3.1. General
Chemicals and solvents were purchased from Sigma-Aldrich GmbH (Steinheim, Germany), Fluka AG (Buchs, Switzerland), TCI EUROPE N.V. (Zwijndrecht, Belgium), ChemPUR GmbH (Karlsruhe, Germany), Merck KGaA (Darmstadt, Germany) and ABCR GmbH (Karlsruhe, Germany) and used as delivered. Anhydrous solvents were purchased from Sigma-Aldrich GmbH (Steinheim, Germany) and stored under argon. Precursors for electrophilic radiofluorination, For-6-(SnMe3)DOPA(Boc)2-OEt, Boc-6-(SnMe3)DOPA(Boc)2-OEt, Boc-6-(SnMe3)mTyr(Boc)-OEt, Boc-2-(SnMe3)Tyr(Boc)-OEt, Boc-4-Boc-3-Me-6-(SnMe3)DOPA-OtBu were purchased from ABX GmbH (Radeberg, Germany) and used as delivered.
3.4. Chemistry
All reactions were carried out with magnetic stirring, if not stated otherwise, and, if air or moisture sensitive, substrates and/or reagents were handled in flame-dried glassware under argon or nitrogen. Organic extracts were dried with anhydrous MgSO4.
Column chromatography: silica gel technical grade (w/Ca, ~0.1%), 60 Å, 230–400 mesh particle size from Sigma-Aldrich GmbH (Steinheim, Germany) was applied for the purification of aryl stannanes. Merck silica gel, grade 60, 230–400 mesh was used for other compounds. Solvent proportions are indicated in a volume/volume ratio.
Thin layer chromatography (TLC) was performed using aluminum finish ALUGRAM® SIL G/UV254 from Macherey-Nagel GmbH (Düren, Germany) or precoated sheets, 0.25 mm Sil G/UV254 from Merck KGaA (Dormstadt, Germany). The chromatograms were viewed under UV light (λ = 254 nm).
3.4.1. Tetrakis(pyridine)copper(II) Bis(trifluoromethanesulfonate)
Copper(II) trifluoromethanesulfonate (5 g, 14 mmol) was dissolved in methanol (25 mL). Pyridine (12 mL, 149 mmol) was added dropwise (exothermic reaction was observed) and the reaction mixture was stirred for 30 min. The mixture was left at ambient temperature for 1 h and thereafter in fridge (at 5 °C) overnight. The blue crystalline precipitate was filtered off, recrystallized from 20% Py in MeOH and dried under a stream of air affording the desired product [
56].
| Yield | 8.5 g, 91% |
| Appearance | blue solid |
| Molecular formula | C22H20CuF6N4O6S2 |
| Molar mass | 678.08042 |
| Anal. | Calcd for C22H20CuF6N4O6S2: C, 38.97; H, 2.97; N, 8.26. Found: C, 39.1± < 0.1; H, 3.16 ± 0.09; N, 8.33 ± 0.01. |
3.4.2. 3-(Benzo[d][1,3]dioxol-5-yl)-1-(3-bromophenyl)-3-hydroxyprop-2-en-1-one—General Procedure 1 (GP1)
To a solution of 1-(benzo[
d][
1,
3]dioxol-5-yl)ethan-1-one (1.5 g, 9.1 mmol), in anhydrous THF (20 mL) was added 1 m LiHMDS in THF (27.3 mL) and the resulting solution was stirred for 1 h at −80 °C. The solution was warmed to room temperature and stirred for 2 h. Thereafter, it was cooled to −80 °C and 3-bromobenzoyl chloride (1.2 mL, 2.0 g, 9.1 mmol) was added dropwise. The solution was allowed to warm to room temperature and stirred for additional 18 h. Afterwards, a saturated solution of NH
4Cl (50 mL) was added, the pH was adjusted to 7.0 and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (100 mL), dried and concentrated under reduced pressure. The residue was purified by column chromatography (Et
2O/petroleum ether, 1:4) affording the title compound [
57].
| Yield | 2.82 g, 89% |
| Appearance | yellow solid |
| Molecular formula | C16H11BrO4 |
| Molar mass | 347.164 |
| TLC | Rf = 0.46 (Et2O/petroleum ether, 1:4) |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 7.88 (q, J = 1.7 Hz, 1H), 7.75–7.60 (m, 1H), 7.53–7.34 (m, 2H), 7.31–7.23 (m, 1H), 7.23–7.08 (m, 1H), 6.75–6.61 (m, 1H), 6.54 (s, 1H), 5.87 (s, 2H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 151.34, 147.91, 136.89, 136.87, 134.57, 129.91, 129.50, 125.19, 122.91, 122.42, 107.89, 106.85, 101.61, 92.48. |
| HRMS | m/z: [M − H]− calcd for C16H10BrO4−: 344.97679; found: 344.97664. Correct isotopic pattern. |
3.4.3. 3-(Benzo[d][1,3]dioxol-5-yl)-1-(3-fluorophenyl)-3-hydroxyprop-2-en-1-one
The title compound was synthesized according to GP1 from 1-(benzo[
d][
1,
3]dioxol-5-yl)ethan-1-one (500 mg, 3 mmol).
| Yield | 735 mg, 84% |
| Appearance | yellow solid |
| Molecular formula | C16H11FO4 |
| Molar mass | 286.2584 |
| TLC | Rf = 0.45 (Et2O/petroleum ether, 1:4) |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 16.80 (s, 1H), 7.74 (dd, J = 7.8, 1.1 Hz, 1H), 7.63 (ddd, J = 10.0, 6.0, 2.2 Hz, 2H), 7.55–7.37 (m, 2H), 7.32–7.16 (m, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.71 (s, 1H), 6.07 (s, 2H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 186.52, 182.20, 165.36, 160.59, 151.79, 148.44, 130.45, 130.29, 130.02, 123.28, 122.76, 122.70, 119.40, 118.98, 114.28, 113.82, 108.41, 107.41, 102.08, 92.88. |
| 19F-NMR | (188 MHz, CDCl3): δ (ppm) = −111.97. |
| HRMS | m/z: [M − H]− calcd for C16H10FO4−: 285.05686; found: 285.05685. |
3.4.4. 3-(Benzo[d][1,3]dioxol-5-yl)-5-(3-bromophenyl)-1H-pyrazole—General Procedure 2 (GP2)
A solution of 3-(benzo[
d][
1,
3]dioxol-5-yl)-1-(3-bromophenyl)-3-hydroxyprop-2-en-1-one (2.4 g, 6.91 mmol) and hydrazine monohydrate (1 mL, 98%, 13.83 mmol, 2 eq.) in ethanol (30 mL) was refluxed for 3 h. Water was added to the clear yellow solution and resulting precipitate was collected by filtration, washed with water and dried under vacuum to provide the title compound [
57].
| Yield | 2 g, 84% |
| Appearance | colorless solid |
| Molecular formula | C16H11BrN2O2 |
| Molar mass | 343.18 |
| TLC | Rf = 0.31 (EtOAc/petroleum ether, 1:4) |
| 1H-NMR | (200 MHz, DMSO-d6 + DCl): δ (ppm) = 7.97 (s, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.34 (t, J = 9.2 Hz, 4H), 6.89 (d, J = 8.6 Hz, 1H), 5.96 (s, 2H). |
| 13C-NMR | (50 MHz, DMSO-d6 + DCl): δ (ppm) = 148.94, 148.55, 147.18, 146.64, 132.67, 131.93, 131.75, 129.01, 125.66, 123.08, 122.73, 121.20, 109.56, 106.84, 102.31, 101.64, 12.33. |
| HRMS | m/z: [M + H]+ calcd for C16H12BrN2O2+: 343.00767; found: 343.00781. Correct isotopic pattern. |
3.4.5. 3-(Benzo[d][1,3]dioxol-5-yl)-5-(3-fluorophenyl)-1H-pyrazole
The title compound was synthesized from 3-(benzo[
d][
1,
3]dioxol-5-yl)-1-(3-fluorophenyl)-3-hydroxyprop-2-en-1-one (335 mg, 1.17 mmol) according to GP2.
| Yield | 735 mg, 84% |
| Appearance | yellow solid |
| Molecular formula | C16H11FN2O2 |
| Molar mass | 282.2744 |
| TLC | Rf = 0.32 (EtOAc/petroleum ether, 1:4) |
| 1H-NMR | (200 MHz, DMSO-d6 + DCl): δ (ppm) = 7.60 (s, 2H), 7.41 (d, J = 6.4 Hz, 1H), 7.28 (d, J = 11.6 Hz, 3H), 7.14 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 5.94 (s, 2H). |
| 13C-NMR | (50 MHz, DMSO-d6 + DCl): δ (ppm) = 165.50, 160.65, 148.95, 148.50, 147.24, 146.84, 146.78, 131.98, 131.80, 131.59, 131.42, 122.78, 122.61, 121.23, 113.53, 113.07, 109.48, 106.79, 102.28, 101.65. |
| 19F-NMR | (188 MHz, DMSO-d6 + DCl): δ (ppm) = −112.07. |
| HRMS | m/z: [M + H]+ calcd for C16H12FN2O2+: 283.08773; found: 283.08775. |
3.4.6. 3-(Benzo[d][1,3]dioxol-5-yl)-5-(3-(trimethylstannyl)phenyl)-1H-pyrazole–General Procedure 3 (GP3)
A flame dried flask containing 3-(benzo[
d][
1,
3]dioxol-5-yl)-5-(3-bromophenyl)-1
H-pyrazole (550 mg, 1.6 mmol) and Pd(PPh
3)
4 (185 mg, 0.16 mmol, 0.1 eq.) were evacuated and purged with argon (three times). Anhydrous 1,4-dioxane (2 mL) followed by hexamethylditin (830 µL, 1.31 g, 4 mmol, 2.5 eq.) was added, and the reaction mixture was heated to 100 °C for 18 h. The black suspension was filtered through a plug of Celite. 1 m TBAF in THF (2 mL) was added to the filtrate; the mixture was stirred for 30 min and diluted with EtOAc (50 mL). The resulting solution was washed with water (50 mL), brine (50 mL), dried and concentrated under reduced pressure. The crude product was purified by column chromatography and by recrystallization from hexane contained a small amount of CH
2Cl
2.
