Four New Compounds Obtained from Cultured Cells of Artemisia annua

Abstract
:1. Introduction
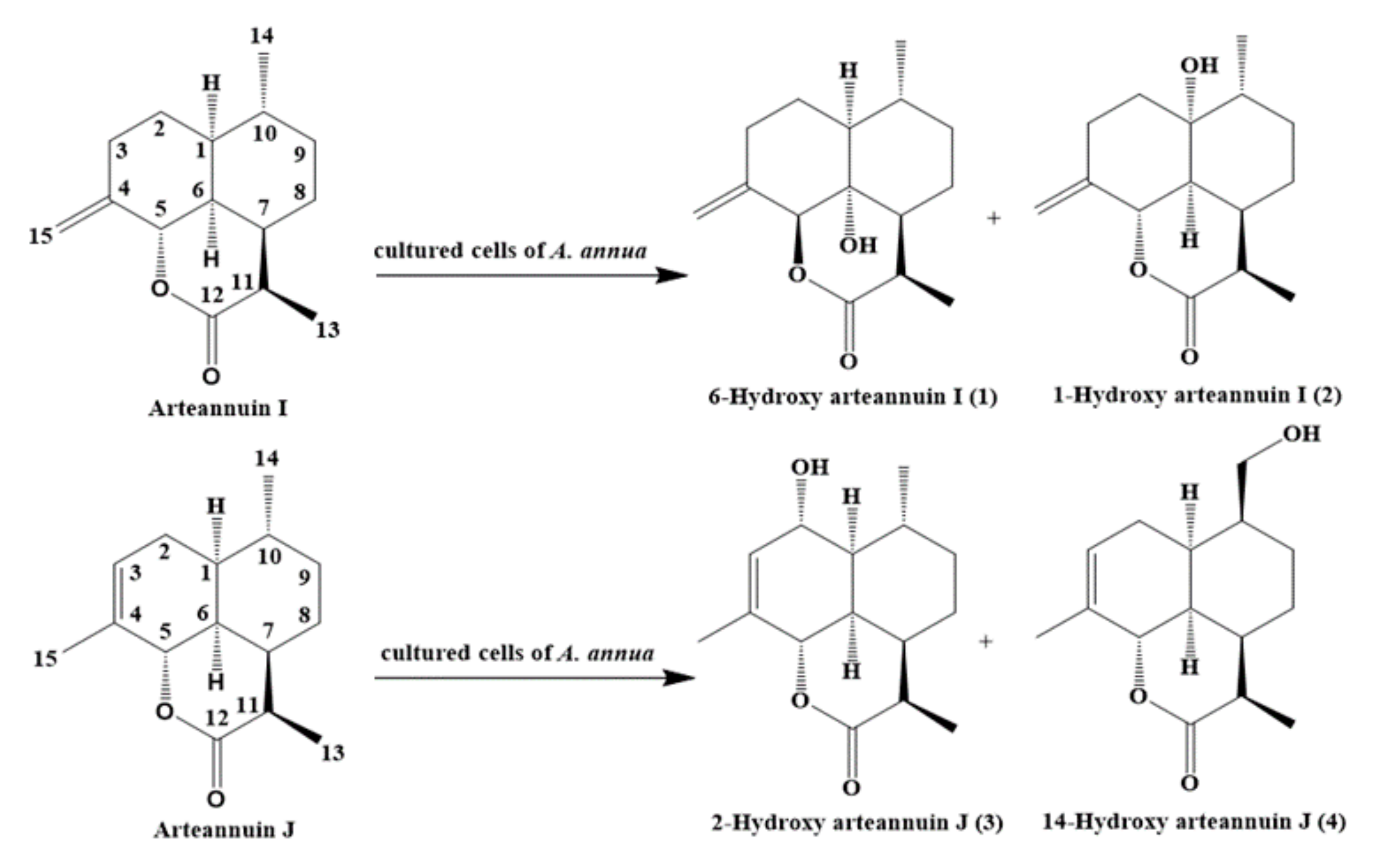
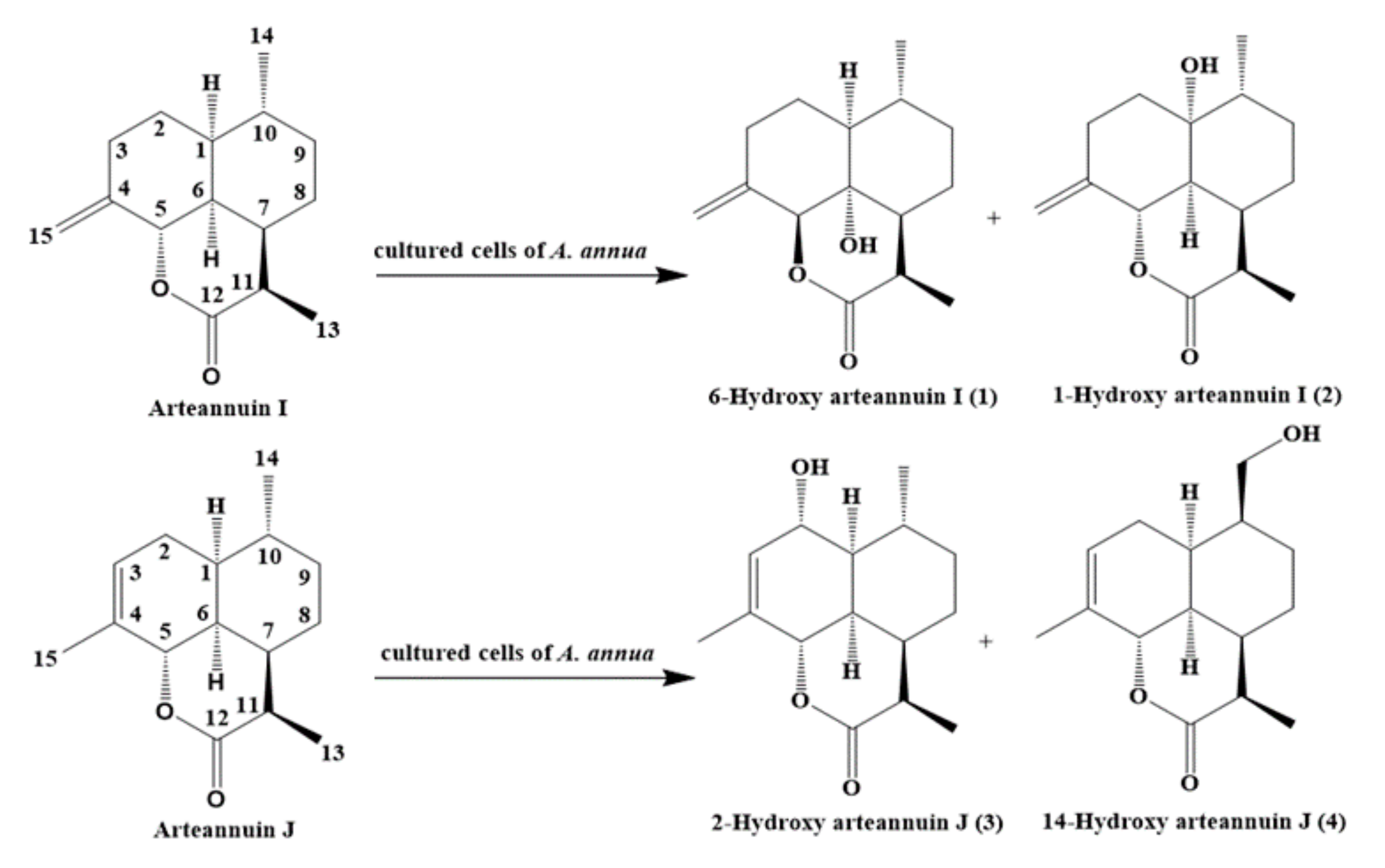
2. Results and Discussion
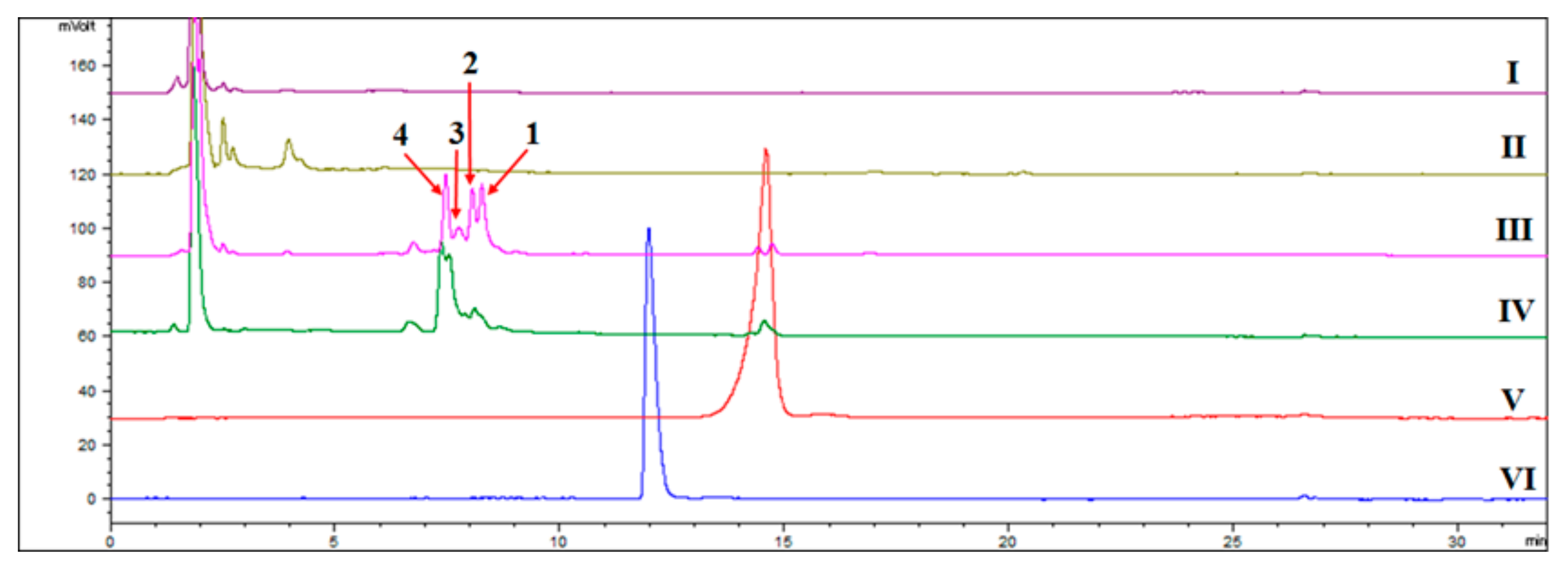
