Novel Mixed-Type Inhibitors of Protein Tyrosine Phosphatase 1B. Kinetic and Computational Studies


Abstract
:1. Introduction
2. Results and Discussion
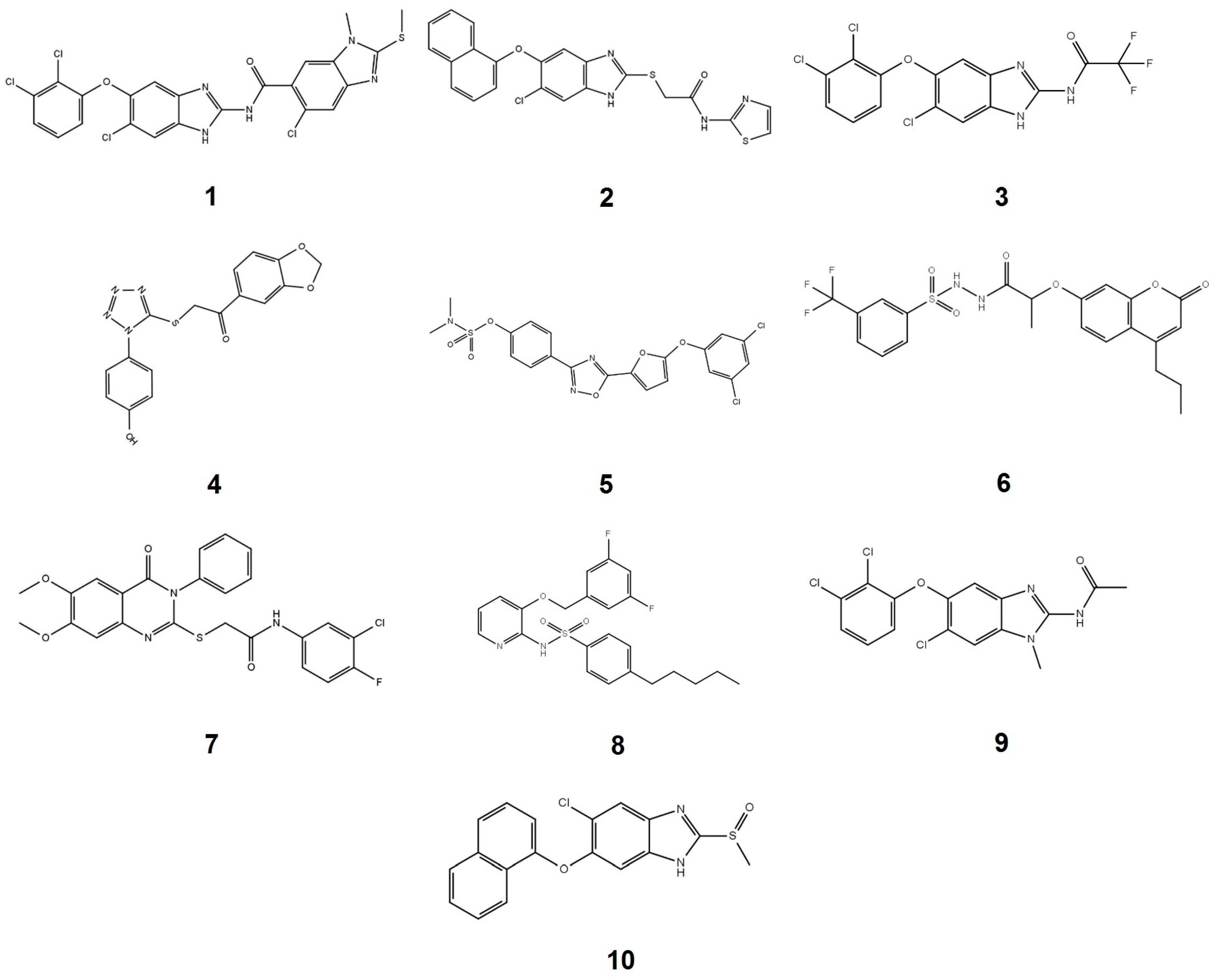
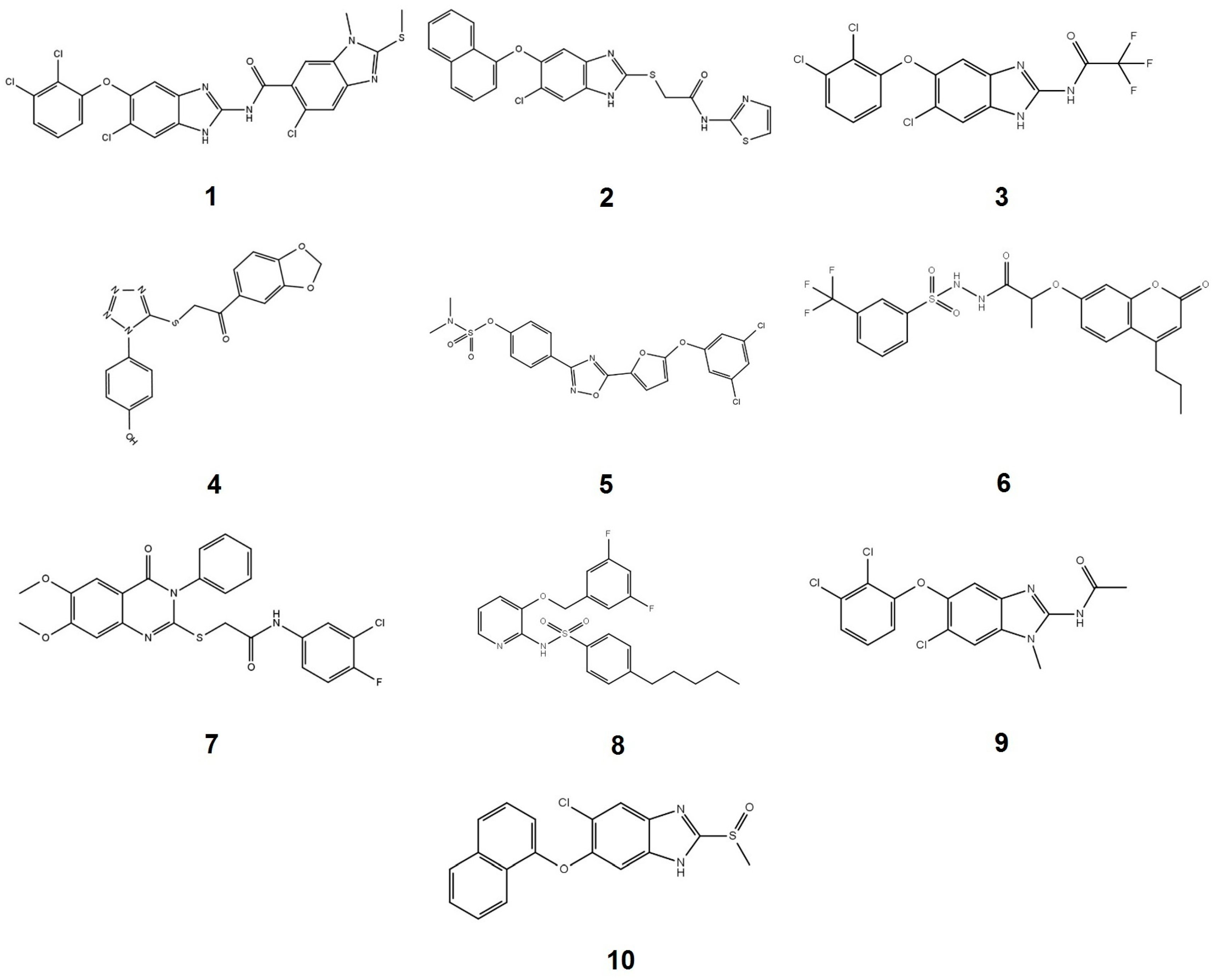
2.1. Compounds Screening
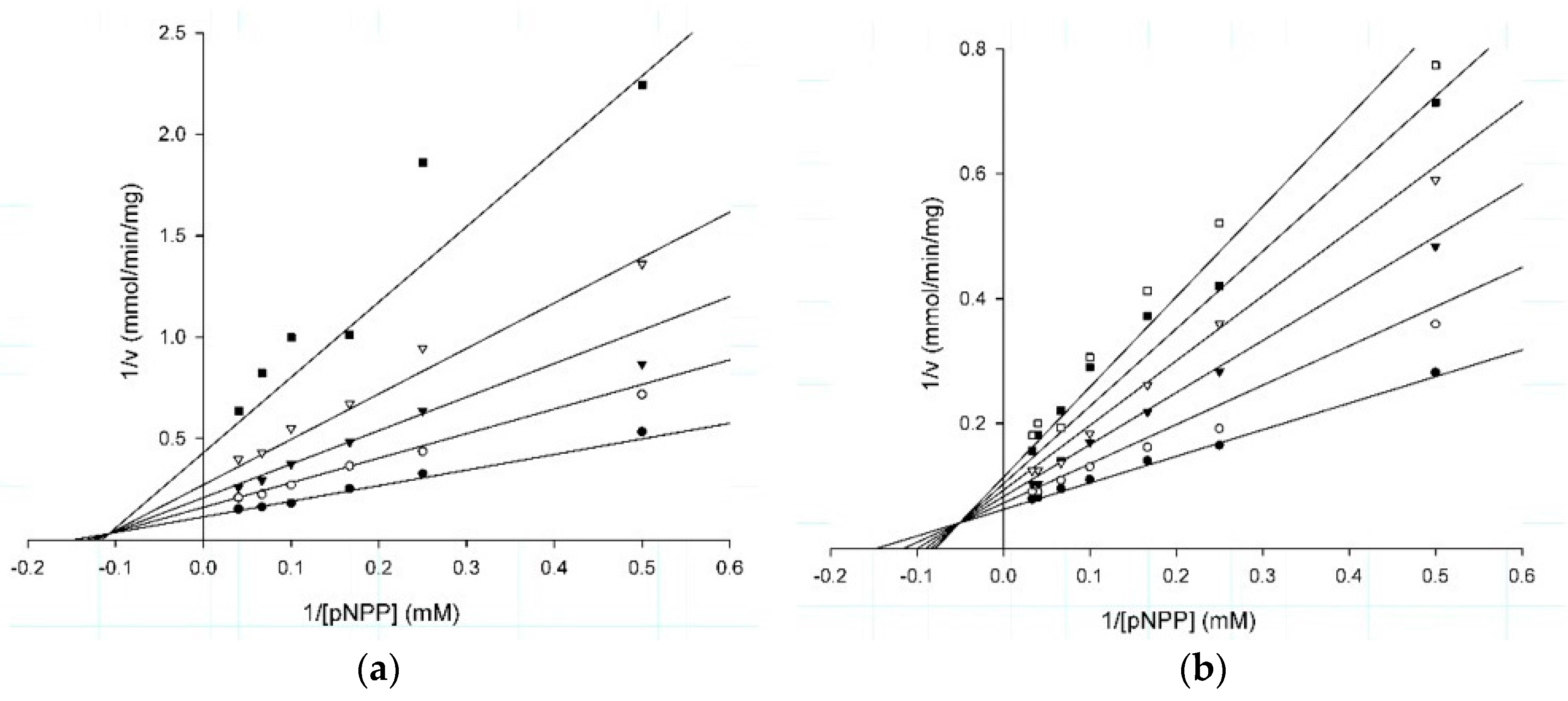
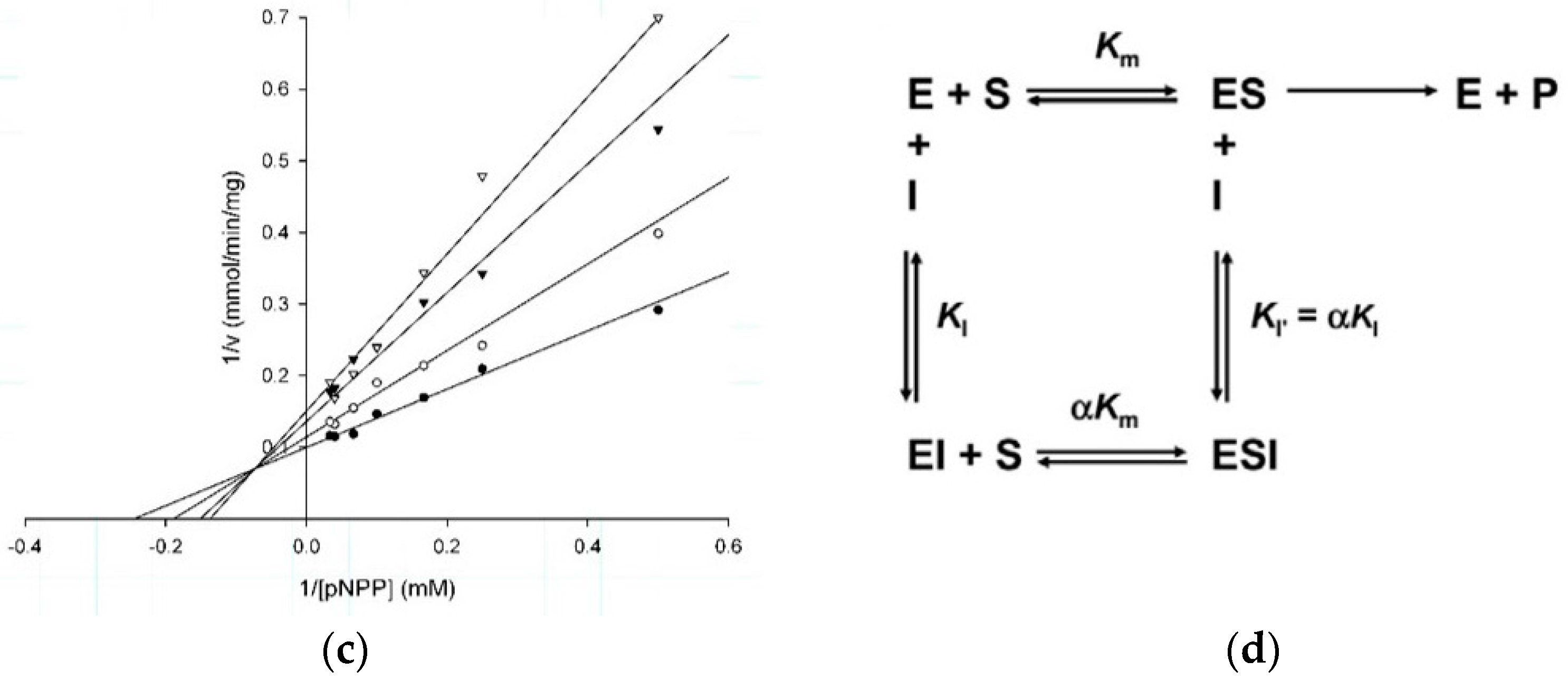
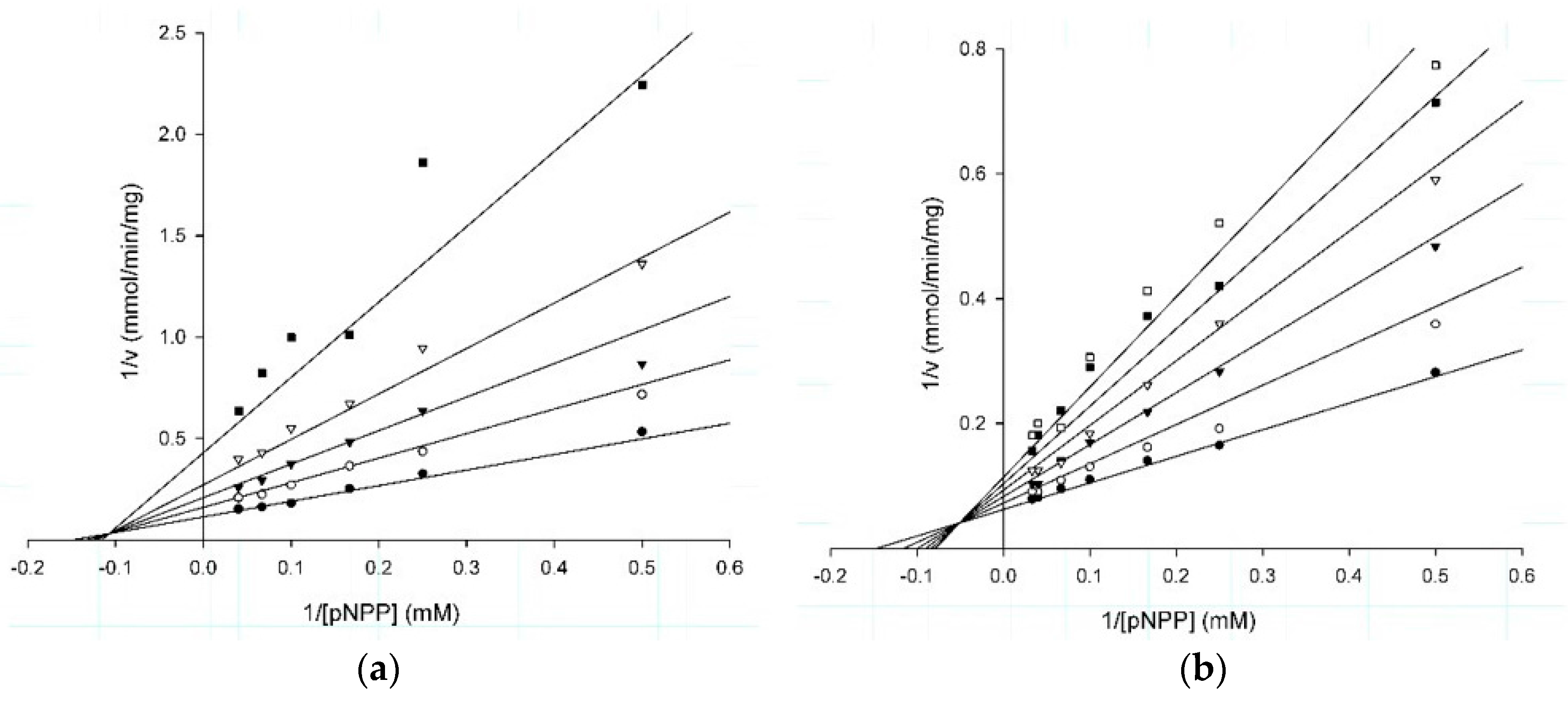
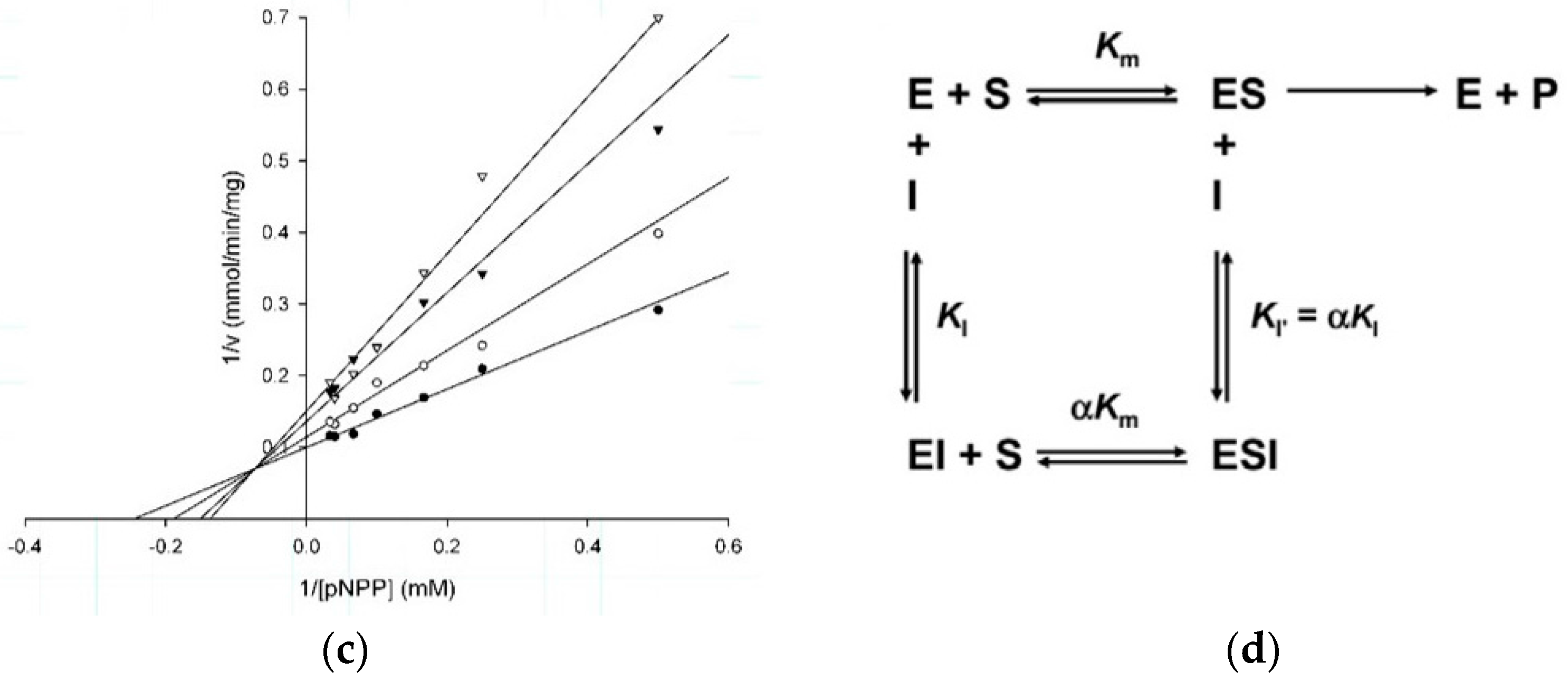
2.2. Kinetic Studies
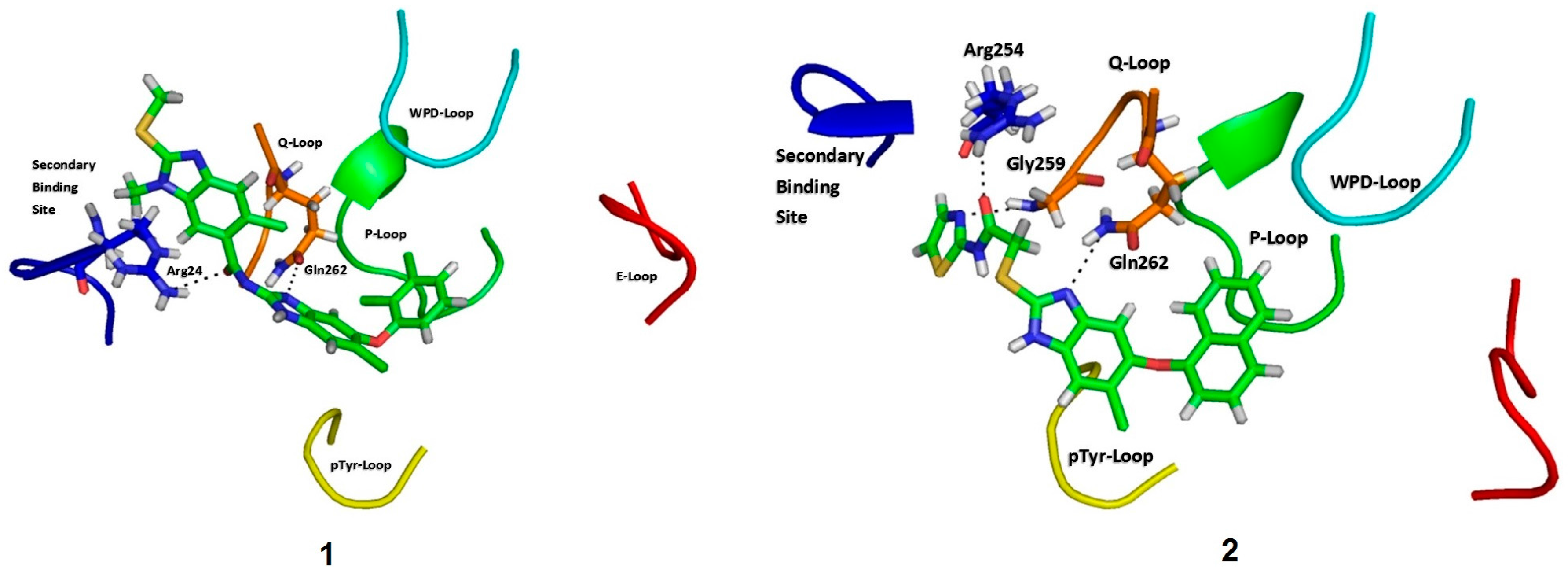
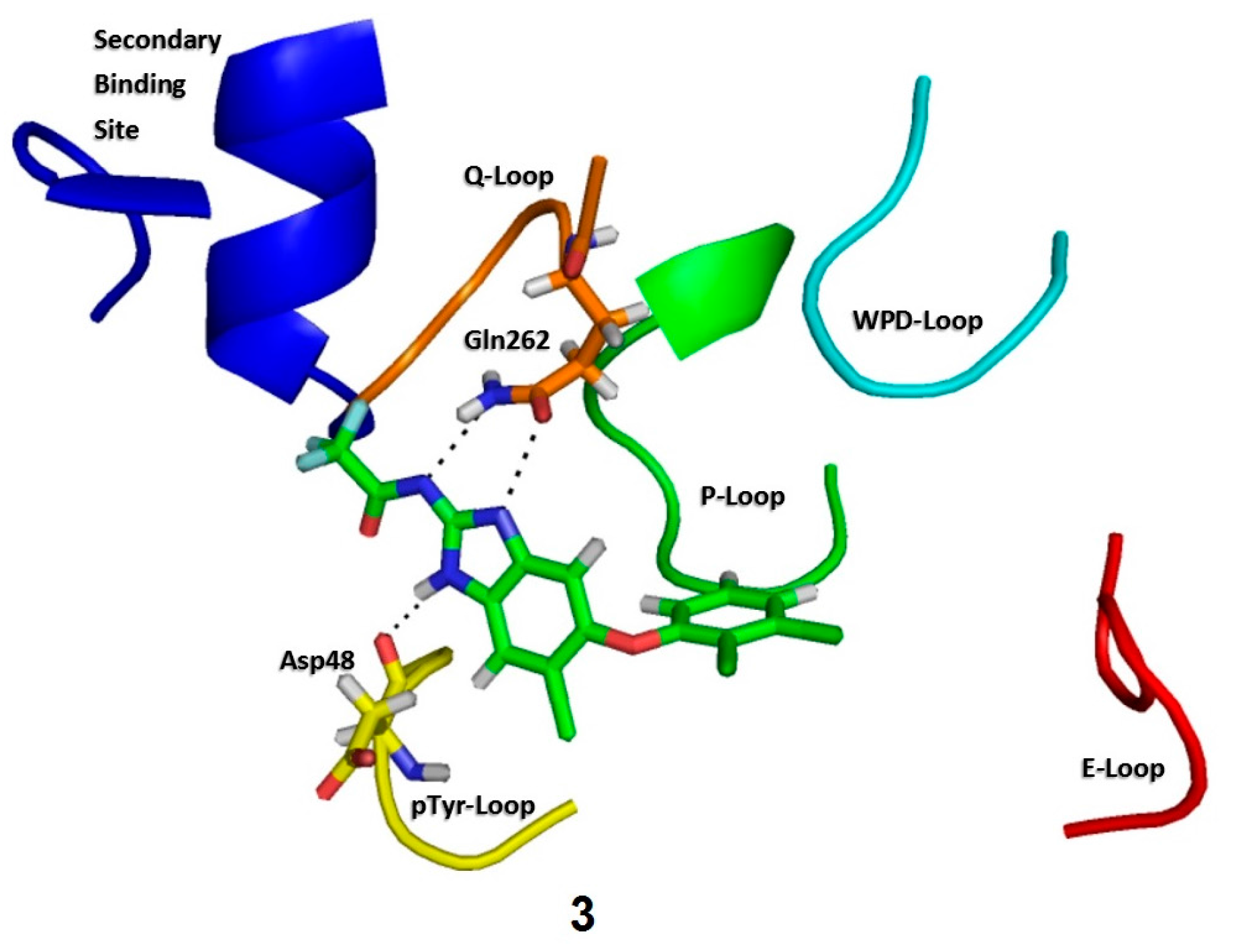
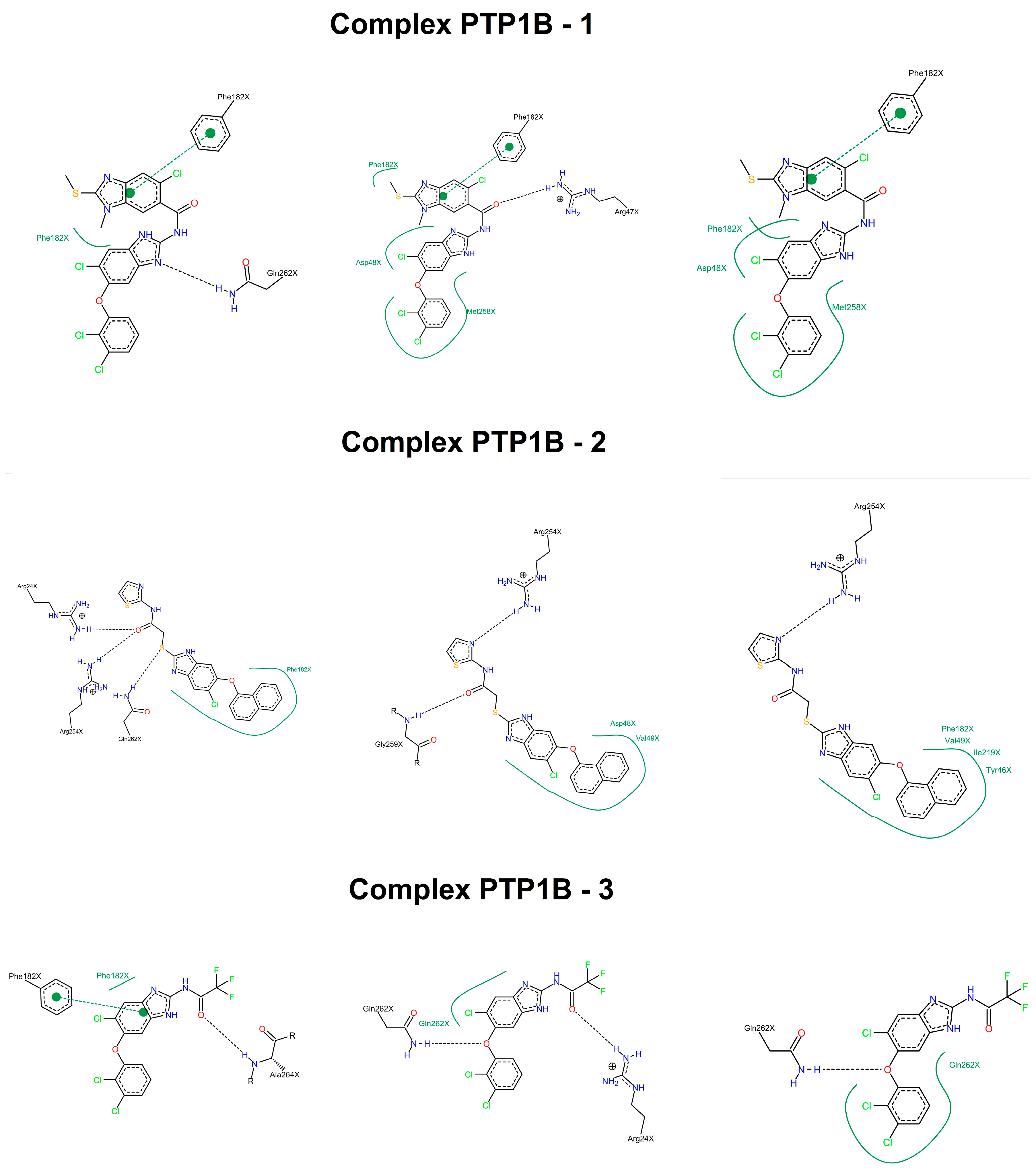
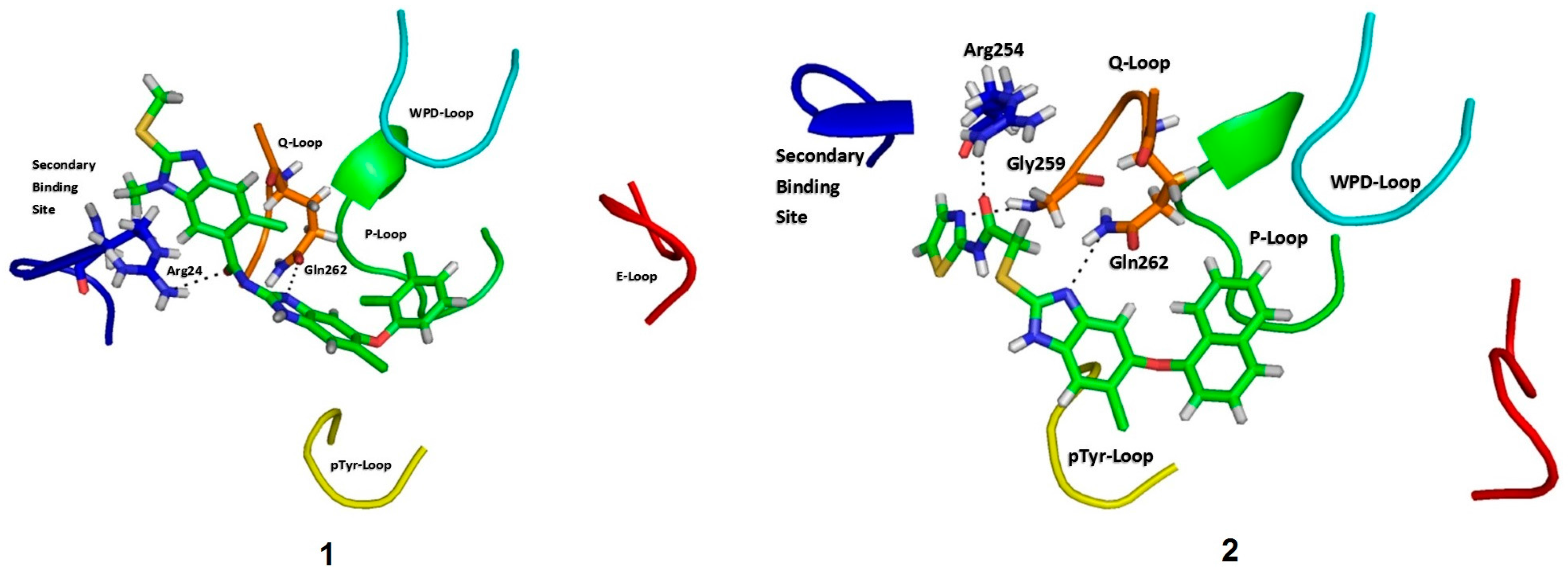
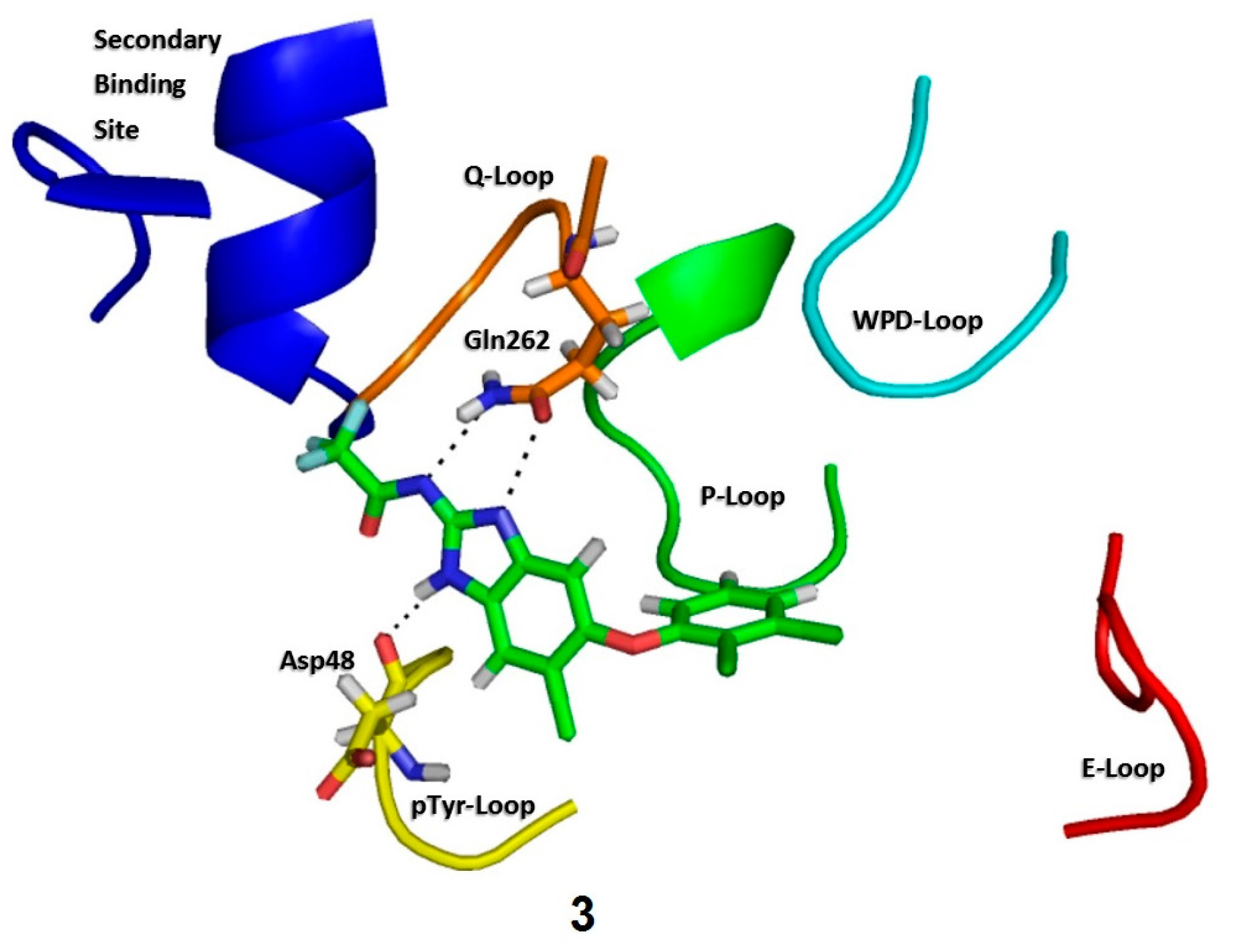
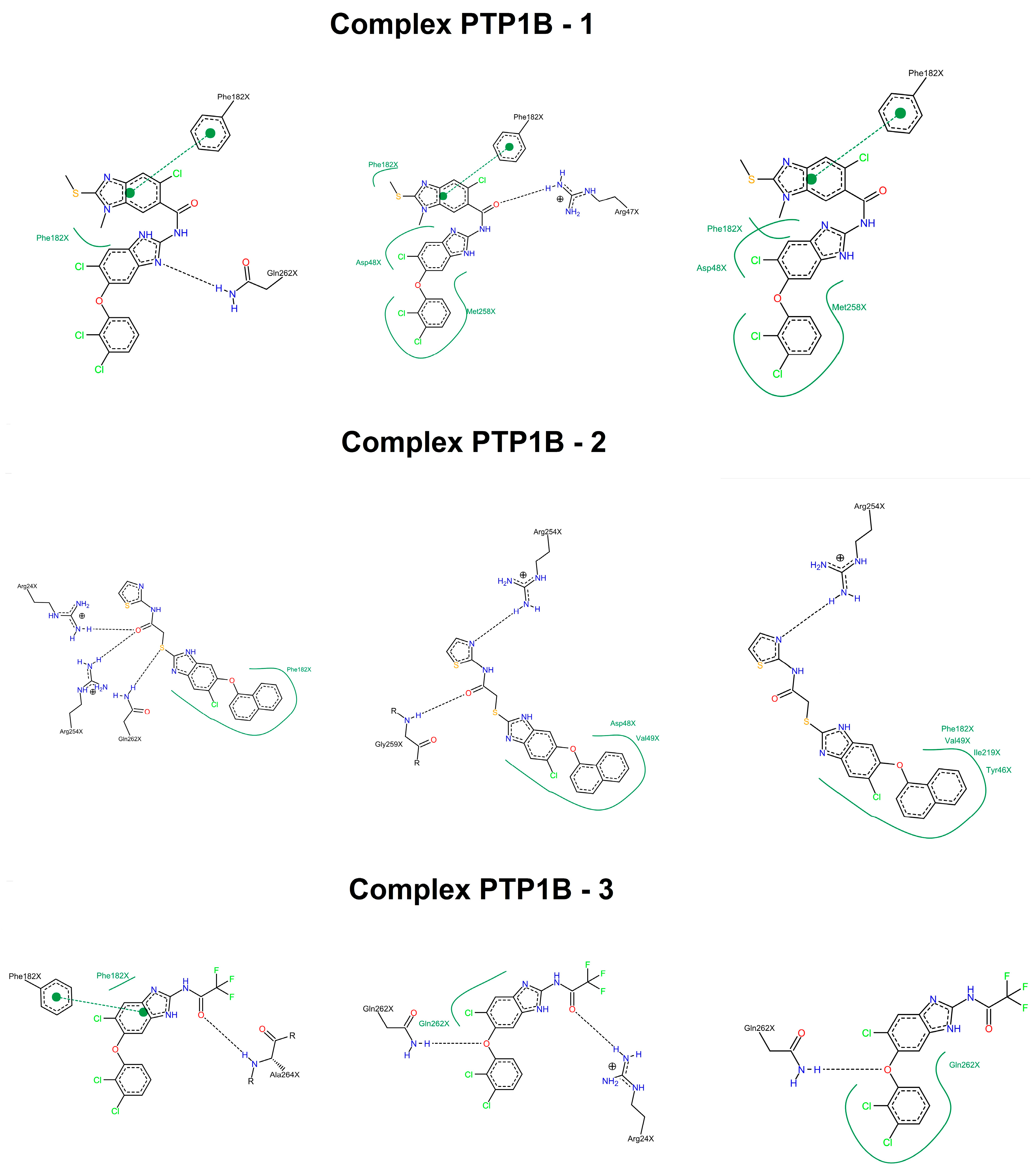
2.3. Molecular Docking
2.4. Molecular Dynamics Simulations
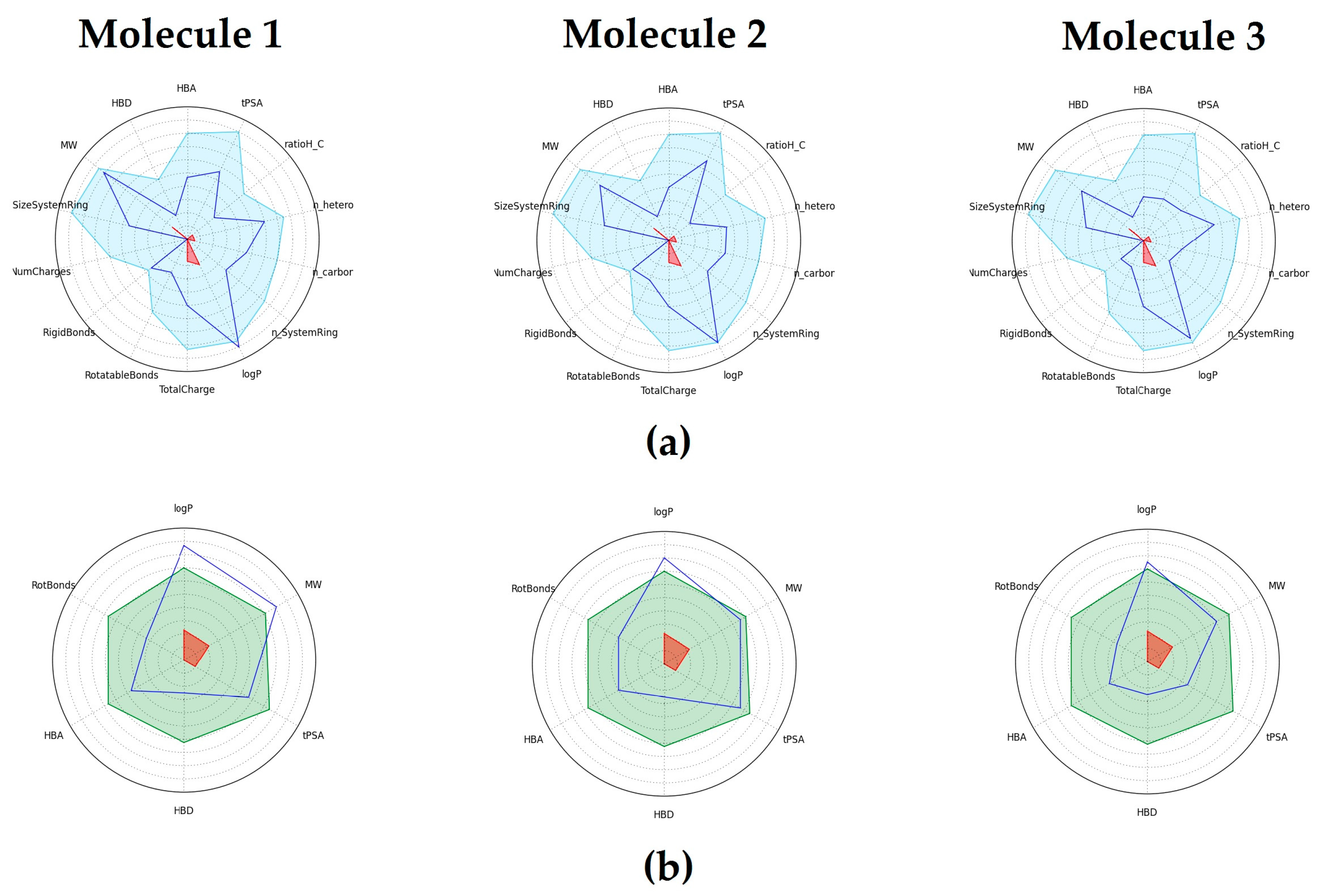
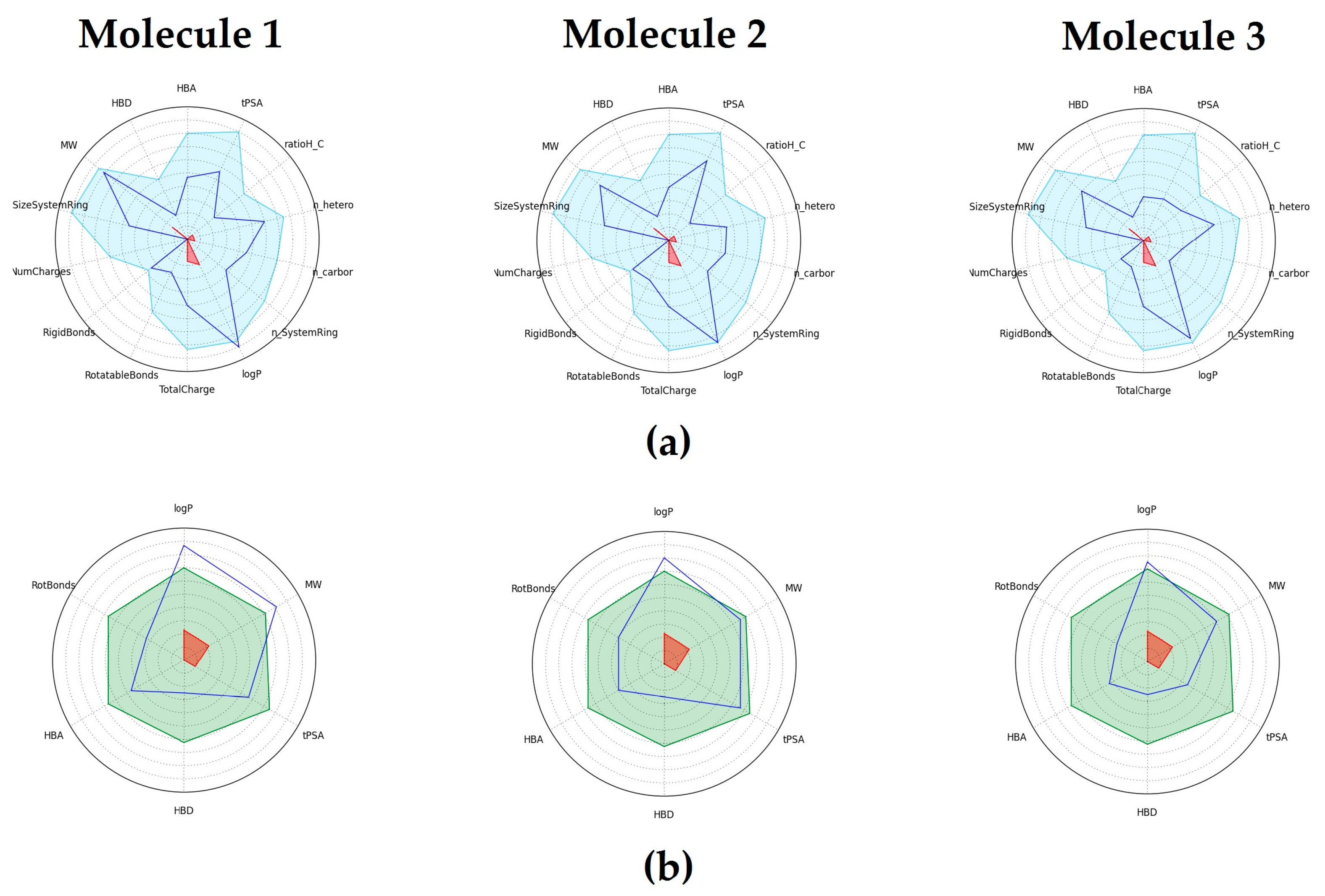
2.5. Physicochemical and Drug-Like properties
2.6. Toxicological Evaluation
3. Materials and Methods
3.1. General Information
3.2. Compounds
3.3. Expression and Purification of PTP1B
3.4. Enzymatic Activity
3.5. Inhibition Assays
3.6. Molecular Docking
3.7. Molecular Dynamic Simulations
3.8. Drug-Like and Toxicological Propierties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 2nd ed.; International Diabetes Federation: Brussels, Belgium, 2003. [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussels, Belgium, 2014. [Google Scholar]
- Bujaidar, E.M.; Juárez, N.G. Revisión de las caracteristicas clinicas, metabólicas y genéticas de la diabetes mellitus. Bioquimia 2003, 28, 14–23. [Google Scholar]
- Gershell, L. Type 2 diabetes market. Nat. Rev. Drug Discov. 2005, 4, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.N.; Sobhia, M.E. Binding and discerning interactions of PTP1B allosteric inhibitors: Novel insights from molecular dynamics simulations. J. Mol. Graph. Model. 2013, 45, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Frittitta, L.; Miscio, G.; Bozzali, M.; Baratta, R.; Centra, M.; Spampinato, D.; Santagati, M.G.; Ercolino, T.; Cisternino, C.; et al. A variation in 3′ UTR of hPTP1B increases specific gene expression and associates with insulin resistance. Am. J. Hum. Genet. 2002, 70, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Bento, J.L.; Palmer, N.D.; Mychaleckyj, J.C.; Lange, L.A.; Langefeld, C.D.; Rich, S.S.; Freedman, B.I.; Bowden, D.W. Association of protein tyrosine phosphatase 1B gene polymorphisms with type 2 diabetes. Diabetes 2004, 53, 3007–3012. [Google Scholar] [CrossRef] [PubMed]
- Asante-Appiah, E.; Kennedy, B.P. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E663–E670. [Google Scholar] [CrossRef] [PubMed]
- Cho, H. Protein Tyrosine Phosphatase 1B (PTP1B) and Obesity, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 91. [Google Scholar]