| Yield | 566 mg, 83% |
| Appearance | colorless solid |
| Molecular formula | C19H20N2O2Sn |
| Molar mass | 427.091 |
| TLC | Rf = 0.30 (EtOAc/petroleum ether, 1:4) |
| 1H-NMR | (200 MHz, DMSO-d6): δ (ppm) = 13.20 (s, 1H), 7.93 (s, 1H), 7.74 (s, 1H), 7.60–7.22 (m, 5H), 7.12 (s, 1H), 6.99 (d, J = 7.7 Hz, 1H), 6.06 (s, 2H), 0.31 (s, 9H). |
| 13C-NMR | (50 MHz, DMSO-d6): δ (ppm) = 147.74, 146.86, 132.25, 128.28, 125.03, 118.82, 108.59, 105.60, 101.12, 99.30, −9.29. |
| HRMS | m/z: [M + H]+ calcd for C19H21N2O2Sn+: 429.06195; found: 429.06286. Correct isotopic pattern. |
3.4.7. Methyl 4-Fluorobenzoate
A solution of 4-fluorobenzoyl chloride (500 µL, 4.2 mmol) in MeOH (20 mL) was stirred at 40 °C for 2 h and concentrated under reduced pressure affording the crude product [
58] which was used without further purification.
| Yield | 440 mg, 67% |
| Appearance | colorless oil |
| Molecular formula | C8H7FO2 |
| Molar mass | 154.1404 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 8.17–7.95 (m, 2H), 7.21–6.97 (m, 2H), 3.92 (s, 3H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 168.41, 166.27, 163.36, 132.34, 132.16, 126.58, 126.52, 115.85, 115.42, 52.31. |
| 19F-NMR | (188 MHz, CDCl3): δ (ppm) = −105.79. |
3.4.8. Methyl 4-(Trimethylstannyl)benzoate
The title compound [
59] was synthesized according to GP3 from methyl 4-iodobenzoate (2 g, 7.6 mmol). The product was purified by column chromatography (Et
2O:PE = 1:9).
| Yield | 1.9 g, 83% |
| Appearance | yellow oil |
| Molecular formula | C11H16O2Sn |
| Molar mass | 298.857 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 8.19–7.86 (m, 2H), 7.79–7.37 (m, 2H), 3.92 (s, 3H), 0.33 (s, 9H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 167.48, 149.69, 135.82, 135.82, 129.86, 128.55, 128.55, 52.08, −9.50. |
3.4.9. 3-(Trimethylstannyl)benzaldehyde (3)
The title compound [
60] was synthesized according to GP3 from 3-bromobenzaldehyde (850 mg, 4.6 mmol) using Pd(PPh
3)
4 (531 mg, 0.5 mmol, 0.1 eq.) and hexamethylditin (1.9 mL, 9.2 mmol, 2 eq.) and purified by column chromatography (Et
2O:PE = 1:9).
| Yield | 900 mg, 73% |
| Appearance | colorless oil |
| Molecular formula | C10H14O3Sn |
| Molar mass | 268.931 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 10.03 (s, 1H), 8.19–7.89 (m, 1H), 7.89–7.64 (m, 2H), 7.52 (t, J = 7.4 Hz, 1H), 0.34 (s, 9H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 193.18, 143.88, 142.05, 137.09, 135.75, 129.87, 128.61, −9.33. |
3.4.10. (2-Methoxyphenyl)trimethylstannane (4)—General Procedure 4 (GP4)
A solution of 2.5 m
nBuLi in hexane (0.52 mL, 1.3 eq.) was added dropwise to a stirring solution of 2-iodoanisol (131 µL, 0.236 g, 1 mmol) in Et
2O (4 mL) at −78 °C and the mixture was stirred at the same temperature for 30 min. Thereafter, a solution of Me
3SnCl (0.24 g, 1.2 mmol, 1.2 eq.) in Et
2O (3 mL) was added dropwise and the reaction mixture was stirred and slowly warmed to ambient temperature for 2 h. 1 m TBAF in THF (1 mL) was added, the mixture was stirred for 30 min, diluted with Et
2O (50 mL) and washed with 10% NaHCO
3 (3 × 10 mL), H
2O (3 × 10 mL), brine (2 × 10 mL), dried and concentrated under reduced pressure. The resulting crude product [
61] was directly used for radiochemical experiments.
| Yield | 150 mg, 55% |
| Appearance | yellow oil |
| Molecular formula | C10H16OSn |
| Molar mass | 270.947 |
| 1H-NMR6 | (200 MHz, CDCl3): δ (ppm) = 7.35 (ddd, J = 9.7, 7.5, 1.7 Hz, 2H), 7.24–6.94 (m, 1H), 6.94–6.72 (m, 1H), 3.80 (s, 3H), 0.27 (s, 9H). |
3.4.11. (3-Methoxyphenyl)trimethylstannane (5)
The title compound [
62] was prepared from 3-iodoanisol (120 µL, 0.236 g, 1 mmol), according to GP4.
| Yield | 180 mg, 66% |
| Appearance | yellow oil |
| Molecular formula | C10H16OSn |
| Molar mass | 270.947 |
| 1H-NMR6 | (200 MHz, CDCl3): δ (ppm) = 7.45–7.18 (m, 1H), 7.18–6.97 (m, 2H), 6.89 (ddd, J = 8.3, 2.7, 1.1 Hz, 1H), 3.85 (s, 3H), 0.33 (s, 9H). |
3.4.12. (4-Methoxyphenyl)trimethylstannane (6a)
The title compound [
7] was prepared from 4-iodoanisol (235 mg, 1 mmol) according to GP4.
| Yield | 190 mg, 70% |
| Appearance | yellow oil |
| Molecular formula | C10H16OSn |
| Molar mass | 270.947 |
| 1H-NMR7 | (200 MHz, CDCl3): δ (ppm) = 7.64–7.20 (m, 2H), 6.94 (ddd, J = 6.5, 4.1, 1.9 Hz, 2H), 3.82 (s, 3H), 0.28 (s, 9H). |
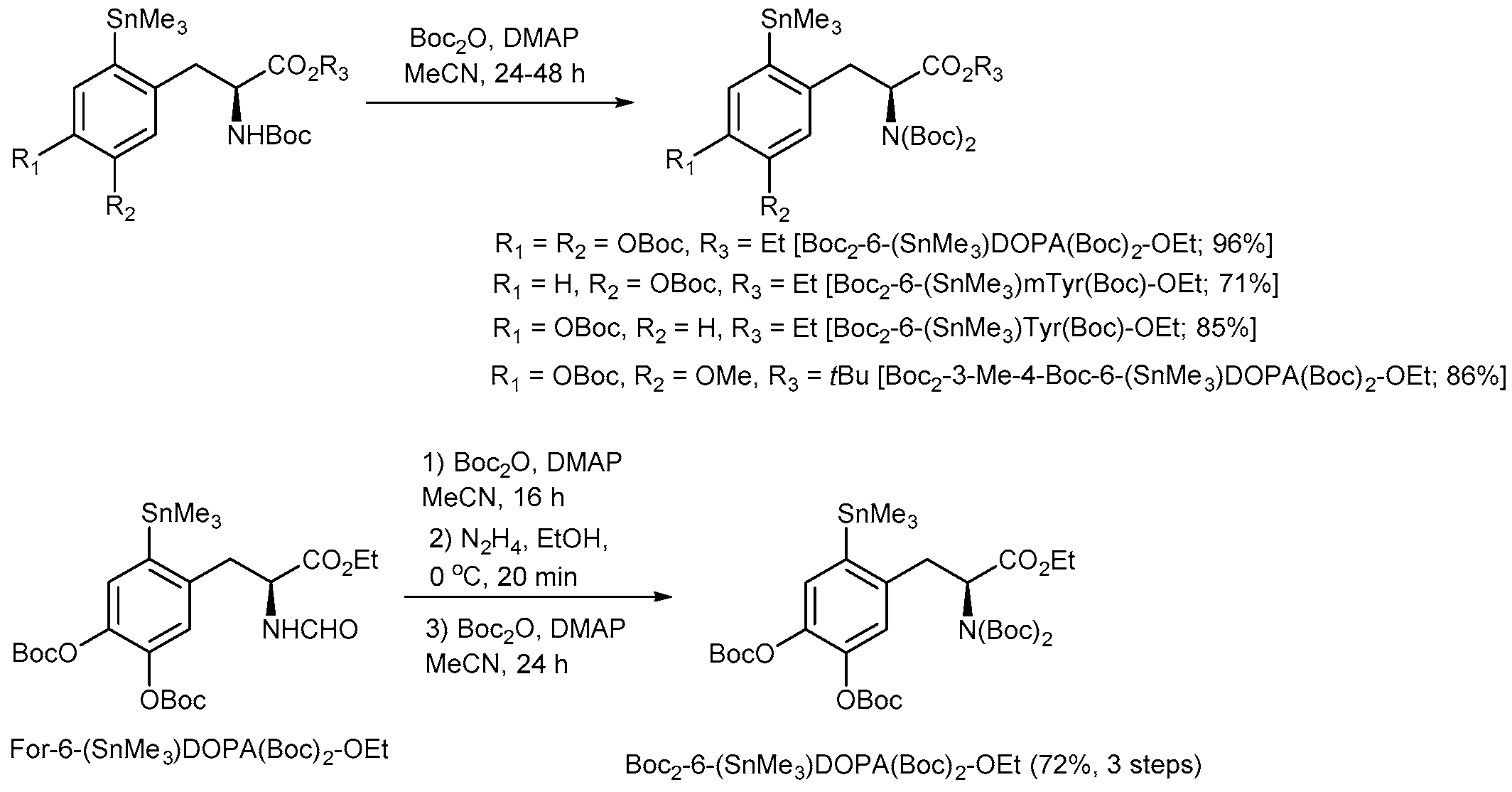
3.4.13. tert-Butyl (S)-2-(Bis(tert-butoxycarbonyl)amino)-3-{4-[(tert-butoxycarbonyl)oxy]-5-methoxy-2-(trimethylstannyl)phenyl}propanoate [Boc2-4-Boc-3-Me-6-(SnMe3)DOPA-OtBu]—General Procedure 5 (GP5)
A solution of tert-butyl (S)-2-[(tert-butoxycarbonyl)amino]-3-{4-[(tert-butoxycarbonyl)oxy]5-methoxy-2-(trimethylstannyl)phenyl}propanoate [Boc-4-Boc-3-Me-6-(SnMe3)DOPA-OtBu] (220 mg, 0.3 mmol) DMAP (17 mg, 0.1 mmol, 0.4 eq.) and di-tert-butyl dicarbonate (229 mg, 1 mmol, 3 eq.) in anhydrous MeCN (3 mL) was stirred at room temperature for 48 h, and then concentrated under vacuum. Purification of the residue by column chromatography (Et2O:PE = 1:9) afforded the title compound.