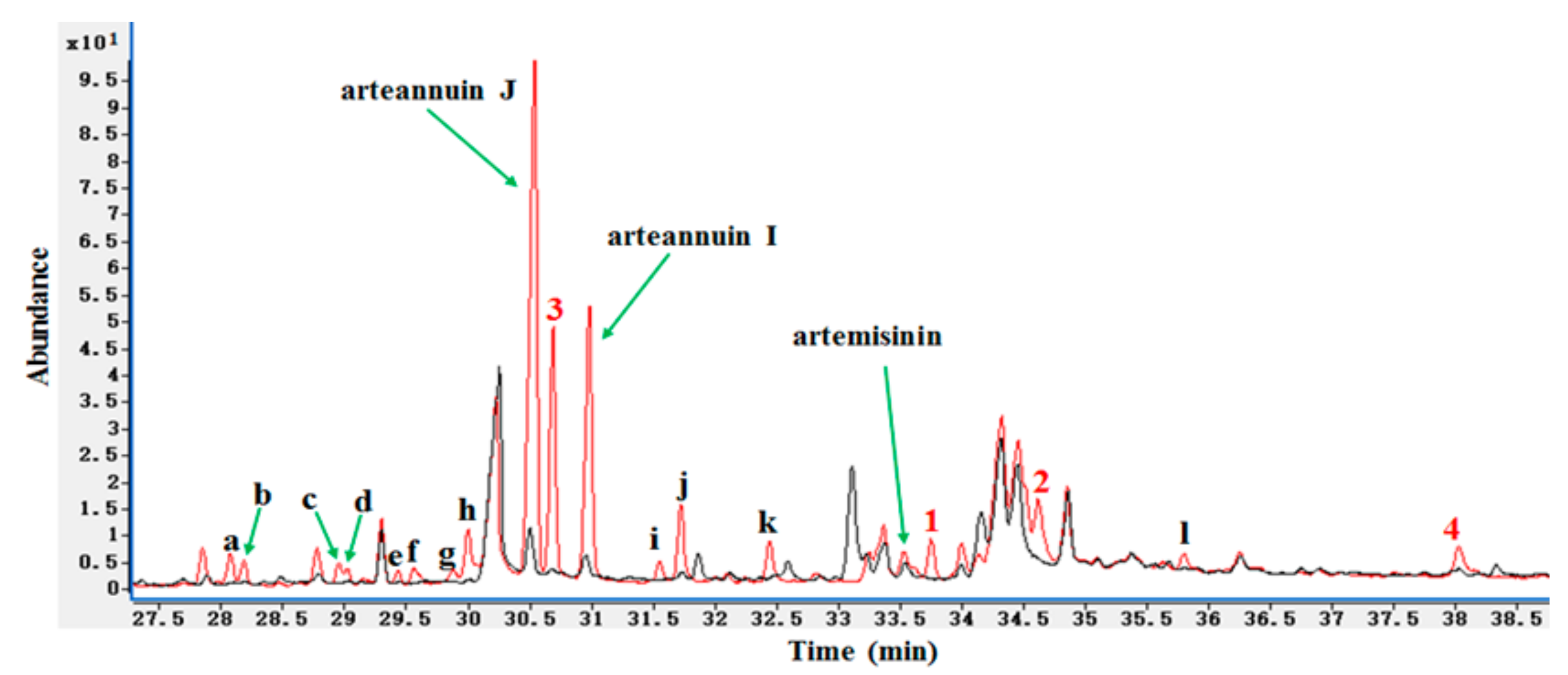
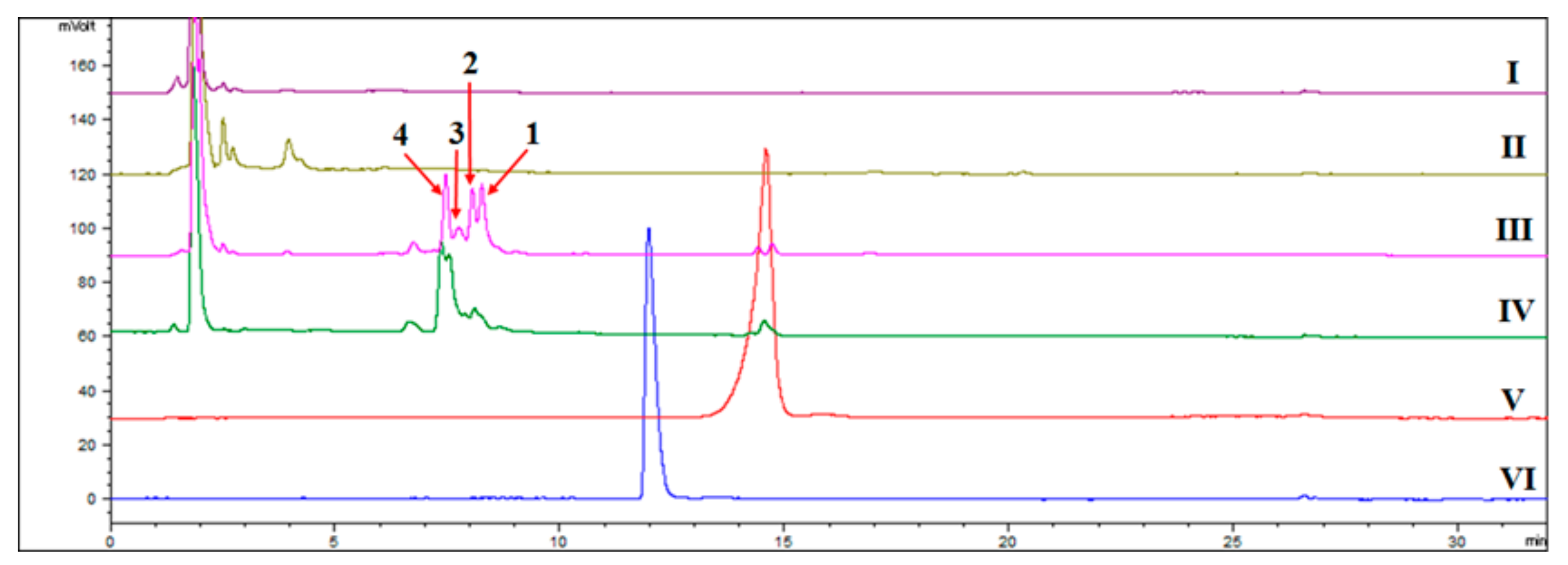
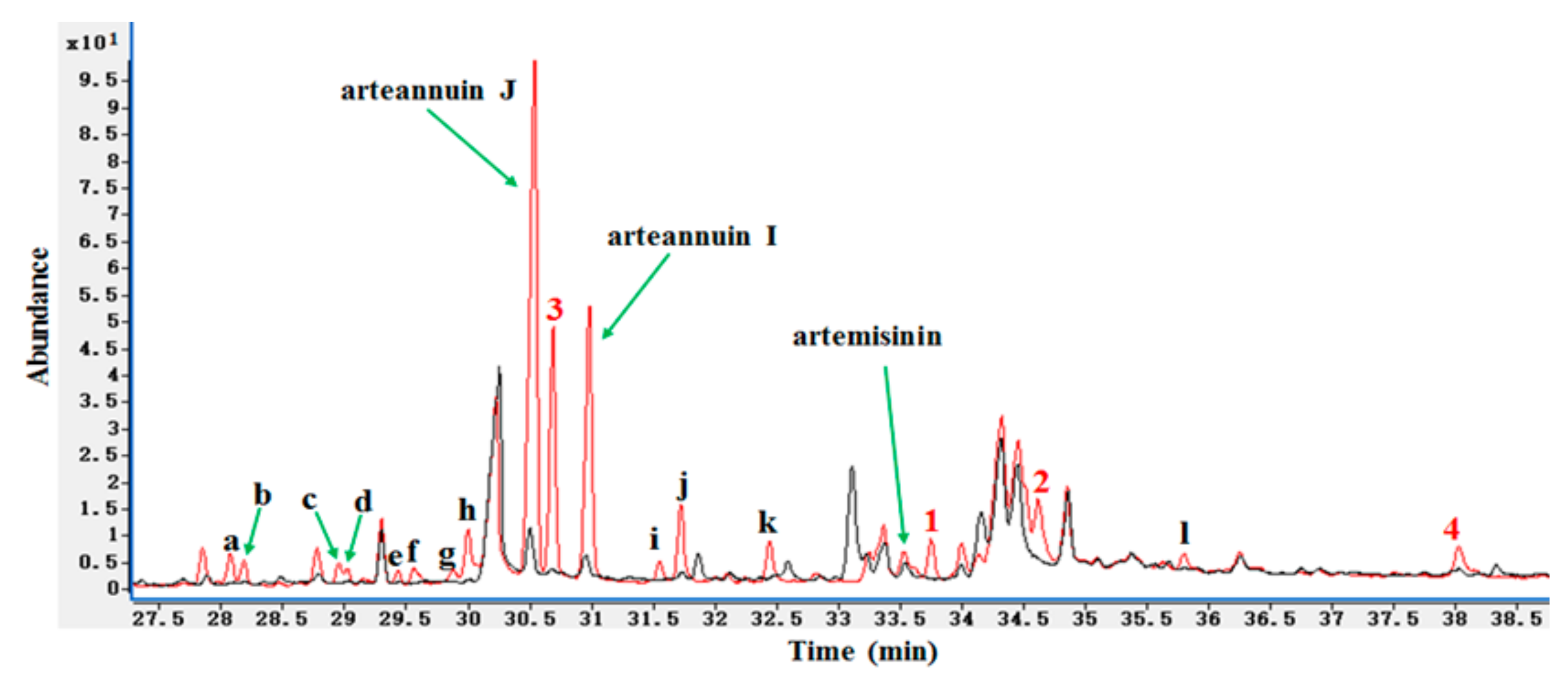
2.1. Detection of the New Products by HPLC-ELSD and GC-MS
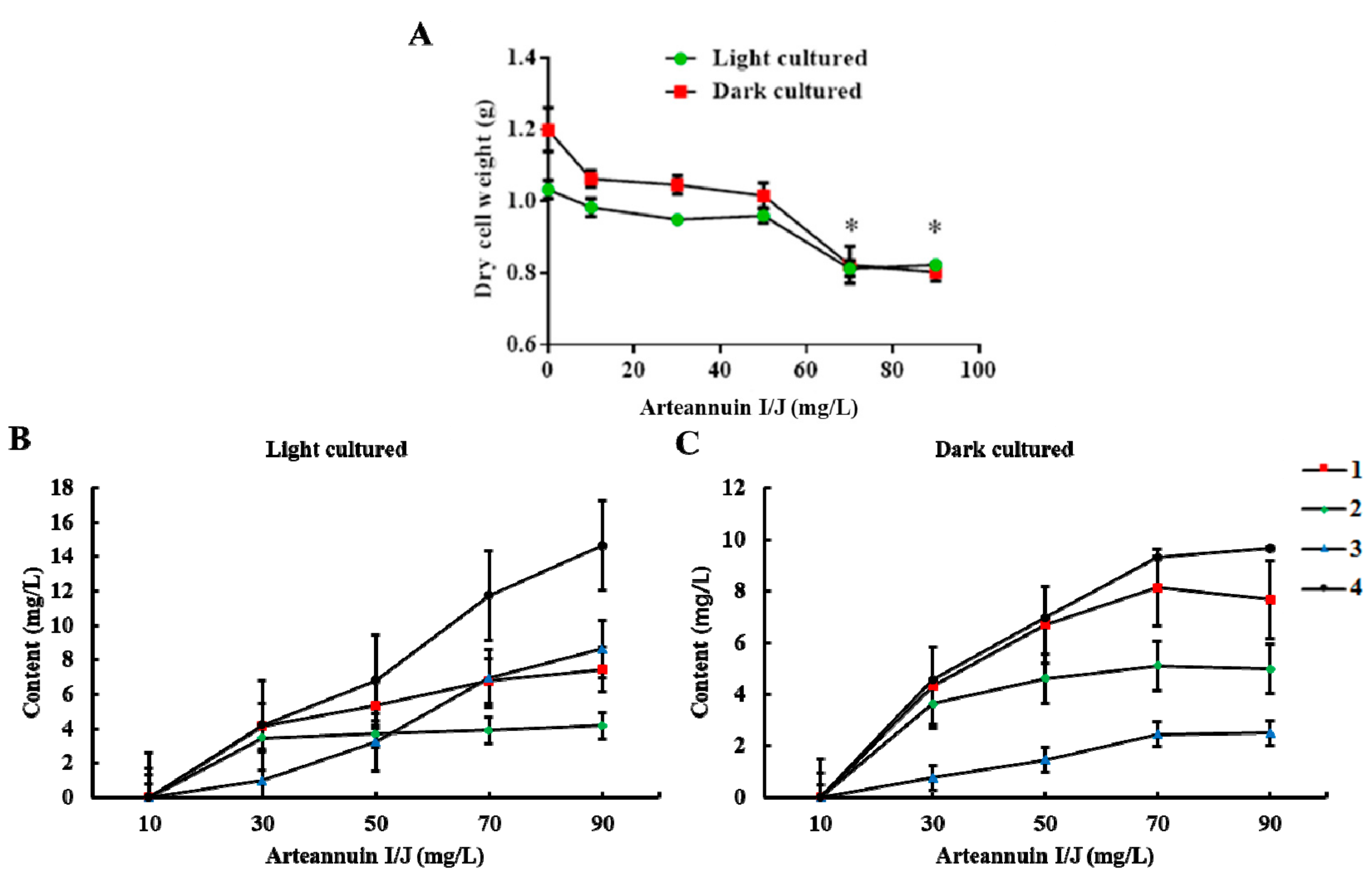