- Bakke, J.; Bettaieb, A.; Nagata, N.; Matsuo, K.; Haj, F.G. Regulation of the SNARE-interacting protein Munc18c tyrosine phosphorylation in adipocytes by protein-tyrosine phosphatase 1B. Cell Commun. Signal. 2013, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Kushner, J.A.; Haj, F.G.; Klaman, L.D.; Dow, M.A.; Kahn, B.B.; Neel, B.G.; White, M.F. Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes 2004, 53, 61–66. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zeng, L.F.; He, Y.; Zhang, S.; Zhang, Z.Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013, 280, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, W.; Liu, X.; Yu, H.; Lu, X.; Jiao, B. Inhibitors of Protein Tyrosine Phosphatase 1B from Marine Natural Products. Chem. Biodivers. 2017, 14, e1600462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-S.; Liang, L.; Guo, Y. Natural products possessing protein tyrosine phosphatase 1B (PTP1B) inhibitory activity found in the last decades. Acta Pharmacol. Sin. 2012, 33, 1217–1245. [Google Scholar] [CrossRef] [PubMed]
- Bharatam, P.V.; Patel, D.S.; Adane, L.; Mittal, A.; Sundriyal, S. Modeling and informatics in designing anti-diabetic agents. Curr. Pharm. Des. 2007, 13, 3518–3530. [Google Scholar] [CrossRef] [PubMed]
- Combs, A.P. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 2010, 53, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Popov, D. Novel protein tyrosine phosphatase 1B inhibitors: Interaction requirements for improved intracellular efficacy in type 2 diabetes mellitus and obesity control. Biochem. Biophys. Res. Commun. 2011, 410, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Abraham, A.; Asano, J.; Breslin, C.; Dick, C.A.J.; Ixkes, U.; Johnston, B.F.; Johnston, D.; Kewnay, J.; Mackay, S.P.; et al. 2-Aryl-3,3,3-trifluoro-2-hydroxypropionic acids: A new class of protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6579–6583. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, G.; Xin, Z.; Serby, M.D.; Pei, Z.; Szczepankiewicz, B.G.; Hajduk, P.J.; Abad-Zapatero, C.; Hutchins, C.W.; Lubben, T.H.; et al. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 5543–5546. [Google Scholar] [CrossRef] [PubMed]
- Black, E.; Breed, J.; Breeze, A.L.; Embrey, K.; Garcia, R.; Gero, T.W.; Godfrey, L.; Kenny, P.W.; Morley, A.D.; Minshull, C.A.; et al. Structure-based design of protein tyrosine phosphatase-1B inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2503–2507. [Google Scholar] [CrossRef] [PubMed]
- Douty, B.; Wayland, B.; Ala, P.J.; Bower, M.J.; Pruitt, J.; Bostrom, L.; Wei, M.; Klabe, R.; Gonneville, L.; Wynn, R.; et al. Isothiazolidinone inhibitors of PTP1B containing imidazoles and imidazolines. Bioorg. Med. Chem. Lett. 2008, 18, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Bhattarai, B.R.; Lee, K.H.; Cho, H. Mono- and disalicylic acid derivatives: PTP1B inhibitors as potential anti-obesity drugs. Bioorg. Med. Chem. 2007, 15, 6535–6548. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, M.; Roller, P.P.; Chen, L.; Zhang, Z.Y.; Ye, B.; Burke, T.R. Potent inhibition of protein-tyrosine phosphatase by phosphotyrosine-mimic containing cyclic peptides. Bioorg. Med. Chem. 1997, 5, 157–163. [Google Scholar] [CrossRef]