| Yield | 255 mg, 86% |
| Appearance | yellow oil |
| Molecular formula | C32H53NO10Sn |
| Molar mass | 730.483 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 7.09 (s, 1H), 6.84–6.68 (m, 1H), 5.29 (s, 3H), 4.88 (dd, J = 9.3, 6.2 Hz, 1H), 3.60–3.36 (m, 2H), 1.53 (s, 9H), 1.48 (s, 9H), 1.38 (s, 18H), 0.30 (s, 9H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 169.01, 152.27, 143.78, 129.41, 114.13, 83.26, 82.84, 81.86, 60.52, 55.74, 28.13, 28.01, 27.77, −8.22. |
| HRMS | m/z: [M + Na]+ calcd for C32H53NNaO10Sn+: 754.25836; found: 754.25918. Correct isotopic pattern. |
3.4.14. Ethyl (S)-3-{4,5-Bis[(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenyl}-2-[bis(tert-butoxycarbonyl)amino]propanoate [Boc2-6-(SnMe3)DOPA(Boc)2-OEt]
The title compound was synthesized according to GP5 from ethyl (S)-3-{4,5-bis[(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenyl}-2-[(tert-butoxycarbonyl)amino]pro-panoate [Boc-6-(SnMe3)DOPA(Boc)2-OEt] (200 mg, 0.3 mmol).
| Yield | 229 mg, 96% |
| Appearance | yellow oil |
| Molecular formula | C34H55NO12Sn |
| Molar mass | 788.519 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 7.23 (s, 1H), 7.15–6.97 (m, 1H), 5.03 (dd, J = 9.8, 4.8 Hz, 1H), 4.21 (ddt, J = 10.3, 7.0, 3.5 Hz, 2H), 3.52–3.18 (m, 2H), 1.52 (d, J = 2.5 Hz, 18H), 1.38 (s, 18H), 1.27 (t, J = 5.8 Hz, 3H), 0.33 (s, 9H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 170.17, 152.04, 150.87, 150.72, 143.22, 142.58, 141.37, 140.61, 130.08, 123.94, 83.54, 83.45, 83.19, 61.57, 59.48, 37.93, 29.80, 27.96, 27.72, 14.26, −8.03. |
| HRMS | m/z: [M + Na − CH2]+ calcd for C34H55NNaO12Sn+: 812.26384; found: 812.26458. Correct isotopic pattern. |
3.4.15. Ethyl (S)-3-{4,5-Bis[(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenyl}-2-[bis(tert-butoxycarbonyl)amino]propanoate [Boc2-6-(SnMe3)DOPA(Boc)2-OEt] from (S)-3-{4,5-Bis[(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenyl}-2-(formylamino)propanoate [For-6-(SnMe3)DOPA(Boc)2-OEt]
A solution of For-6-(SnMe3)DOPA(Boc)2-OEt (0.765 g, 1.24 mmol), DMAP (17 mg, 0.14 mmol) and Boc2O (1.08 g, 4.95 mmol) in anhydrous MeCN (4 mL) was incubated for 16 h at ambient temperature. Thereafter, the reaction mixture was diluted with Et2O (30 mL). N,N-3-(Dimethylamino)-1-propylamine (0.62 mL, 0.506 g, 4.95 mmol) was added, the mixture was incubated at ambient temperature for 10 min, washed with 1 m NaHSO4 (3 × 10 mL), H2O (3 × 10 mL), brine (2 × 10 mL), dried and concentrated under reduced pressure to give the crude Boc,For-6-(SnMe3)DOPA(Boc)2-OEt (0.91 g, 100%) which was immediately used for the next step.
A solution of N2H4·H2O (140 µL, 140 mg, 2.36 mmol) in MeOH (1 mL) was added dropwise to an ice-cold solution of Boc,For-6-(SnMe3)DOPA(Boc)2-OEt (0.91 g, max. 1.24 mol) in MeOH (7.7 mL) and the reaction mixture was stirred for 20 min. Et2O (50 mL) was added and the resulting solution was washed with 1 M NaHSO4 (3 × 10 mL), H2O (3 × 10 mL), brine (2 × 10 mL), dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc:hexane = 1:3) to give Boc-6-(SnMe3)DOPA(Boc)2-OEt (0.64 g, 75% over two steps) as a colorless foam which was immediately used for the next step. Rf = 0.38, EtOAc:hexane = 1:3
Boc2-6-(SnMe3)DOPA(Boc)2-OEt (0.71 g, 72% over three steps) was prepared according to GP4 from Boc-6-(SnMe3)DOPA(Boc)2-OEt (0.64 g, 0.93 mmol) using Boc2O (1.08 g, 4.95 mmol) and DMAP (16 mg, 0.13 mmol) and purified by column chromatography (EtOAc:hexane = 1:5). Rf = 0.29, EtOAc:hexane = 1:5.
3.4.16. Ethyl (S)-2-[Bis(tert-butoxycarbonyl)amino]-3{5-[(tert-butoxycarbonyl)oxy]-2(trimethylstannyl)phenyl}propanoate [Boc-6-(SnMe3)-m-Tyr(Boc)-OEt]
The title compound was synthesized from ethyl (S)-2-[(tert-butoxycarbonyl)amino]-3-{5-[(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenyl}propanoate (300 mg, 0.5 mmol) according to GP5.
| Yield | 250 mg, 71% |
| Appearance | yellow oil |
| Molecular formula | C29H47NO9Sn |
| Molar mass | 672.403 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 7.37 (t, J = 8.4 Hz, 1H), 7.12–6.97 (m, 1H), 6.92 (d, J = 2.2 Hz, 1H), 5.02 (dd, J = 10.4, 4.5 Hz, 1H), 4.22 (qd, J = 7.1, 2.7 Hz, 2H), 3.59–3.20 (m, 2H), 1.53 (s, 9H), 1.38 (s, 18H), 1.28 (s, 3H), 0.32 (s, 9H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 170.21, 151.98, 151.90, 151.72, 146.34, 140.06, 137.19, 122.32, 118.86, 83.34, 83.09, 61.58, 59.69, 38.42, 29.82, 27.96, 14.28, −8.10. |
| HRMS | m/z: [M + Na]+ calcd. for C29H47NNaO9Sn+: 696.21650; found: 696.21766. Correct isotopic pattern. |
3.4.17. Ethyl (S)-2-[Bis(tert-butoxycarbonyl)amino]-3-[4-(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenylpropanoate [Boc2-2-(SnMe3)Tyr(Boc)-OEt]
The title compound was synthesized from ethyl (S)-2-[(tert-butoxycarbonyl)amino]-3-[4-(tert-butoxycarbonyl)oxy]-2-(trimethylstannyl)phenylpropanoate [Boc2-2-(SnMe3)Tyr(Boc)-OEt] (100 mg, 0.2 mmol) according to GP5.
| Yield | 117 mg, 85% |
| Appearance | yellow oil |
| Molecular formula | C29H47NO9Sn |
| Molar mass | 672.403 |
| 1H-NMR | (200 MHz, CDCl3): δ (ppm) = 7.17 (d, J = 2.3 Hz, 1H), 7.12–6.84 (m, 1H), 4.98 (dd, J = 9.7, 5.1 Hz, 1H), 4.21 (qd, J = 7.2, 1.9 Hz, 1H), 3.35 (dd, J = 7.2, 4.2 Hz, 1H), 1.54 (s, 1H), 1.36 (s, 2H), 1.27 (s, 1H), 0.33 (s, 1H). |
| 13C-NMR | (50 MHz, CDCl3): δ (ppm) = 170.19, 151.95, 151.81, 149.48, 144.66, 142.01, 130.20, 128.49, 121.39, 83.39, 83.11, 61.54, 59.89, 37.87, 29.79, 27.97, 27.80, 14.26, −8.10. |
| HRMS | m/z: [M + Na]+ calcd for C29H47NNaO9Sn+: 696.21650; found: 696.21700. Correct isotopic pattern. |
3.5. Radiochemistry
3.5.1. General Procedures
All radiosyntheses were carried out using anhydrous DMA and nBuOH stored over molecular sieves (available from “Acros”, Geel, Belgium, or “Aldrich”). Cu(OTf)2(py)4 was stored under ambient conditions without any precautions.
[18F]Fluoride was produced by the 18O(p,n)18F reaction by bombardment of enriched [18O]water with 16.5 MeV protons using a BC1710 cyclotron (The Japan Steel Works Ltd., Shinagawa, Japan) at the INM-5 (Forschungszentrum Jülich).
All radiolabeling experiments were carried out under ambient or synthetic air. Each radiochemical experiment was carried out at least in triplicates if not otherwise mentioned. Standard deviations (SD) were calculated by the least-square method. All experiments were carried out by using one-pot procedure. Before the determination of radiochemical conversions (RCCs), reaction mixtures were always diluted with H2O (1–4 mL) to dissolve any 18F-fluoride adsorbed onto the reaction vessel walls. The loss of radioactivity on the vessel walls did not exceed 13 ± 2% from the starting activity (n > 100). All radiochemical yields (RCYs) are decay corrected and radiochemical purities (RCPs) were determined after purification.
3.5.2. Processing [18F]Fluoride
Aqueous [18F]fluoride was loaded onto an anion-exchange resin (e.g., QMA cartridge). It should be noted that aqueous [18F]fluoride was loaded onto the cartridge from the male side, whereas flushing, washing and 18F− elution were carried out from the female side. If the QMA cartridge had been loaded, flushed and eluted from the female side only, sometimes a significant amount of [18F]fluoride remained on the resin (this is probably because QMA-light (46 mg) cartridges have a single frit on the male side but four frits on the female side).
3.5.3. High-Performance Liquid Chromatography (HPLC)
For manual radiosyntheses the following HPLC system was used:
Ultimate® 3000 HPLC system from Thermo Scientific (Sunnyvale, CA, USA) with Ultimate® 3000 LPG-3400A pump, Ultimate® 3000 VWD-3100 UV/Vis detector and γ-detector Gabi Star from Raytest GmbH (Straubenhardt, Germany) were used. The volume of injection was 20 μL.
Columns:
Chromolith® SpeedROD RP-18 endcapped 50 × 4.6 mm, Merck KGaA (Darmstadt, Germany).
ProntoSIL C18 ace-EPS 125 × 4.6 mm, Bischoff Analysentechnik und -geräte GmbH (Leonberg, Germany).
Gemini® 5 µm C18 110 Å, 250 × 4.6 mm, Phenomenex Inc. (Aschaffenburg, Germany).
Gemini® 5 µm C18 110 Å, 250×10 mm, Phenomenex Inc. (Aschaffenburg, Germany).
For automated syntheses the following system was used:
WellChrom Spectro-photometer K-2501 UV/Vis detector, BlueShadow Pump 80P from KNAUER Wissenschaftliche Geräte GmbH, Berlin, Germany and AD 1422 PIN-photodiode and scintillator detector from Eckert & Ziegler Strahlen- und Medizintechnik AG, Berlin, Germany was connected directly to the automated module.
Columns:
Synergi™ 4 µm Hydro-RP 80 Å, 250 × 10 mm, Phenomenex Inc. (Aschaffenburg, Germany).
Synergi™ 4 µm Hydro-RP 80 Å, 150 × 21.2 mm, AXIA™, Phenomenex Inc. (Aschaffenburg, Germany).