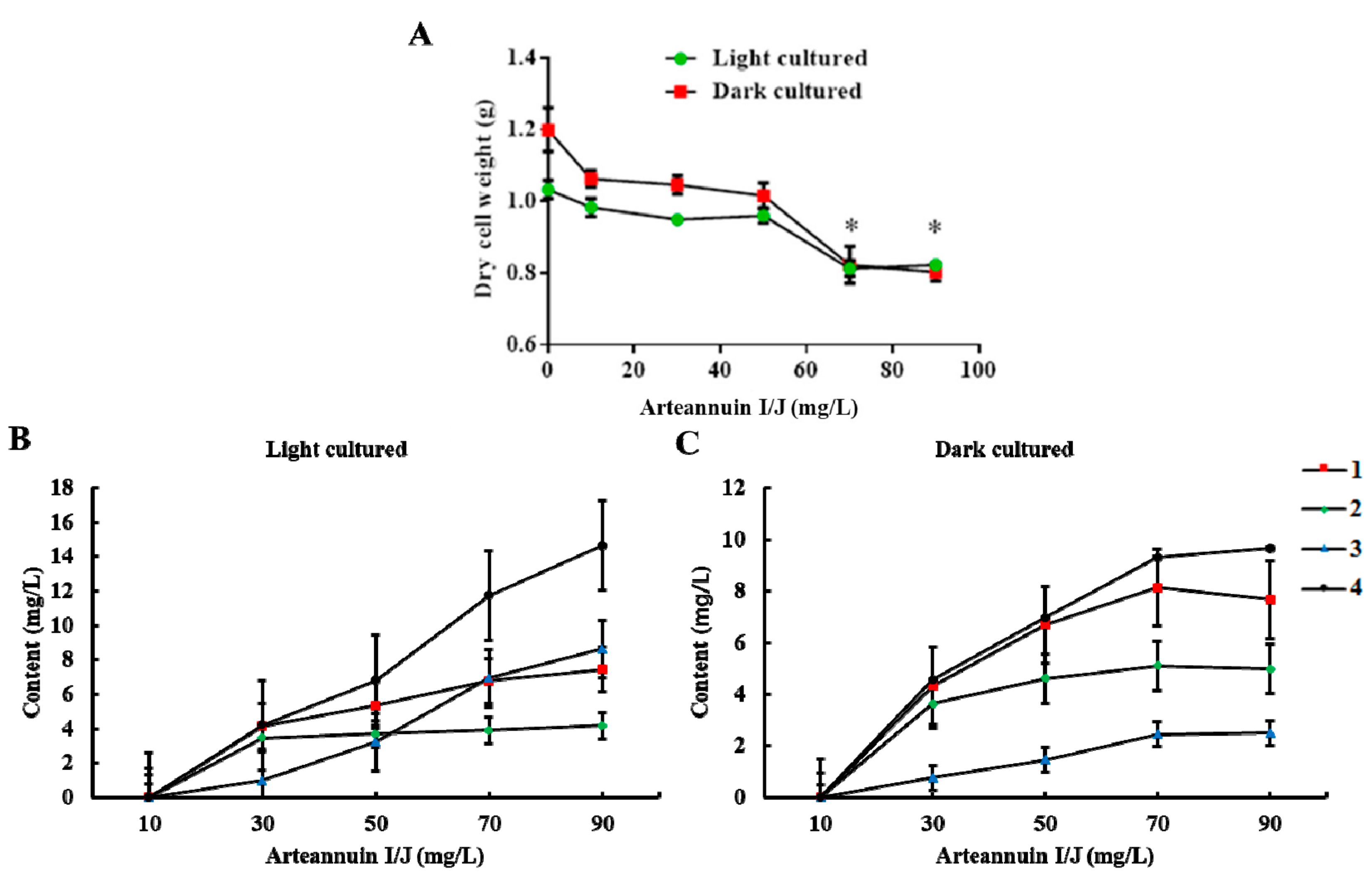
2.2. Effects of Arteannuin I/J Dosage on Cell Growth and the Content of New Products
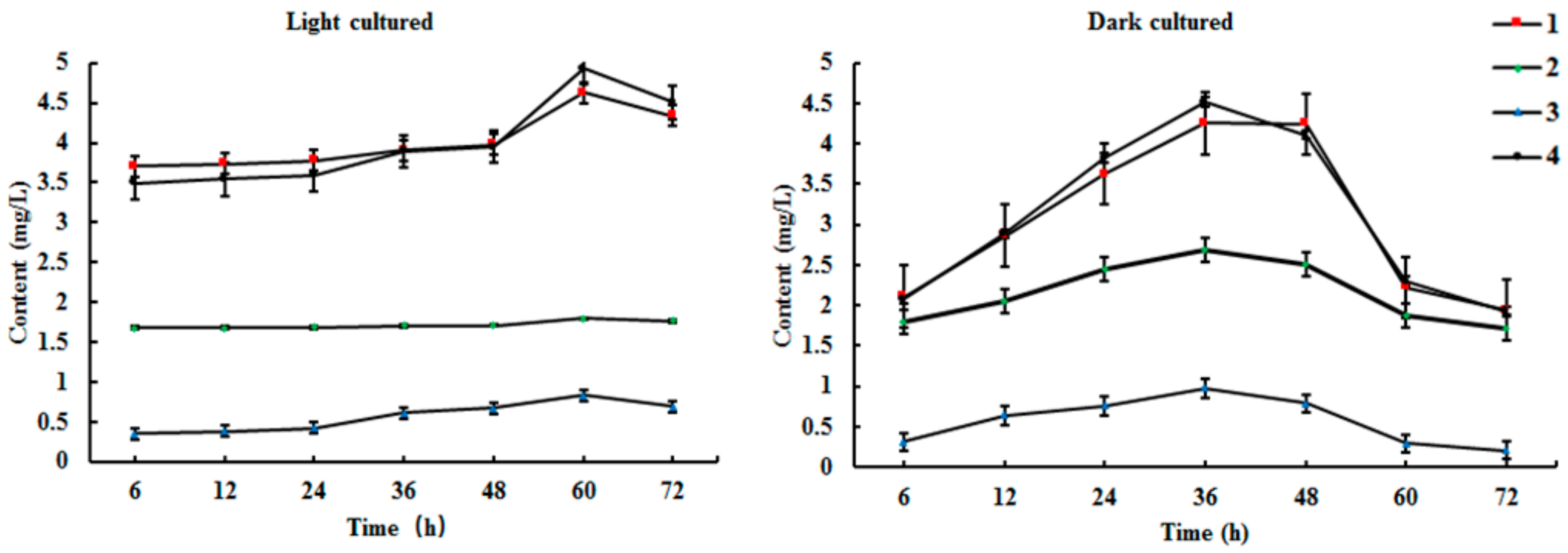
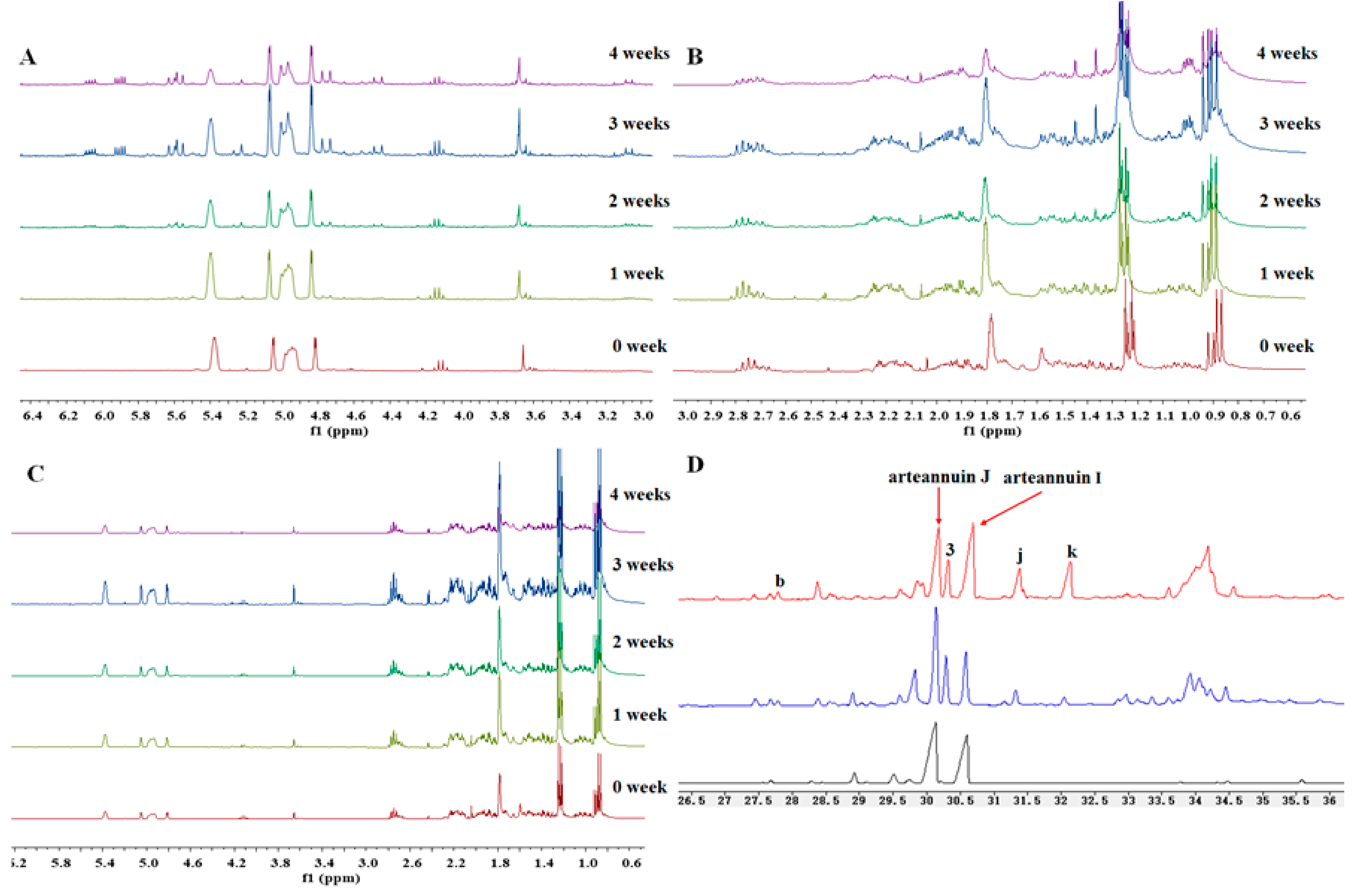