- Burke, T.R.; Yao, Z.J.; Zhao, H.; Milne, G.W.A.; Wu, L.; Zhang, Z.Y.; Voigt, J.H. Enantioselective synthesis of nonphosphorus-containing phosphotyrosyl mimetics and their use in the preparation of tyrosine phosphatase inhibitory peptides. Tetrahedron 1998, 54, 9981–9994. [Google Scholar] [CrossRef]
- Wilson, D.P.; Wan, Z.K.; Xu, W.X.; Kirincich, S.J.; Follows, B.C.; Joseph-McCarthy, D.; Foreman, K.; Moretto, A.; Wu, J.; Zhu, M.; et al. Structure-based optimization of protein tyrosine phosphatase 1B inhibitors: From the active site to the second phosphotyrosine binding site. J. Med. Chem. 2007, 50, 4681–4698. [Google Scholar] [CrossRef] [PubMed]
- Combs, A.P.; Yue, E.W.; Bower, M.; Ala, P.J.; Wayland, B.; Douty, B.; Takvorian, A.; Polam, P.; Wasserman, Z.; Zhu, W.; et al. Structure-based design and discovery of protein tyrosine phosphatase inhibitors incorporating novel isothiazolidinone heterocyclic phosphotyrosine mimetics. J. Med. Chem. 2005, 48, 6544–6548. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zhang, Y.; Wang, F.; Zheng, M.; Zhang, X.; Luo, X.; Shen, X.; Jiang, H.; Liu, H. Novel thiophene derivatives as PTP1B inhibitors with selectivity and cellular activity. Bioorg. Med. Chem. 2010, 18, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lee, W.; Kim, S.-N.; Yoon, G.; Cheon, S.H. Design, synthesis, and evaluation of bromo-retrochalcone derivatives as protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.; Choi, Y.H.; Sohn, J.H.; Ahn, J.S.; Yim, J.H.; Lee, H.K.; Oh, H. Ohioensins F and G: Protein tyrosine phosphatase 1B inhibitory benzonaphthoxanthenones from the Antarctic moss polytrichastrum alpinum. Bioorg. Med. Chem. Lett. 2008, 18, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.F.; Møller, K.B.; Pedersen, A.K.; Peters, G.H.; Petersen, A.S.; Andersen, H.S.; Branner, S.; Mortensen, S.B.; Møller, N.P.H. Structure determination of T cell protein-tyrosine phosphatase. J. Biol. Chem. 2002, 277, 19982–19990. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Peter, G.; Andersen, H.S.; Tonks, N.K.; Møller, P.H.; et al. Structural and Evolutionary Relationships among Protein Tyrosine Phosphatase Domains Structural and Evolutionary Relationships among Protein Tyrosine Phosphatase Domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Wang, L.J.; Shi, D.Y. The design strategy of selective PTP1B inhibitors over TCPTP. Bioorg. Med. Chem. 2016, 24, 3343–3352. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Ling, H.; Zhang, M.; Shen, J.; Li, Q. Discovery of novel, potent, selective and cellular active ADC type PTP1B inhibitors via fragment-docking-oriented de novel design. Bioorg. Med. Chem. 2015, 23, 4891–4898. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Du, Y.; Song, L.; Shen, J.; Li, Q. Novel, potent, selective and cellular active ABC type PTP1B inhibitors containing (methanesulfonyl-phenyl-amino)-acetic acid methyl ester phosphotyrosine mimetic. Bioorg. Med. Chem. 2015, 23, 7079–7088. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Hart, S.G.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Zhang, S.E.; Nie, F.; Yang, Y.; Tang, Y.B.; Yin, W.; Tian, J.Y.; Ye, F.; Xiao, Z. Discovery of novel PTP1B inhibitors via pharmacophore-oriented scaffold hopping from Ertiprotafib. Bioorg. Med. Chem. Lett. 2013, 23, 6217–6222. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.M.; Havel, P.J.; Levin, A.A.; Bremer, A.A.; Stanhope, K.L.; Butler, M.; Booten, S.L.; Graham, J.L.; McKay, R.A.; Murray, S.F.; et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology 2009, 150, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Ala, P.J.; Gonneville, L.; Hillman, M.; Becker-Pasha, M.; Yue, E.W.; Douty, B.; Wayland, B.; Polam, P.; Crawley, M.L.; McLaughlin, E.; et al. Structural insights into the design of nonpeptidic isothiazolidinone-containing inhibitors of protein-tyrosine phosphatase 1B. J. Biol. Chem. 2006, 281, 38013–38021. [Google Scholar] [CrossRef] [PubMed]
- Combs, A.P.; Zhu, W.; Crawley, M.L.; Glass, B.; Polam, P.; Sparks, R.B.; Modi, D.; Takvorian, A.; McLaughlin, E.; Yue, E.W.; et al. Potent benzimidazole sulfonamide protein tyrosine phosphatase 1B inhibitors containing the heterocyclic (S)-isothiazolidinone phosphotyrosine mimetic. J. Med. Chem. 2006, 49, 3774–3789. [Google Scholar] [CrossRef] [PubMed]
- Sparks, R.B.; Polam, P.; Zhu, W.; Crawley, M.L.; Takvorian, A.; McLaughlin, E.; Wei, M.; Ala, P.J.; Gonneville, L.; Taylor, N.; et al. Benzothiazole benzimidazole (S)-isothiazolidinone derivatives as protein tyrosine phosphatase-1B inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs2: Free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Molsoft. Available online: http://molsoft.com/mprop/ (accessed on 5 June 2017).
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley Sons: New York, NY, USA, 1993. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- EMBL-EBI The European Bioinformatics Institute. Available online: http://www.ebi.ac.uk/ (accessed on 7 December 2017).
- Haftchenary, S.; Ball, D.P.; Aubry, I.; Landry, M.; Shahani, V.M.; Fletcher, S.; Page, B.D.G.; Jouk, A.O.; Tremblay, M.L.; Gunning, P.T. Identification of a potent salicylic acid-based inhibitor of tyrosine phosphatase PTP1B. Med. Chem. Commun. 2013, 4, 987–992. [Google Scholar] [CrossRef]