UV and radioactivity detectors were connected in series, giving a time delay of 0.1–0.9 min depending on the flow rate. 18F-Labeled compounds were identified by co-injection of the unlabeled reference compounds. The completeness of the radioactivity elution was controlled by analyzing of the same sample amount choosing a column bypass.
3.5.4. Determination of the Enantiomeric Purity
The enantiomeric purity of radiolabeled amino acids was determined using chiral HPLC. Conditions: column: CROWNPAK®CR(+) 150 × 4.6 mm 5 µm (Daicel Corporation, Osaka, Japan); eluent: 0.1 M HClO4 or 5% MeOH in 0.1 M HClO4; flow rate: 1.0 mL/min.
3.5.5. Automated Radiosyntheses
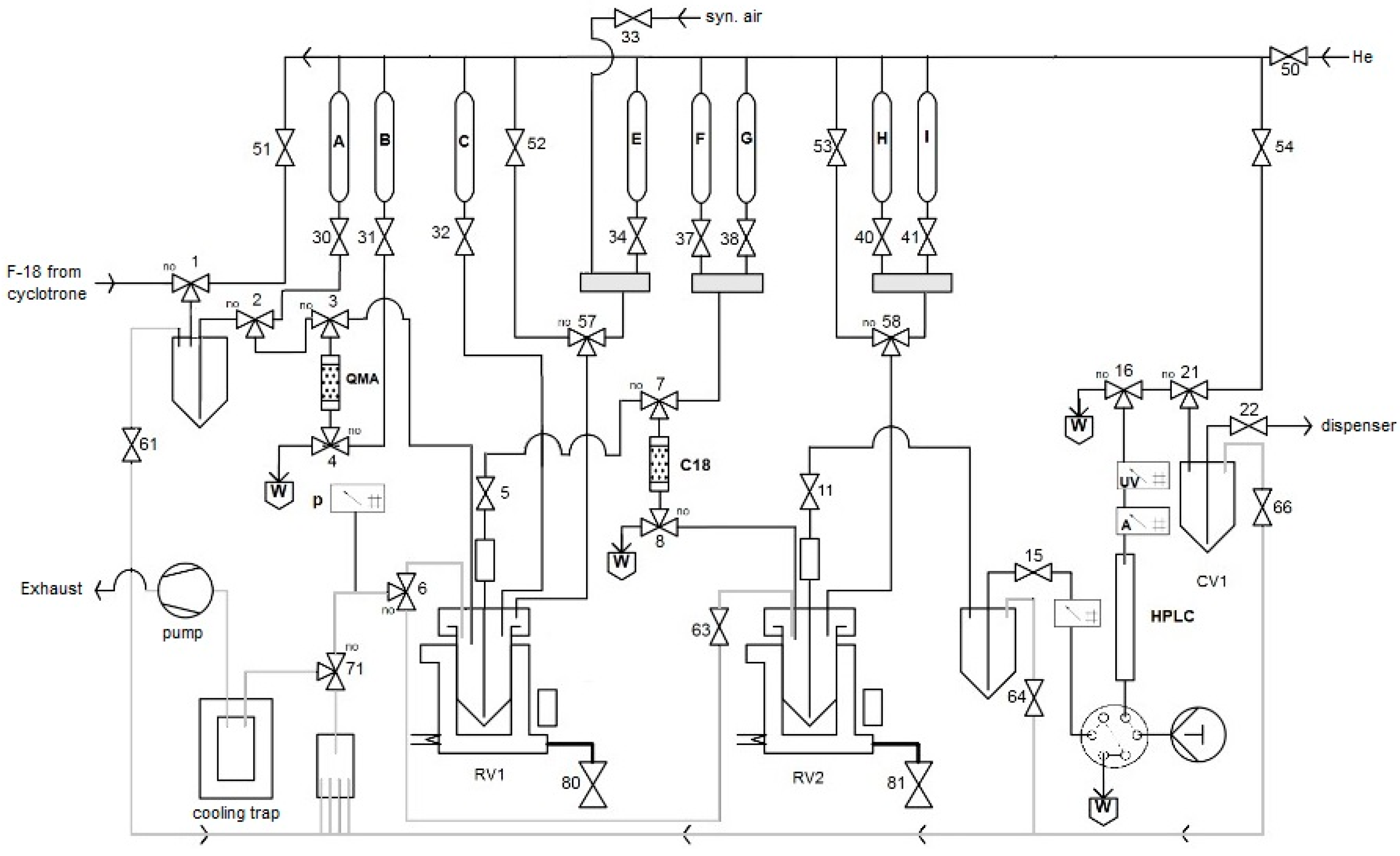
All automated radiosyntheses were carried out in a home-made synthesis module. FFKM valves (Christian Bürkert GmbH&Co. KG, Ingelfingen, Germany) were applied. All connections between the valves were made using PTFE tubes and PEEK fittings. The flow scheme for the preparation of radiolabeled amino acids is depicted in
Figure 13 and
Figure S1. Synthetic air and He (Westfalen AG, Muenster, Germany) were used as operating gases.
3.5.6. Miscellaneous Information
Radioactivity was measured with a CRC®-55tR Dose Calibrator from Capintec, Inc. (Florham Park, NJ, USA) or the Curiementor 2 from PTW GmbH (Freiburg, Germany).
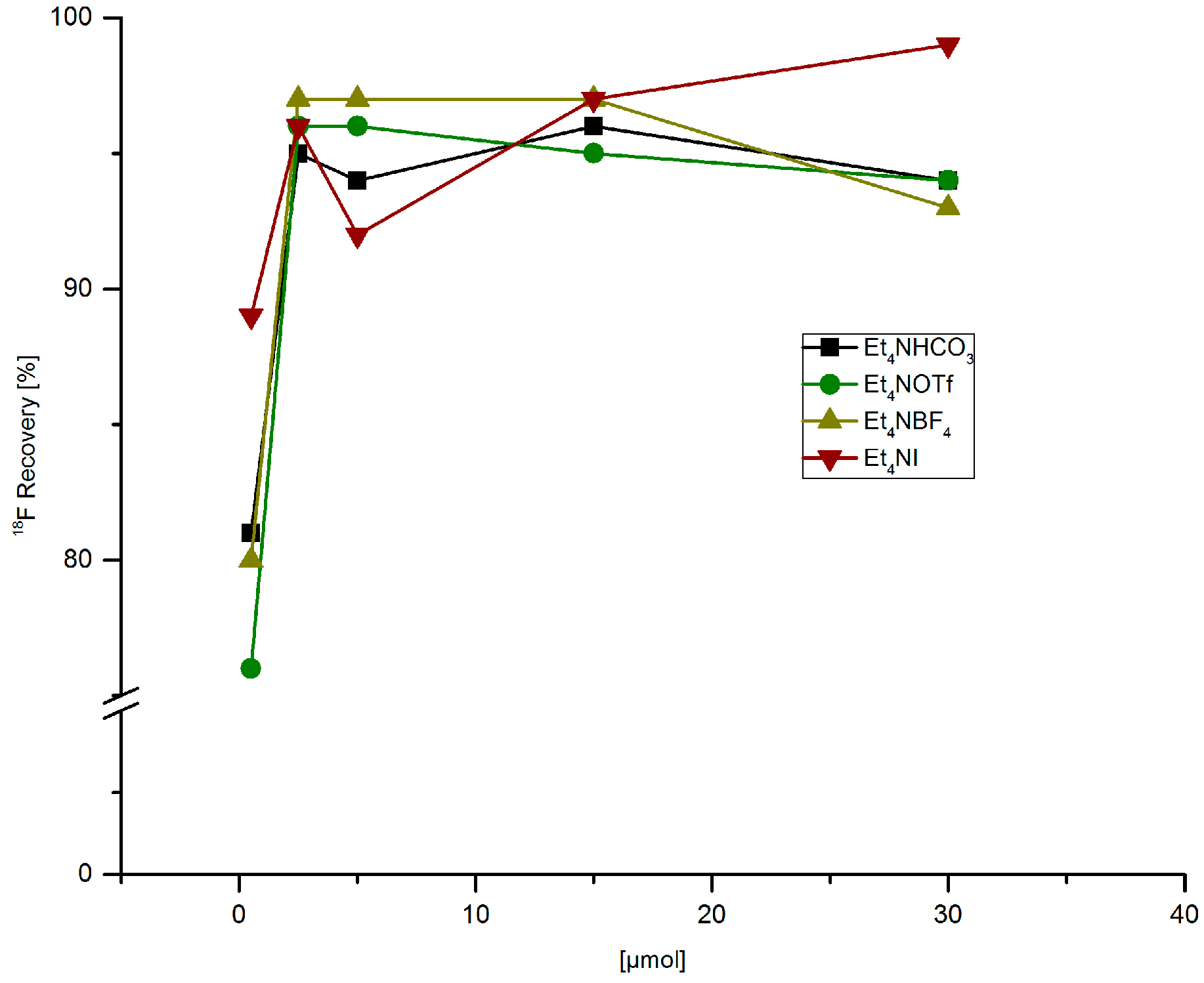
3.5.7. Recovery of 18F− from Anion Exchange Resin with MeOH Solutions of Different Tetramethylammonium Salts
[18F]Fluoride (~50 MBq) was fixed on QMA-CO3 cartridge from the male side, the cartridge was washed with MeOH (1 mL) in the same direction. Finally, [18F]fluoride was eluted with a solution of Et4NX in MeOH (500 µL) from the female side.