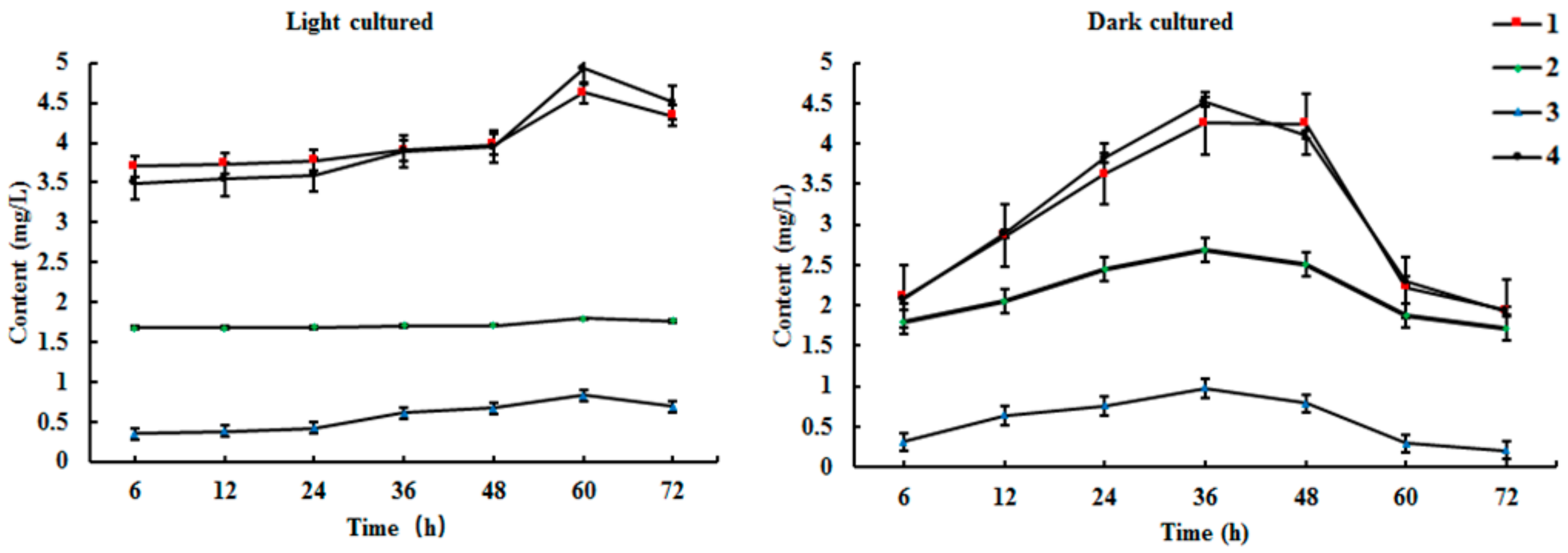
2.3. Time Course of Biosynthesis Reaction
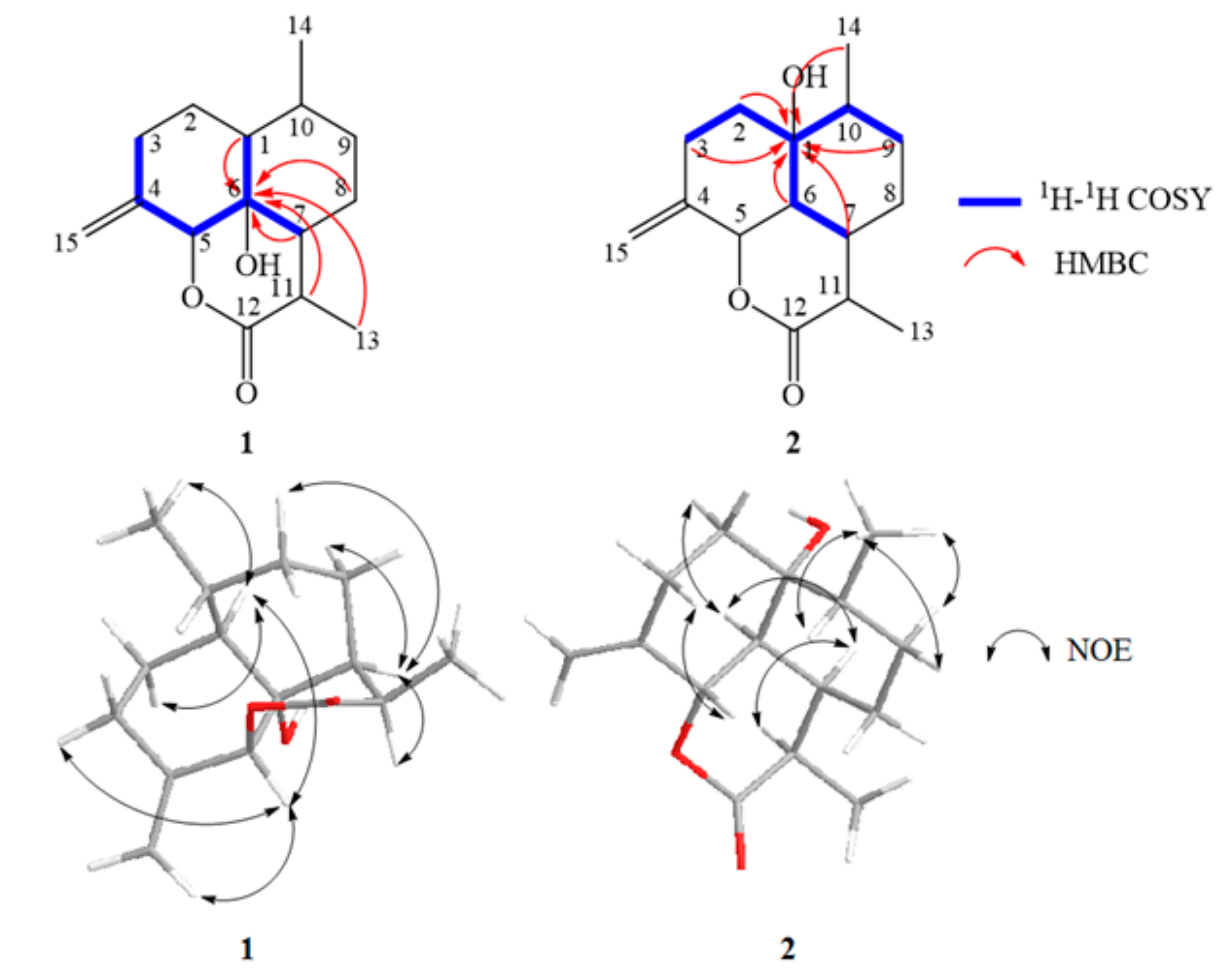
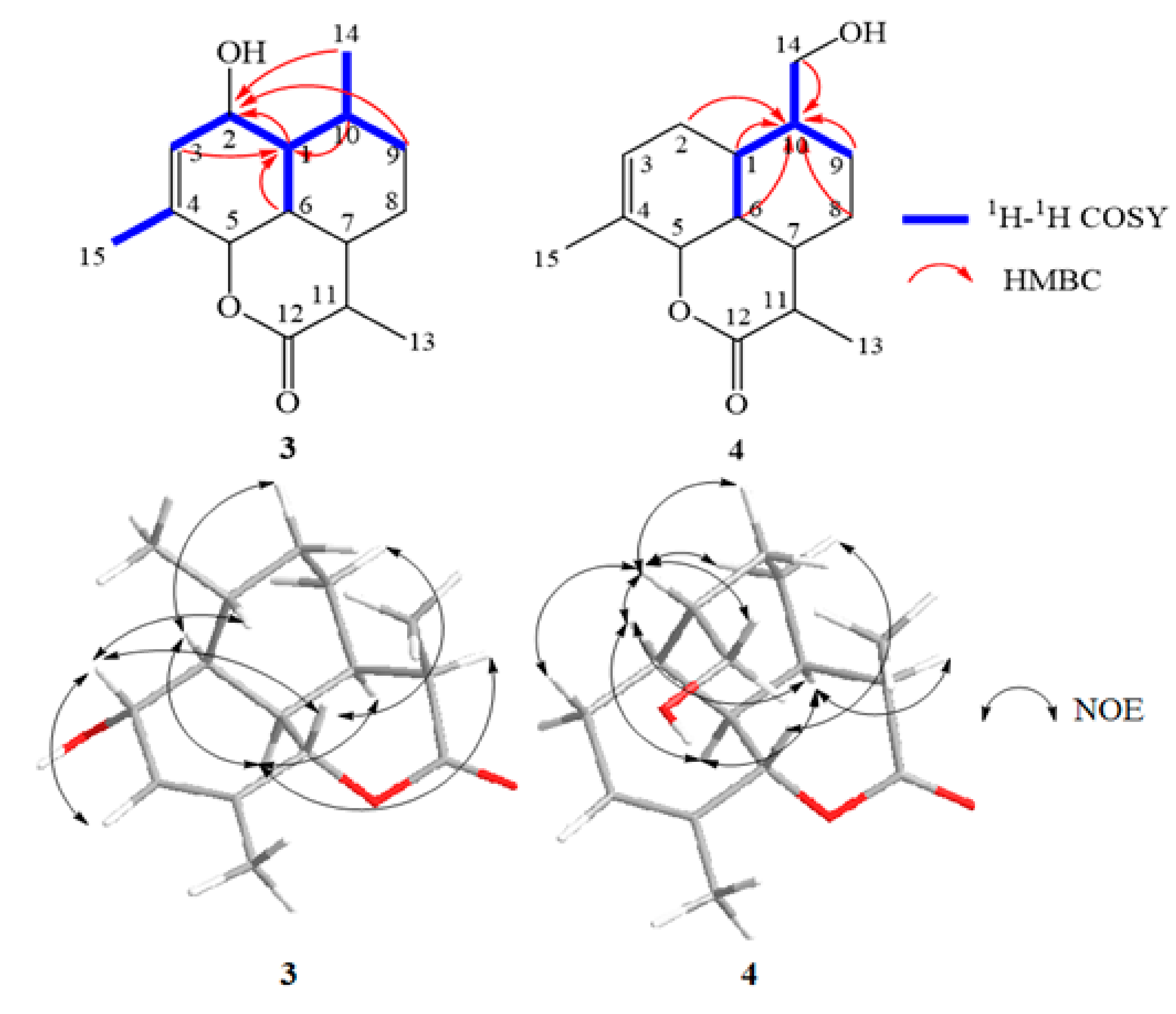
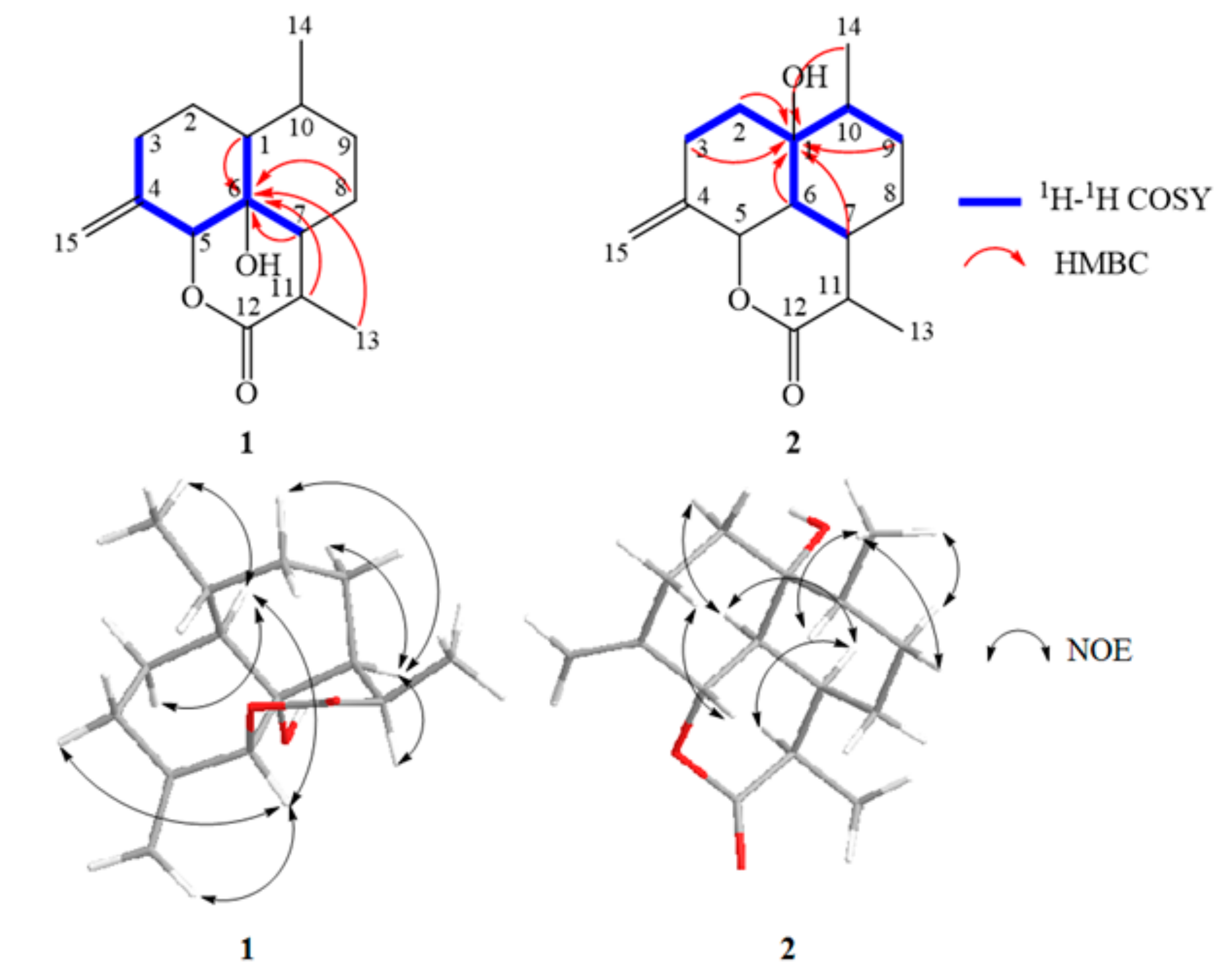
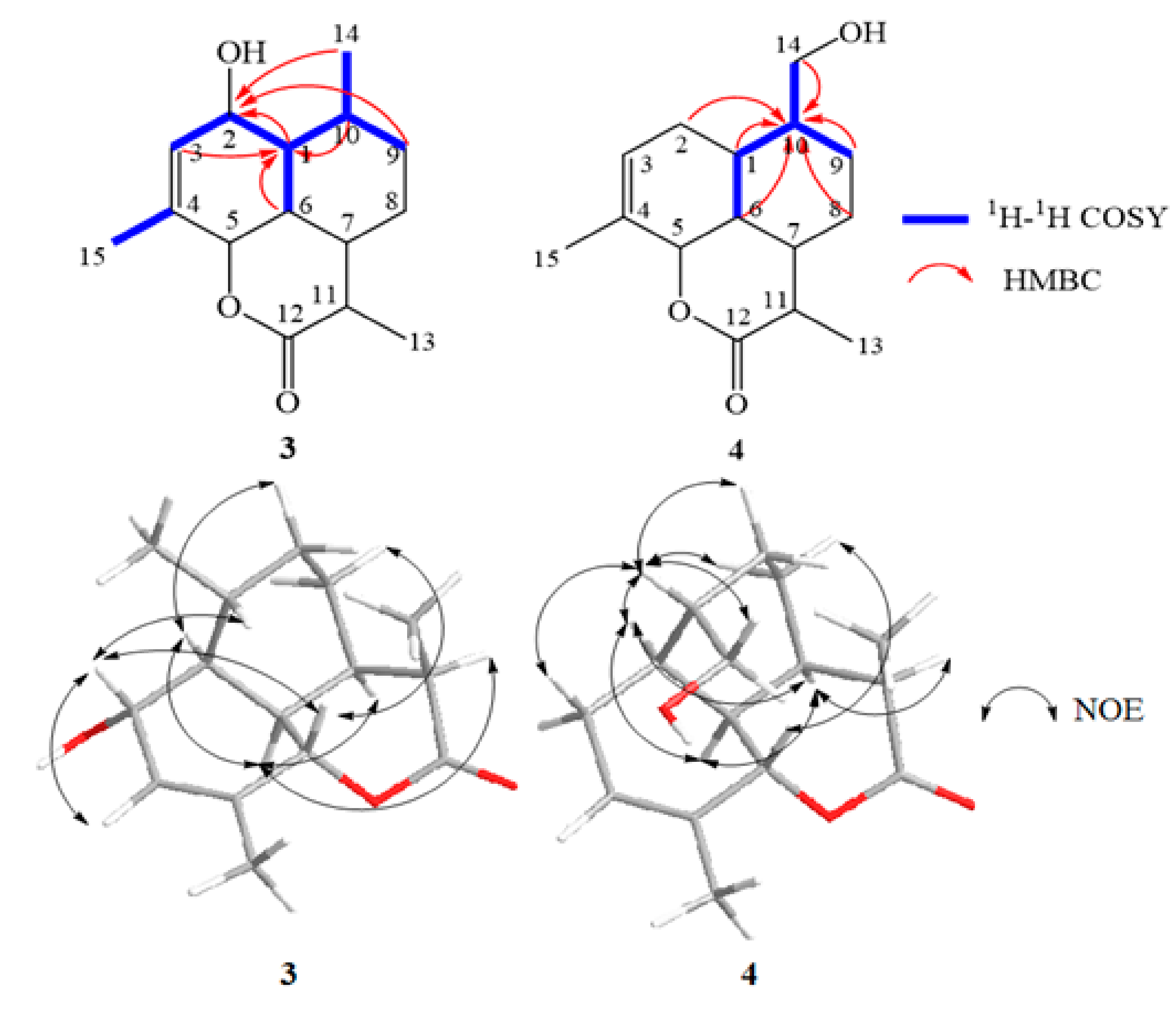
2.4. Structural Elucidation of the New Products
2.5. Detection of Spontaneous Autoxidation Products of Arteannuin I/J
3. Materials and Methods
3.1. General
3.2. Chemical Synthesis of Arteannuin I/J from Dihydroartemisinic Acid
3.3. Plant Cell Cultures
3.4. Detection of Secondary Metabolites after Feeding of Arteannuin I/J
3.5. Isolation and Structure Elucidation of Compound 1–4
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2016; Working Papers; World Health Organization: Geneva, Switzerland, 2016; Volume 30, pp. 189–206. ISBN 978-92-4-151171-1. Available online: https://malariaworld.org/blog/world-malaria-report-2016 (accessed on 12 December 2017).
