to FTY720. EPMA J. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Balliu, M.; Cellai, C.; Lulli, M.; Laurenzana, A.; Torre, E.; Vannucchi, A.M.; Paoletti, F. HDAC1 controls CIP2A transcription in human colorectal cancer cells. Oncotarget 2016, 7, 25862–25871. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Cheng, R.Y.; Vitek, T.M.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Bareford, M.D.; Hamed, H.A.; Allegood, J.; Cruickshanks, N.; Poklepovic, A.; Park, M.A.; Ogretmen, B.; Spiegel, S.; Grant, S.; Dent, P. Sorafenib and pemetrexed toxicity in cancer cells is mediated via SRC-ERK signaling. Cancer Biol. Ther. 2012, 13, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Liu, C.Y.; Chang, K.C.; Chu, P.Y.; Shiau, C.W.; Chen, K.F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.A.; Koo, J.S.; Park, J.S.; Park, M.Y.; Jeong, A.L.; Oh, K.S.; Yang, Y. Estradiol enhances CIP2A expression by the activation of p70 S6 kinase. Endocr. Relat. Cancer 2014, 21, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Wang, Y.C.; Lin, P.L.; Cheng, Y.W.; Chen, C.Y.; Wu, T.C.; Lee, H. IL-10 promotes tumor aggressiveness via upregulation of CIP2A transcription in lung adenocarcinoma. Clin. Cancer Res. 2013, 19, 4092–4103. [Google Scholar] [CrossRef] [PubMed]
- Böckelman, C.; Lassus, H.; Hemmes, A.; Leminen, A.; Westermarck, J.; Haglund, C.; Bützow, R.; Ristimäki, A. Prognostic role of CIP2A expression in serous ovarian cancer. Br. J. Cancer 2011, 105, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Liu, G.; Dong, J.; Jin, Y. Clinical implications of CIP2A protein expression in breast cancer. Med. Oncol. 2013, 30. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [PubMed]
- Soofiyani, S.R.; Hejazi, M.S.; Baradaran, B. The role of CIP2A in cancer: A review and update. Biomed. Pharmacother. 2017, 96, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Anazawa, Y.; Nakagawa, H.; Furihara, M.; Ashida, S.; Tamura, K.; Yoshioka, H.; Shuin, T.; Fujioka, T.; Katagiri, T.; Nakamura, Y. PCOTH, a novel gene overexpressed in prostate cancers, promotes prostate cancer cell growth through phosphorylation of oncoprotein TAF-Ibeta/SET. Cancer Res. 2005, 65, 4578–4586. [Google Scholar] [CrossRef] [PubMed]
- Irie, A.; Harada, K.; Araki, N.; Nishimura, Y. Phosphorylation of SET protein at Ser171 by protein kinase D2 diminishes its inhibitory effect on protein phosphatase 2A. PLoS ONE 2012, 7, e51242. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.J.; Ohkubo, N.; Oddo, J.; Van Kanegan, M.J.; Neil, J.; Li, F.; Colton, C.A.; Vitek, M.P. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J. Immunol. 2011, 186, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Perl, A.; Tohme, R.; Kiselar, J.; Kastrinsky, D.B.; Zaware, N.; Izadmehr, S.; Mazhar, S.; Wiredja, D.D.; O’Connor, C.M.; et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Investig. 2017, 127, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Cole, K.S.; Marotti, J.D.; Hu, R.; Schnitt, S.J.; Tamimi, R.M. Androgen receptor expression in breast cancer in relation to molecular phenotype: Results from the Nurses’ Health Study. Mod. Pathol. 2011, 24, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Corn, P.G.; Michaelson, M.D.; Hammers, H.J.; Alumkal, J.J.; Ryan, C.J.; Bruce, J.Y.; Moran, S.; Lee, S.Y.; Lin, H.M.; et al. Phase II study of single-agent orteronel (TAK-700) in patients with nonmetastatic castration-resistant prostate cancer and rising prostate-specific antigen. Clin. Cancer Res. 2014, 20, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Wang, D.; Chuang, H.C.; Wei, S.; Kulp, S.K.; Chen, C.S. α-Tocopheryl succinate and derivatives mediate the transcriptional repression of androgen receptor in prostate cancer cells by targeting the PP2A-JNK-Sp1-signaling axis. Carcinogenesis 2009, 30, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Singh, S.; Srivastava, S.K.; Honkanen, R.E.; Reed, E.; Singh, A.P. Modulation of protein phosphatase 2A activity alters androgen-independent growth of prostate cancer cells: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhao, L.; Feng, X.; Xu, C.; Li, C.; Niu, Y. Lin28A activates androgen receptor via regulation of c-myc and promotes malignancy of ER-/Her2+ breast cancer. Oncotarget 2016, 7, 60407–60418. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Li, D.; Zhang, B.; Qi, Y.; Diao, Y.; Zhen, Y.; Shu, X. PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer. Molecules 2017, 22, 2277. https://doi.org/10.3390/molecules22122277
Zhao H, Li D, Zhang B, Qi Y, Diao Y, Zhen Y, Shu X. PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer. Molecules. 2017; 22(12):2277. https://doi.org/10.3390/molecules22122277
Chicago/Turabian StyleZhao, Henan, Duojiao Li, Baojing Zhang, Yan Qi, Yunpeng Diao, Yuhong Zhen, and Xiaohong Shu. 2017. "PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer" Molecules 22, no. 12: 2277. https://doi.org/10.3390/molecules22122277
APA StyleZhao, H., Li, D., Zhang, B., Qi, Y., Diao, Y., Zhen, Y., & Shu, X. (2017). PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer. Molecules, 22(12), 2277. https://doi.org/10.3390/molecules22122277