3.5.8. 18F-Recovery and RCCs of [18F]FPh Using Different Salts in nBuOH
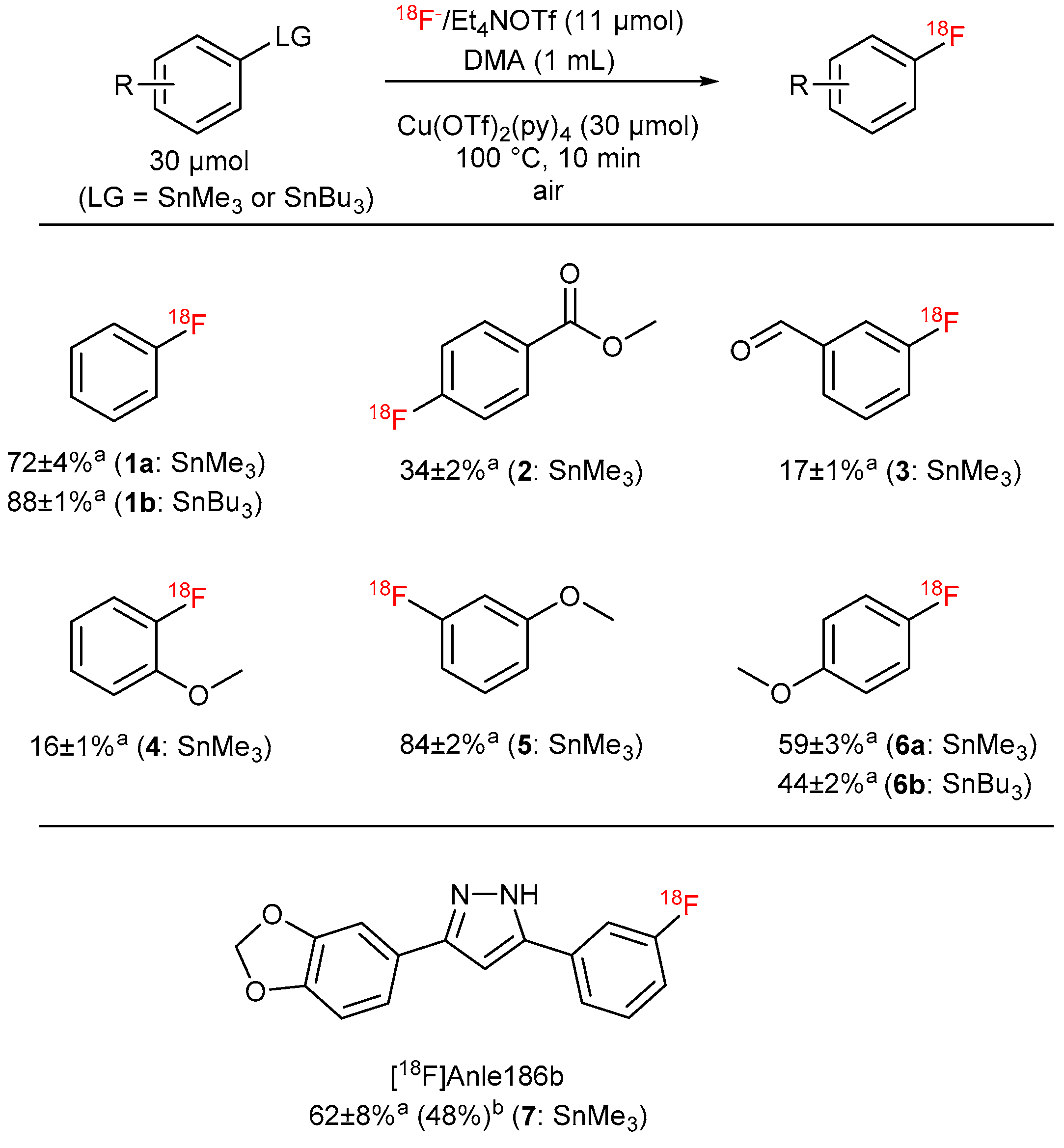
[18F]Fluoride (~50 MBq) was recovered from QMA-CO3 cartridge with a solution of the respective salt (11 µmol) in nBuOH (300 µL). A solution PhSnMe3 (14.5 mg, 60 µmol), Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA (700 µL) was added, the reaction mixture was heated at 100 °C for 10 min under air, diluted with H2O (1 mL) and analyzed by HPLC.
3.5.9. Dependence of [18F]Fluoride Recovery and 18F-Incorporation Yields on the Type of an Anion Exchange Cartridge
[18F]Fluoride (~50 MBq) was eluted from the respective anion exchange cartridge with a solution of Et4NOTf (3.1 mg, 11 µmol) nBuOH (300 µL). A solution PhSnMe3 (14.5 mg, 60 µmol), Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA (700 µL) was added, the reaction mixture was heated at 100 °C for 10 min under air, diluted with H2O (1 mL) and analyzed by HPLC.
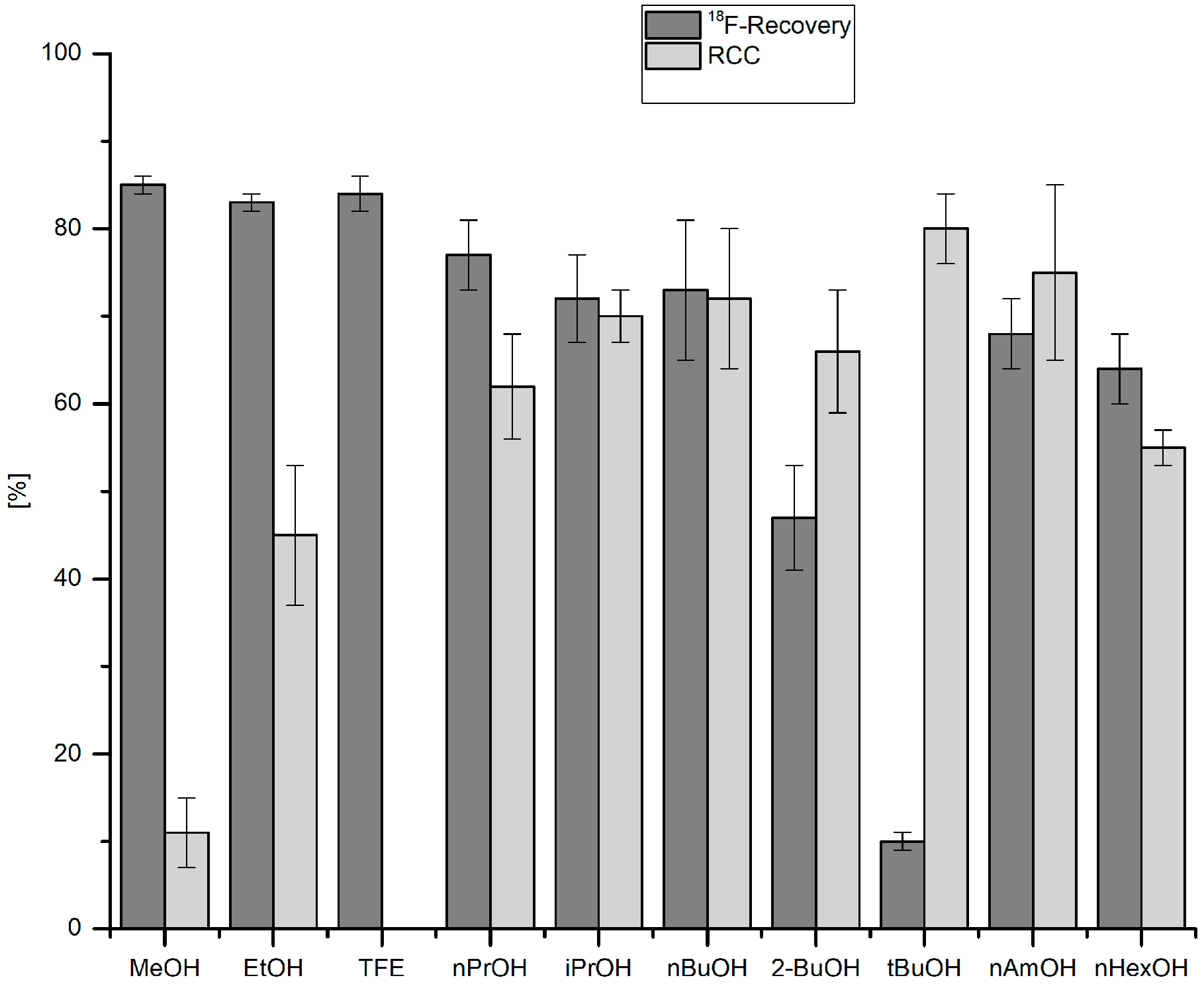
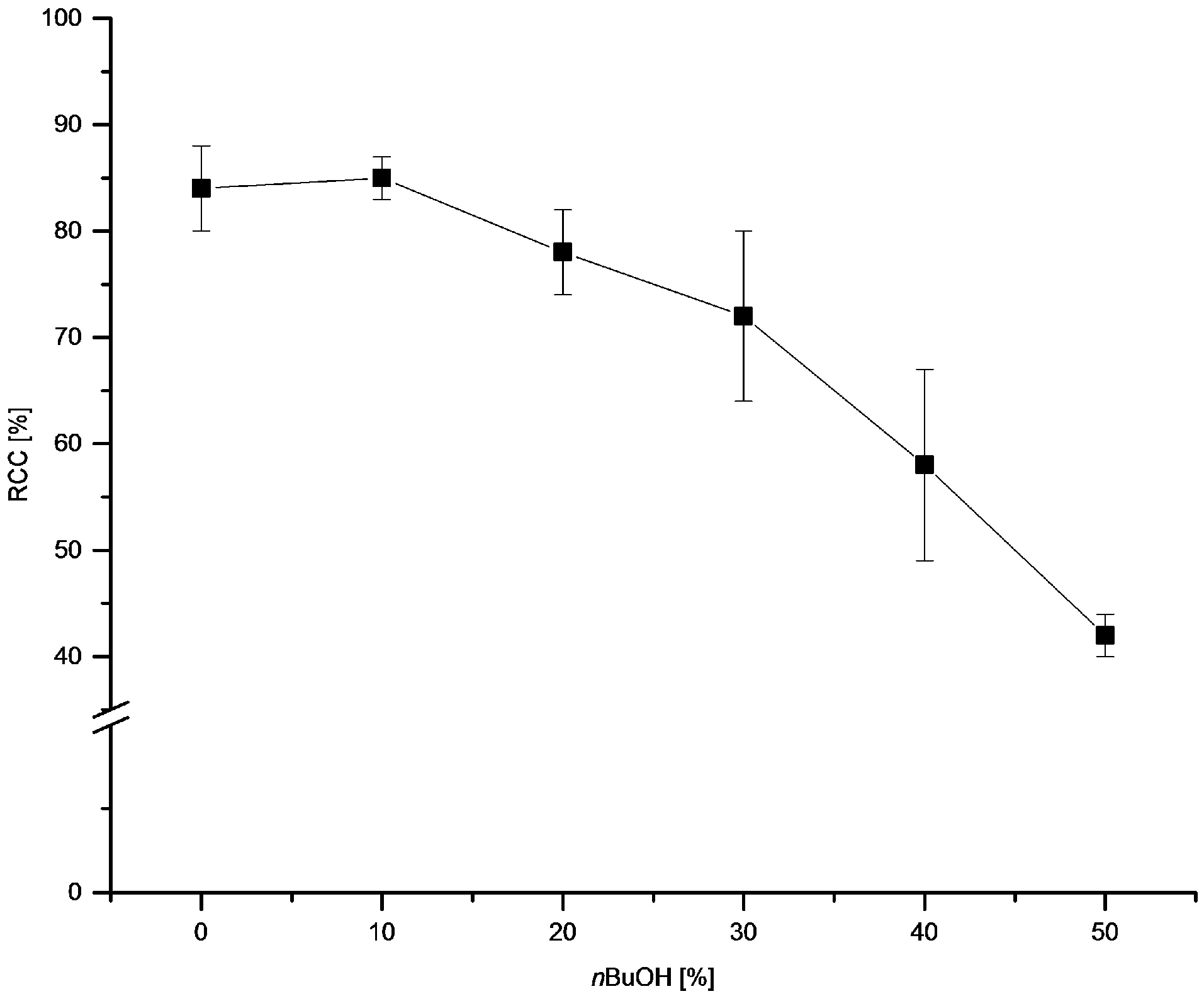
3.5.10. Effect of Alcohol on 18F-Recovery and 18F-Fluorodestannylation
18F− (50–150 MBq) was eluted into the reaction vial with a solution of Et4NOTf (3.1 mg, 11 µmol) in the corresponding anhydrous alcohol (300 µL); to this solution a solution trimethyl(phenyl)tin (14.5 mg, 60 µmol), Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA (700 µL) was added, the reaction mixture was heated at 100 °C for 10 min under air, diluted with H2O (1 mL) and analyzed by HPLC.