- Raj, B.; Kafle, B.; Hwang, J.; Wook, S.; Lee, K.; Park, H.; Han, I.; Cho, H. Novel thiazolidinedione derivatives with anti-obesity effects : Dual action as PTP1B inhibitors and PPAR activators. Bioorg. Med. Chem. Lett. 2010, 20, 6758–6763. [Google Scholar] [CrossRef]
- Cheung, A.W.; Banner, B.; Bose, J.; Kim, K.; Li, S.; Marcopulos, N.; Orzechowski, L.; Sergi, J.A.; Thakkar, K.C.; Wang, B.; et al. 7-Phenyl-pyrido[2,3-d]pyrimidine-2,4-diamines: Novel and highly selective protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7518–7522. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Espinosa, J.J.; Rios, M.Y.; López-Martínez, S.; López-Vallejo, F.; Medina-Franco, J.L.; Paoli, P.; Camici, G.; Navarrete-Vázquez, G.; Ortiz-Andrade, R.; Estrada-Soto, S. Antidiabetic activity of some pentacyclic acid triterpenoids, role of PTP–1B: In vitro, in silico, and in vivo approaches. Eur. J. Med. Chem. 2011, 46, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, Y.; Jin, K.; Gao, L.; Luo, T.; Sheng, L.; Shao, X.; Li, J. Synthesis and biological evaluation of novel thiadiazole amides as potent Cdc25B and PTP1B inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4125–4128. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, S.; Higai, K.; Sasaki, T.; Asada, Y.; Ohshima, S.; Koike, K. Evaluation of licorice flavonoids as protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5836–5839. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vazquez, G.; Paoli, P.; León-Rivera, I.; Villalobos-Molina, R.; Medina-Franco, J.L.; Ortiz-Andrade, R.; Estrada-Soto, S.; Camici, G.; Diaz-Coutiño, D.; Gallardo-Ortiz, I.; et al. Synthesis, in vitro and computational studies of protein tyrosine phosphatase 1B inhibition of a small library of 2-arylsulfonylaminobenzothiazoles with antihyperglycemic activity. Bioorg. Med. Chem. 2009, 17, 3332–3341. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vazquez, G.; Ramírez-Martínez, M.; Estrada-Soto, S.; Nava-Zuazo, C.; Paoli, P.; Camici, G.; Escalante-García, J.; Medina-Franco, J.L.; López-Vallejo, F.; Ortiz-Andrade, R. Synthesis, in vitro and in silico screening of ethyl 2-(6-substituted benzo[d]thiazol-2-ylamino)-2-oxoacetates as protein-tyrosine phosphatase 1B inhibitors. Eur. J. Med. Chem. 2012, 53, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Ottanà, R.; Maccari, R.; Mortier, J.; Caselli, A.; Amuso, S.; Camici, G.; Rotondo, A.; Wolber, G.; Paoli, P. Synthesis, biological activity and structure–activity relationships of new benzoic acid-based protein tyrosine phosphatase inhibitors endowed with insulinomimetic effects in mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem. 2014, 71, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Ahmed, V.; Hill, B.; Ahmed, Z.; Taylor, S.D. A re-examination of the difluoromethylenesulfonic acid group as a phosphotyrosine mimic for PTP1B inhibition. Bioorg. Med. Chem. 2008, 16, 6764–6777. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Zhao, B.T.; Ali, M.Y.; Choi, J.S.; Rhyu, D.Y.; Min, B.S.; Woo, M.H. Insulin-mimetic selaginellins from selaginella tamariscina with protein tyrosine phosphatase 1B (PTP1B) inhibitory activity. J. Nat. Prod. 2015, 78, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Ji, D.J.; Han, Y.R.; Choi, J.S.; Rhyu, D.Y.; Min, B.S.; Woo, M.H. Selaginellin and biflavonoids as protein tyrosine phosphatase 1B inhibitors from Selaginella tamariscina and their glucose uptake stimulatory effects. Bioorg. Med. Chem. 2015, 23, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Espinosa, J.J.; Rios, M.Y.; Paoli, P.; Flores-Morales, V.; Camici, G.; Rosa-Lugo, V.D.L.; Hidalgo-Figueroa, S.; Navarrete-Vázquez, G.; Estrada-Soto, S. Synthesis of oleanolic acid derivatives: In vitro, in vivo and in silico studies for PTP-1B inhibition. Eur. J. Med. Chem. 2014, 87, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Thrasher, J. Pharmacologic Management of Type 2 Diabetes Mellitus: Available Therapies. Am. J. Med. 2017, 130, S4–S17. [Google Scholar] [CrossRef] [PubMed]
- Safavi, M.; Foroumadi, A.; Abdollahi, M. The importance of synthetic drugs for type 2 diabetes drug discovery. Expert Opin. Drug Discov. 2013, 8, 1339–1363. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, S.R.; Evdokimov, A.G.; Colson, A.O.; Fairweather, N.T.; Neuman, J.J.; Maier, M.B.; Gray, J.L.; Gerwe, G.S.; Stake, G.E.; Howard, B.W.; et al. 1,2,3,4-Tetrahydroisoquinolinyl sulfamic acids as phosphatase PTP1B inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1574–1578. [Google Scholar] [CrossRef] [PubMed]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Deora, G.S.; Rathore, V.; Tanwar, O.; Rawat, A.K.; Srivastava, A.K.; Jain, D. Identification of ZINC02765569: A potent inhibitor of PTP1B by vHTS. Med. Chem. Res. 2013, 22, 28–34. [Google Scholar] [CrossRef]
- Rakse, M.; Karthikeyan, C.; Deora, G.S.; Moorthy, N.S.H.N.; Rathore, V.; Rawat, A.K.; Srivastava, A.K.; Trivedi, P. Design, synthesis and molecular modelling studies of novel 3-acetamido-4-methyl benzoic acid derivatives as inhibitors of protein tyrosine phosphatase 1B. Eur. J. Med. Chem. 2013, 70, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, M.; Zhao, Y.; Gordon, S.; Zhang, Z. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1998, 273, 26368. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Develop ment Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Valdez, J.; Cedillo, R.; Hernández-Campos, A.; Yépez, L.; Hernández-Luis, F.; Navarrete-Vázquez, G.; Tapia, A.; Cortés, R.; Hernández, M.; Castillo, R. Synthesis and antiparasitic activity of 1H-benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 2221–2224. [Google Scholar] [CrossRef]