- Thwing, J.; Eisele, T.P.; Steketee, R.W. Protective efficacy of malaria case management and intermittent preventive treatment for preventing malaria mortality in children: A systematic review for the Lives Saved Tool. BMC Public Health 2011, 11 (Suppl. S3), 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abdin, M.Z.; Israr, M.; Rehman, R.U.; Jain, S.K. Artemisinin, a novel antimalarial drug: Biochemical and molecular approaches for enhanced production. Planta Med. 2003, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Y.; Cook, S.P. A concise synthesis of (+)-artemisinin. J. Am. Chem. Soc. 2012, 134, 13577–13579. [Google Scholar] [CrossRef] [PubMed]
- Corsello, M.A.; Garg, N.K. Synthetic chemistry fuels interdisciplinary approaches to the production of artemisinin. Nat. Prod. Rep. 2015, 32, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; BenJamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Yan, T.X.; Fu, X.Q.; Tang, K.X. Transcriptional regulation of artemisinin biosynthesis in Artemisia annua L. Sci. Bull. 2016, 61, 18–25. [Google Scholar] [CrossRef]
- Ma, D.M.; Li, G.; Zhu, Y.; Xie, D.Y. Overexpression and Suppression of Artemisia annua 4-Hydroxy-3-Methylbut-2-enyl Diphosphate Reductase 1 Gene (AaHDR1) Differentially Regulate Artemisinin and Terpenoid Biosynthesis. Front. Plant Sci. 2017, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yuan, M.; Zhang, Q.; Zhu, Y.M.; Yong, L.; Wang, W.; Qi, Y.; Guo, D.J. Chemotype-dependent Metabolic Response to Methyl Jasmonate Elicitation in Artemisia annua. Planta Med. 2011, 77, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Wallaart, T.E.; Pras, N.; Beekman, A.C.; Quax, W.J. Seasonal Variation of Artemisinin and its Biosynthetic Precursors in Plants of Artemisia annua of Different Geographical Origin: Proof for the Existence of Chemotypes. Planta Med. 2000, 66, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; Dixit, V.K. Yield enhancement strategies for artemisinin production by suspension cultures of Artemisia annua. Bioresour. Technol. 2008, 99, 4609–4614. [Google Scholar] [CrossRef] [PubMed]
- Sy, L.K.; Brown, G.D.; Haynes, R. A novel endoperoxide and related sesquiterpenes from Artemisia annua which are possibly derived from allylic hydroperoxides. Tetrahedron 1998, 29, 4345–4356. [Google Scholar] [CrossRef]
- Brown, G.D.; Sy, L.K. In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron 2004, 60, 1139–1159. [Google Scholar] [CrossRef]
- Zhu, J.H.; Yang, J.Z.; Zeng, Z.H.; Zhang, W.J.; Song, L.Y.; Wen, W.; Yu, R.M. Inducing Effect of Dihydroartemisinic Acid in the Biosynthesis of Artemisinins with Cultured Cells of Artemisia annua by Enhancing the Expression of Genes. Sci. World J. 2014, 293190. [Google Scholar] [CrossRef]
- Sy, L.K.; Brown, G.D. The mechanism of the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron 2002, 58, 897–908. [Google Scholar] [CrossRef]
- Nakamura, M.; Satoh, T.; Tanaka, S.I.; Mochizuki, N.; Yokota, T.; Nagatani, A. Activation of the cytochrome P450 gene, CYP72C1, reduces the levels of active brassinosteroids in vivo. J. Exp. Bot. 2005, 56, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Nakazawa, M.; Shibata, K.; Yokota, T.; Ishikawa, A.; Suzuki, K.; Kawashima, M.; Ichikawa, T.; Shimada, H.; Matsui, M. shk1-D, A dwarf Arabidopsis mutant caused by activation of the CYP72C1 gene, has altered brassinosteroid levels. Plant J. 2005, 42, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Janocha, S.; Schmitz, D.; Bernhardt, R. Terpene hydroxylation with microbial cytochrome P450 monooxygenases. Adv. Biochem. Eng. Biotechnol. 2015, 148, 215–250. [Google Scholar] [PubMed]
- Zhang, X.; Yang, Z.; Xu, P.; Ding, R. Synthesis of (15-14C) dihydroartemisilacton. Hejishu 1993, 16, 759–761. Available online: https://www.researchgate.net/publication/298495013_Synthesis_of_15-14C_dihydroartemisilacton (accessed on 12 December 2017).
- Vlad, P.F.; Ungur, N.D.; Aricu, A.N.; Andreeva, I.Y. Regioselective dehydration of axial and equatorial tertiary alcohols with α-methyl group in the cyclohexane ring by Swern’s reagent. Russ. Chem. Bull. 1997, 46, 767–770. [Google Scholar] [CrossRef]
Sample Availability: Sample of the arteannuin I/J is available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | ||
|---|---|---|---|---|
| δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | |
| 1 | 1.76, m | 27.75 | 73.36 | |
| 2α | 1.42, m | 28.44 | 1.40, m | 37.74 |
| 2β | 1.69, m | 2.16, m | ||
| 3α | 2.29, m | 29.85 | 2.34, m | 29.57 |
| 3β | 1.24, m | 2.06, m | ||
| 4 | 145.35 | 144.39 | ||
| 5 | 4.76, d (12.1) | 76.79 | 4.91, d (12.3) | 76.17 |
| 6 | 71.71 | 1.87, dd (11.8, 3.9) | 51.29 | |
| 7 | 1.82, d (4.2) | 51.20 | 2.39, dd (9.46, 3.6) | 34.72 |
| 8α | 2.02, m | 28.22 | 1.78, m | 22.51 |
| 8β | 1.52, d (3.8) | 1.30, m | ||
| 9α | 1.73, dt (5.6, 2.8) | 30.15 | 1.46, dd (7.6, 5.1) | 29.82 |
| 9β | 2.20, m | 1.68, m | ||
| 10 | 2.00, m | 38.64 | 1.93, d (6.5) | 31.56 |
| 11 | 2.57, d (7.1) | 48.73 | 2.64, p (7.2) | 40.23 |
| 12 | 172.79 | 174.33 | ||
| 13 | 1.29, d (7.2) | 8.90 | 1.24, d (7.2) | 13.54 |
| 14 | 0.94, d (6.2) | 19.90 | 0.92, d (6.6) | 14.36 |
| 15a | 5.08, s | 105.77 | 4.88, d (1.5) | 106.69 |
| 15b | 4.85, d (1.7) | 5.13, d (1.2) | ||
| OH | 1.58, s | 1.60, s |
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | |
| 1 | 1.58, d (4.0) | 50.27 | 2.19, dt (10.8, 3.7) | 40.40 |
| 2 | 4.18, d (4.1) | 65.48 | 2.25, (overlap) | 27.12 |
| 3 | 5.57, m | 124.16 | 5.39, m | 122.77 |
| 4 | 145.35 | 132.08 | ||
| 5 | 4.84, d (11.1) | 74.92 | 4.95, d (10.8) | 75.28 |
| 6 | 2.38, dt (11.0, 3.6) | 35.88 | 1.87, m | 36.23 |
| 7 | 1.94, m | 38.64 | 1.96, m | 38.73 |
| 8α | 1.76, m | 23.11 | 1.83, m | 22.76 |
| 8β | 1.33, dd (9.9, 4.1) | 1.39, dd (13.2, 3.8) | ||
| 9α | 1.07, dd (12.0, 4.2) | 34.96 | 1.25, d (7.3) | 29.31 |
| 9β | 1.86, (overlap) | 2.01, m | ||
| 10 | 1.29, m | 28.09 | 1.52, m | 36.77 |
| 11 | 2.78, p (7.2) | 40.87 | 2.77, p (7.2) | 40.80 |
| 12 | 175.14 | 175.43 | ||
| 13 | 1.25, d (7.3) | 13.45 | 1.25, d (7.3) | 13.48 |
| 14a | 0.98, d (6.2) | 20.17 | 3.69, dd (10.8, 3.3) | 65.67 |
| 14b | 3.53, dd (10.8, 5.8) | |||
| 15 | 1.86, (overlap) | 18.44 | 1.79, dd (2.4, 1.2) | 18.43 |
| OH | 1.60, m | 1.60, m |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Xiao, P.; Qian, M.; Chen, C.; Liang, C.; Zi, J.; Yu, R. Four New Compounds Obtained from Cultured Cells of Artemisia annua. Molecules 2017, 22, 2264. https://doi.org/10.3390/molecules22122264
Zhu J, Xiao P, Qian M, Chen C, Liang C, Zi J, Yu R. Four New Compounds Obtained from Cultured Cells of Artemisia annua. Molecules. 2017; 22(12):2264. https://doi.org/10.3390/molecules22122264
Chicago/Turabian StyleZhu, Jianhua, Peijie Xiao, Minghua Qian, Chang Chen, Chuxin Liang, Jiachen Zi, and Rongmin Yu. 2017. "Four New Compounds Obtained from Cultured Cells of Artemisia annua" Molecules 22, no. 12: 2264. https://doi.org/10.3390/molecules22122264
APA StyleZhu, J., Xiao, P., Qian, M., Chen, C., Liang, C., Zi, J., & Yu, R. (2017). Four New Compounds Obtained from Cultured Cells of Artemisia annua. Molecules, 22(12), 2264. https://doi.org/10.3390/molecules22122264