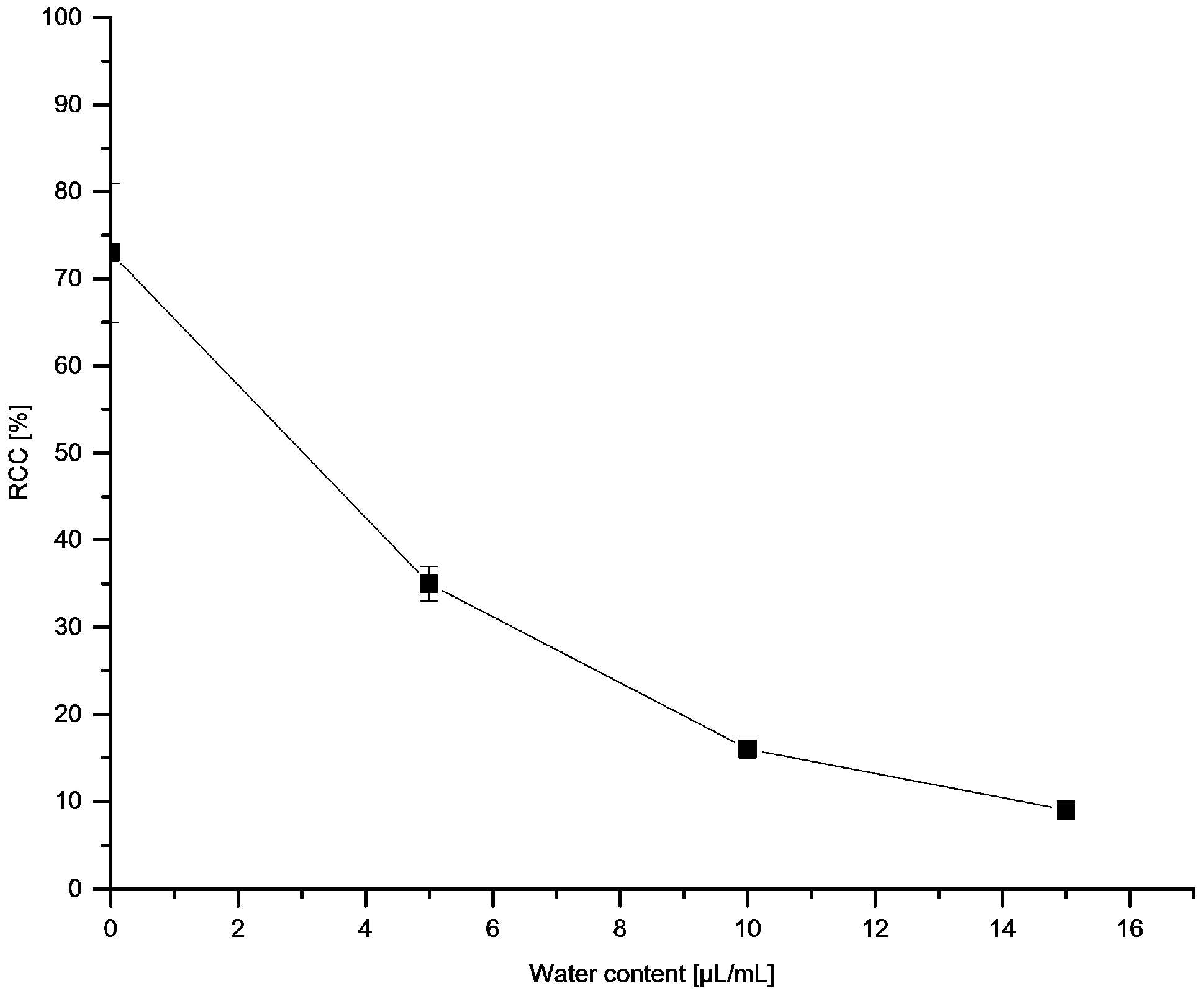
3.5.11. Effect of Water on [18F]Fluorodestannylation
[18F]Fluoride (~50 MBq) was eluted from the respective anion exchange cartridge with a solution of Et4NOTf (3.1 mg, 11 µmol) in nBuOH (300 µL). A solution of PhSnMe3 (14.5 mg, 60 µmol), Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA (700 µL) containing H2O was added, the reaction mixture was heated at 100 °C for 10 min under air, diluted with H2O (1 mL) and analyzed by HPLC.
3.5.12. Dependency of RCC on Alcohol Content
[18F]Fluoride (~50 MBq) was eluted from QMA-CO3 with a solution of Et4NOTf (3.1 mg, 11 µmol) in MeOH (500 µL), MeOH was evaporated at 100 °C under a flow of air within 2–3 min. A solution of PhSnMe3 (14.5 mg, 60 µmol), Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA/nBuOH (1 mL) was added, the reaction mixture was heated at 100 °C for 10 min under air, diluted with H2O (1 mL) and analyzed by HPLC.
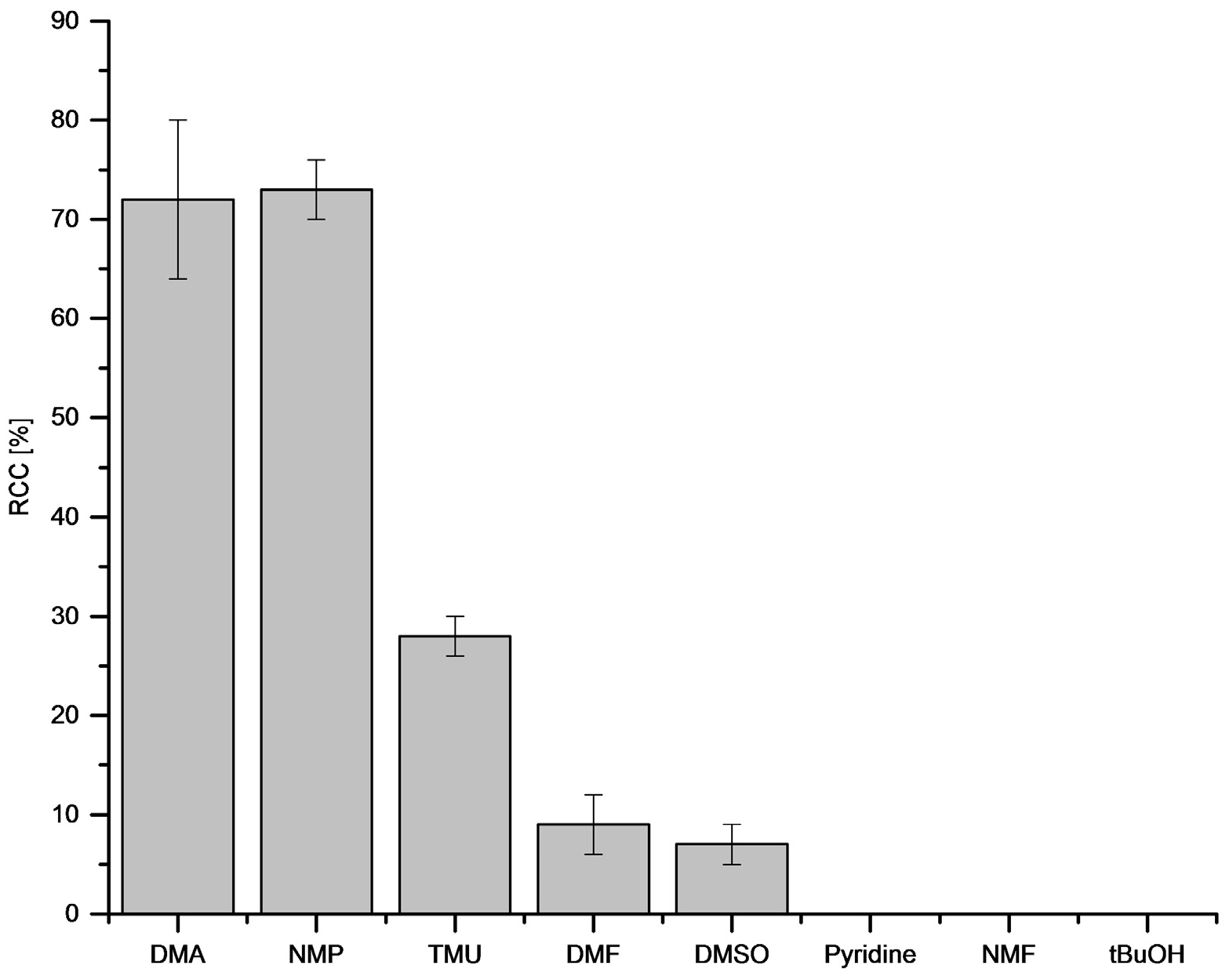
3.5.13. Optimization of Aprotic Solvent
[18F]Fluoride (~50 MBq) was eluted from QMA-CO3 with a solution of Et4NOTf (3.1 mg, 11 µmol) in (300 µL) in nBuOH. A solution of PhSnMe3 (14.5 mg, 60 µmol) and Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in the appropriate solvent was added and the reaction mixture was heated at 100 °C for 10 min, cooled down, diluted with H2O (1 mL) and analyzed by HPLC.