- Flores-Carrillo, P.; Velázquez-López, J.M.; Aguayo-Ortiz, R.; Hernández-Campos, A.; Trejo-Soto, P.J.; Yépez-Mulia, L.; Castillo, R. Synthesis, antiprotozoal activity, and chemoinformatic analysis of 2-(methylthio)-1H-benzimidazole-5-carboxamide derivatives: Identification of new selective giardicidal and trichomonicidal compounds. Eur. J. Med. Chem. 2017, 137, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Campos, A.; Ibarra-Velarde, F.; Vera-Montenegro, Y.; Rivera-Fernández, N.; Castillo, R. Synthesis and Fasciolicidal Activity of 5-Chloro-2-methylthio-6-(1-naphthyloxy)-1H-benzimidazole. Chem. Pharm. Bull. 2002, 50, 649–652. [Google Scholar] [CrossRef]
- Velázquez-López, J.M.; Hernández-Campos, A.; Yépez-Mulia, L.; Téllez-Valencia, A.; Flores-Carrillo, P.; Nieto-Meneses, R.; Castillo, R. Synthesis and trypanocidal activity of novel benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 4377–4381. [Google Scholar] [CrossRef] [PubMed]
- Soria-Arteche, O.; Castillo, R.; Hernández-Campos, A.; Hurtado-de la Peña, M.; Gabriel Navarrete-Vázquez, G.; Medina-Franco, J.L.; Gómez-Flores, K. Studies on the Selective S-Oxidation of Albendazole, Fenbendazole, Triclabendazole, and other Benzimidazole Sulfides. J. Mex. Chem. Soc. 2005, 49, 353–358. [Google Scholar]
- Goldstein, B.J.; Bittner-Kowalczyk, A.; White, M.F.; Harbeck, M. Tyrosine Dephosphorylation and Deactivation of Insulin Receptor Substrate-1 by Protein-tyrosine Phosphatase 1B. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Valencia, A.; Najera, H.; Sampedro, J.; Aguirre, B.; Olivares, V.R.A. Diseño de fármacos antiparasitarios. Inhibición especie específica de la triosafosfato isomerasa de Leishmanía mexicana. Boletín Inf. los Serv. Salud del Estado Hidalgo 2006, 29, 2–3. [Google Scholar]
- Schrödinger Release 2014-1: LigPrep, version 2.9; Schrödinger: New York, NY, USA, 2014.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, M.A.; Atatreh, N.; Bichenkova, E.V.; Bryce, R.A. Protein tyrosine phosphatases: Ligand interaction analysis and optimisation of virtual screening. J. Mol. Graph. Model. 2014, 52C, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG—A tool for highthroughput crystallography of protein—Ligand complexes. Acta Crystallogr. 2004, 60, 1355–1363. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Source, O.; Discovery, D.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | MW a | HBD a | HBA a | LogP a | Drug Likeness b | Binding Energy (Kcal/mol) | % Inhibition (200 µM) |
|---|---|---|---|---|---|---|---|
| 1 | 567.27 | 2 | 7 | 6.68 | 1.07 | −4.20 | 100 |
| 2 | 466.96 | 2 | 6 | 6.00 | 0.86 | −4.99 | 99 |
| 3 | 424.59 | 2 | 5 | 5.53 | 0.66 | −4.47 | 92 |
| 4 | 356.36 | 1 | 8 | 2.94 | -0.49 | −3.70 | 88 |
| 5 | 496.32 | 0 | 9 | 5.13 | 0.18 | −4.02 | 85 |
| 6 | 498.47 | 2 | 8 | 4.26 | -0.12 | −5.24 | 84 |
| 7 | 499.94 | 1 | 7 | 4.95 | 0.41 | −3.49 | 80 |
| 8 | 446.51 | 1 | 5 | 5.80 | 0.05 | −3.98 | 74 |