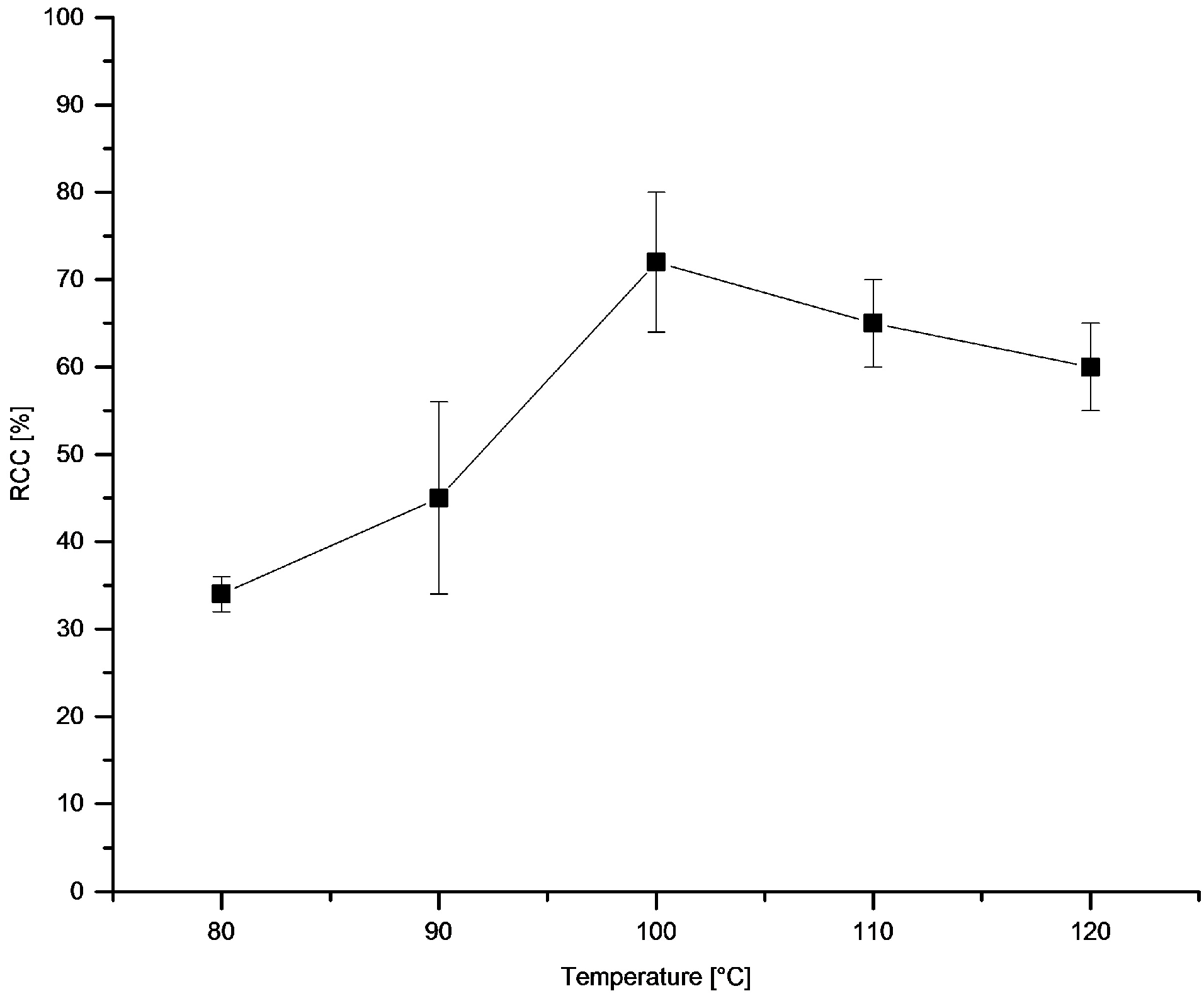
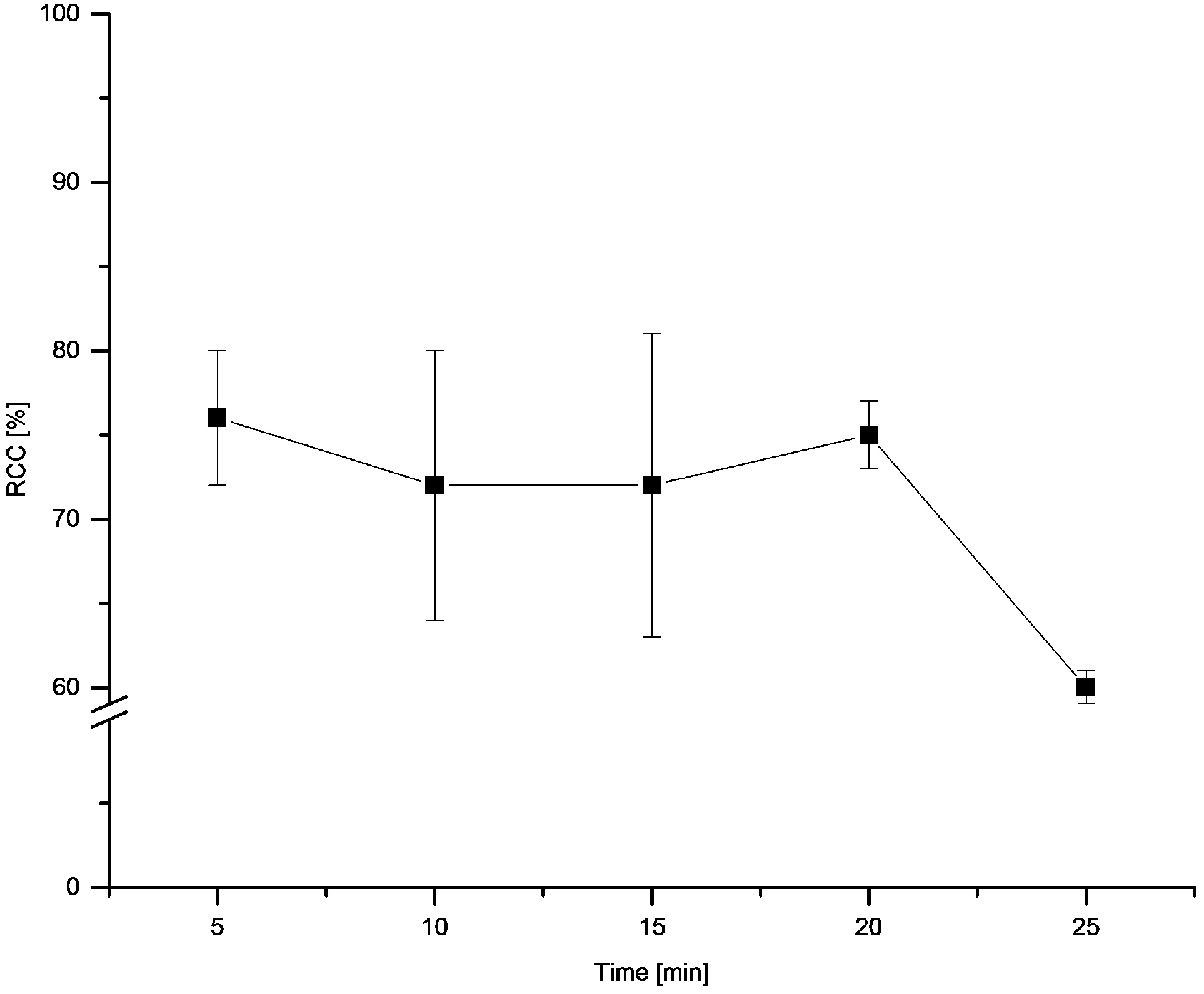
3.5.14. Dependence of RCCs on Temperature and on Time
[18F]Fluoride (~50 MBq) was eluted from QMA-CO3 with a solution of Et4NOTf (3.1 mg, 11 µmol) in nBuOH (300 µL). A solution of PhSnMe3 (14.5 mg, 60 µmol) and Cu(py)4(OTf)2 (20.3 mg, 30 µmol) in DMA (700 µL) was added the reaction mixture was heated at given temperature for 10 min or at 100 °C for given time, cooled down, diluted with H2O (1 mL) and analyzed by HPLC.
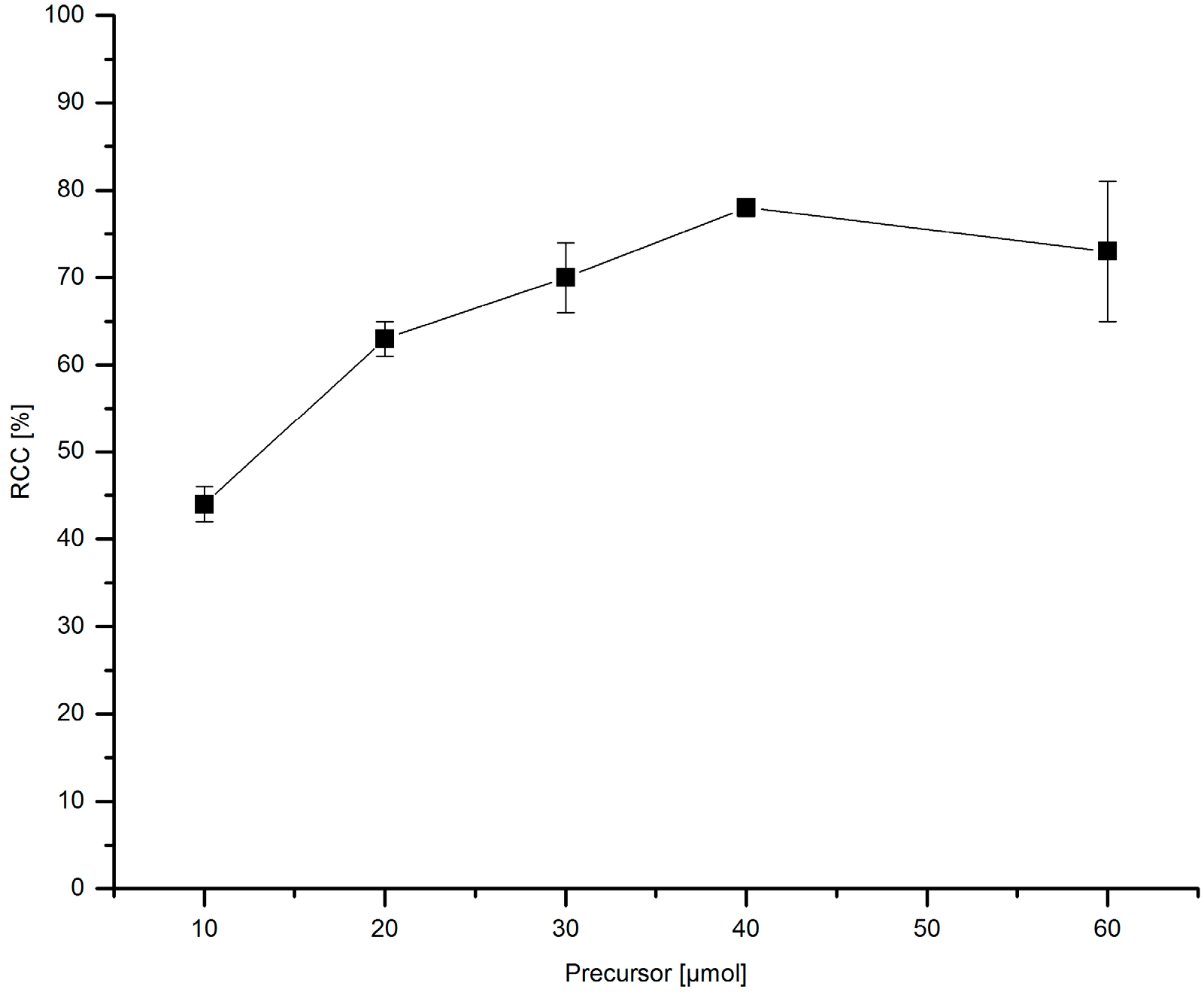
3.5.15. Dependence of 18F-Incorporation Rate on the Precursor Amount
[18F]Fluoride (~50 MBq) was eluted from QMA-CO3 with a solution of Et4NOTf (3.1 mg, 11 µmol) in nBuOH. A solution of given amount of PhSnMe3 and Cu(py)4(OTf)2 (20.3 mg, 30 µmol), the mixture was heated under air at 100 °C for 10 min, cooled down, diluted with H2O (1 mL) and analyzed by HPLC.