| 9 | 384.64 | 1 | 5 | 4.36 | 0.76 | −3.99 | 70 |
| 10 | 356.83 | 1 | 4 | 4.35 | 0.08 | −4.01 | 65 |
| Molecule | Ki (µM) | IC50 (µM) | α | Vmax (µmol/min/mg) | Km (mM) | Inhibition Type |
|---|---|---|---|---|---|---|
| 1 | 5.2 | 7.5 | 1.4 | 8.8 | 6.7 | Mixed |
| 2 | 4.2 | 8.4 | 2.9 | 16 | 6.8 | Mixed |
| 3 | 41.3 | 31.3 | 3.3 | 10 | 4.1 | Mixed |
| Complex | Energy (kcal/mol) | Hydrogen Bonds | |||||
|---|---|---|---|---|---|---|---|
| Van der Waals Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy | ΔG Binding | Range | Average | |
| PTP1B-1 | −47.56 | −17.97 | 32.34 | −4.17 | −37.36 | 0–3 | 3 |
| PTP1B-2 | −35.46 | −27.01 | 33.57 | −3.54 | −32.43 | 0–5 | 4 |
| PTP1B-3 | −29.50 | −3.63 | 12.33 | −2.93 | −23.74 | 0–4 | 2 |
| Molecule | LD50 a (mg/kg) | Toxicity Class a | Toxic Frag. a | Toxicity Targets a | Mutagenic b | Tumorigenic b | Reprod. Effec. b | Irritant b | Drug Likeness c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1600 | 4 | None | No Binding | Low | None | High | High | 1.07 |
| 2 | 1000 | 4 | None | No Binding | None | None | None | None | 0.86 |
| 3 | 1600 | 4 | None | No Binding | Low | None | High | High | 0.66 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarabia-Sánchez, M.J.; Trejo-Soto, P.J.; Velázquez-López, J.M.; Carvente-García, C.; Castillo, R.; Hernández-Campos, A.; Avitia-Domínguez, C.; Enríquez-Mendiola, D.; Sierra-Campos, E.; Valdez-Solana, M.; et al. Novel Mixed-Type Inhibitors of Protein Tyrosine Phosphatase 1B. Kinetic and Computational Studies. Molecules 2017, 22, 2262. https://doi.org/10.3390/molecules22122262
Sarabia-Sánchez MJ, Trejo-Soto PJ, Velázquez-López JM, Carvente-García C, Castillo R, Hernández-Campos A, Avitia-Domínguez C, Enríquez-Mendiola D, Sierra-Campos E, Valdez-Solana M, et al. Novel Mixed-Type Inhibitors of Protein Tyrosine Phosphatase 1B. Kinetic and Computational Studies. Molecules. 2017; 22(12):2262. https://doi.org/10.3390/molecules22122262
Chicago/Turabian StyleSarabia-Sánchez, Marie Jazmín, Pedro Josué Trejo-Soto, José Miguel Velázquez-López, Carlos Carvente-García, Rafael Castillo, Alicia Hernández-Campos, Claudia Avitia-Domínguez, Daniel Enríquez-Mendiola, Erick Sierra-Campos, Mónica Valdez-Solana, and et al. 2017. "Novel Mixed-Type Inhibitors of Protein Tyrosine Phosphatase 1B. Kinetic and Computational Studies" Molecules 22, no. 12: 2262. https://doi.org/10.3390/molecules22122262
APA StyleSarabia-Sánchez, M. J., Trejo-Soto, P. J., Velázquez-López, J. M., Carvente-García, C., Castillo, R., Hernández-Campos, A., Avitia-Domínguez, C., Enríquez-Mendiola, D., Sierra-Campos, E., Valdez-Solana, M., Salas-Pacheco, J. M., & Téllez-Valencia, A. (2017). Novel Mixed-Type Inhibitors of Protein Tyrosine Phosphatase 1B. Kinetic and Computational Studies. Molecules, 22(12), 2262. https://doi.org/10.3390/molecules22122262