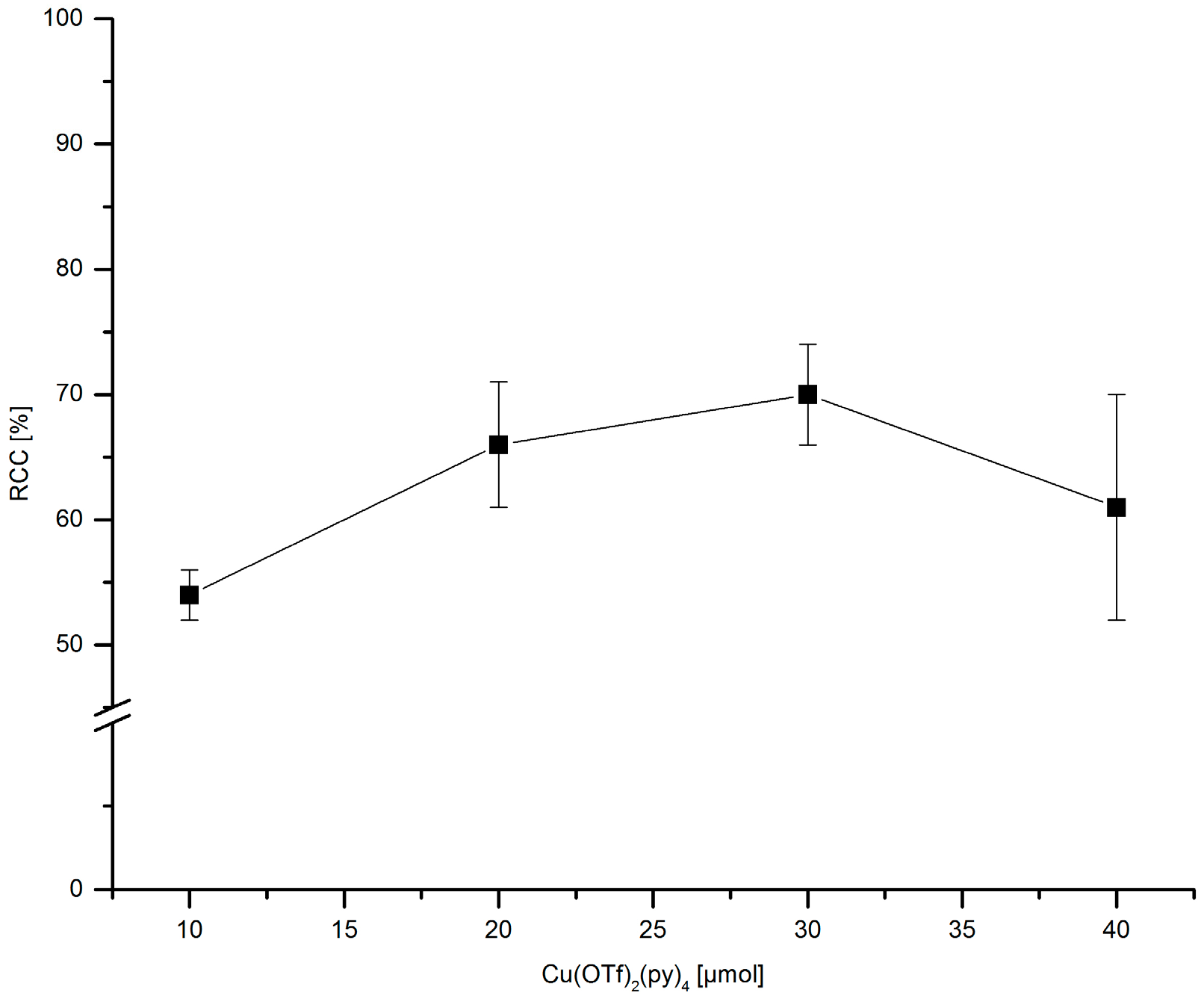
3.5.16. Dependence of 18F-Incorporation Rate on the Cu(py)4(OTf)2 Amount
[18F]Fluoride (~50 MBq) was eluted from QMA-CO3 with a solution of Et4NOTf (3.1 mg, 11 µmol) in nBuOH. A solution of PhSnMe3 (7.2 mg, 30 µmol) and given amount of Cu(py)4(OTf)2 in DMA (700 µL) was added, the mixture was heated under air at 100 °C for 10 min, cooled down, diluted with H2O (1 mL) and analyzed by HPLC.
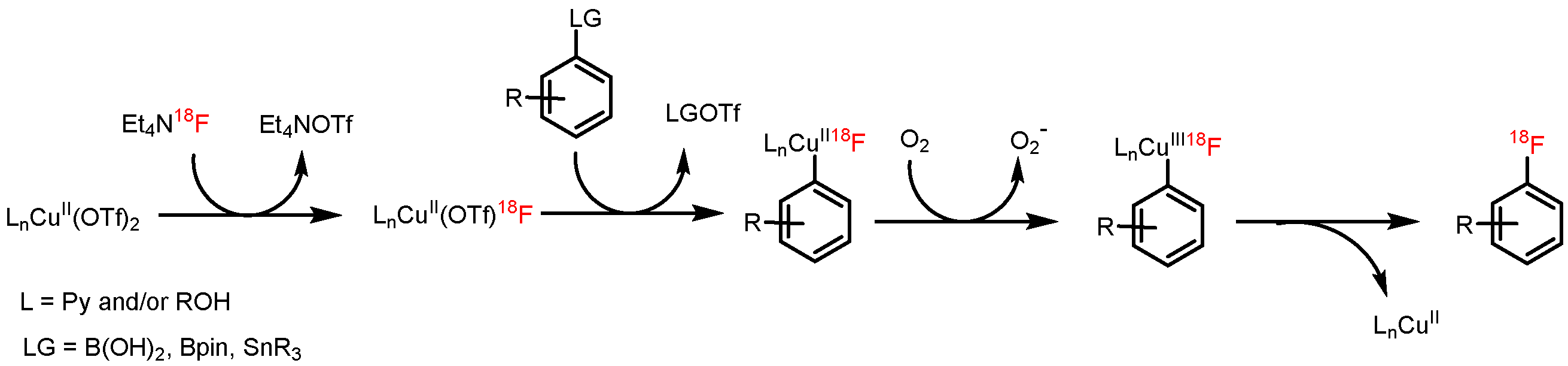
3.5.17. Optimized Procedure for 18F-Fluorodestannylation—General Procedure (GP6)
[18F]Fluoride (50–100 MBq) was loaded on an anion exchange cartridge (QMA-CO3, preconditioned with 1 mL water and dried with air) from the male side. The cartridge was rinsed with MeOH (1 mL) and dried with air, then [18F]fluoride was eluted with a methanolic solution (500 µL) of Et4NOTf (2.79 mg, 10 µmol). Methanol was removed under reduced pressure (600 mBar) in a stream of argon at 100 °C within 3 min. Afterwards, the pressure was reduced to 50 mBar and the reaction vial was purged with air. A solution of the corresponding precursor (30 µmol) and Cu(OTf)2(py)4 (20.3 mg, 30 µmol) in DMA (1 mL) was added, the reaction mixture was stirred at 100 °C for 10 min and cooled down to room temperature in an ice bath. The reaction mixture was quenched with water (4 mL) and analyzed by HPLC.
3.5.18. Manual Synthesis of Radiolabeled Amino Acids—General Procedure 7 (GP7)
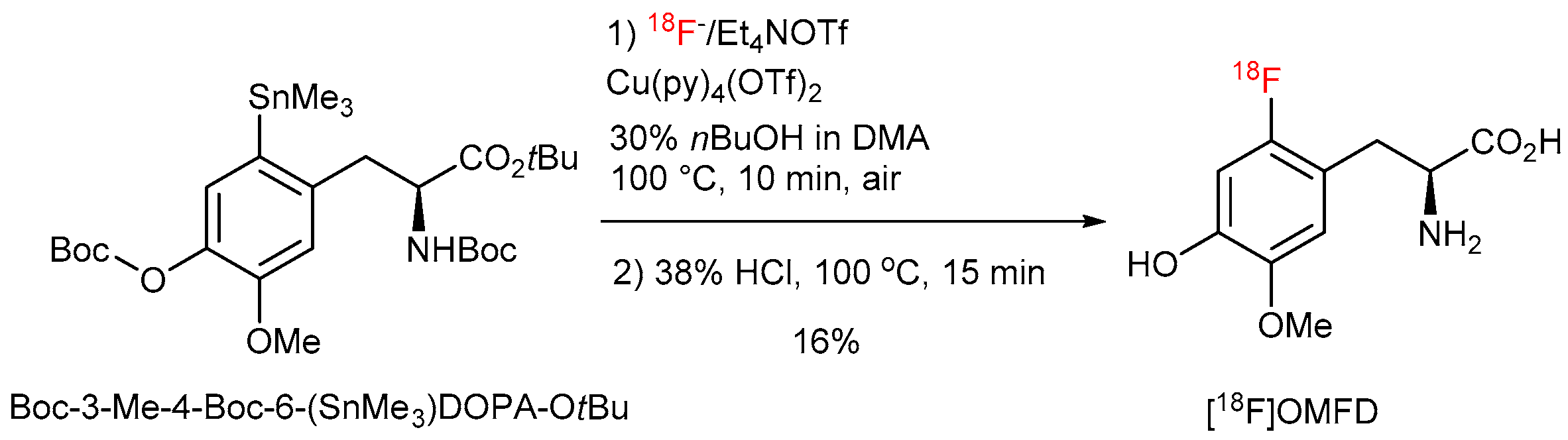
[18F]Fluoride (200–300 MBq) was loaded onto an anion exchange cartridge (QMA-CO3 preconditioned with 1 mL water and dried with air) from the male side. The cartridge was washed with MeOH (1 mL) and dried with air. Thereafter, [18F]fluoride was eluted into the reaction vial using a solution of Et4NOTf (2.79 mg, 10 µmol) in MeOH (500 µL). MeOH was removed under reduced pressure (600 mBar) using a stream of air at 100 °C within 5 min. A solution of Cu(OTf)2(py)4 (40.7 mg, 60 µmol) and the corresponding precursor (30 µmol) in DMA (1 mL) was added. The reaction mixture was stirred at 100 °C for 10 min, and cooled down to room temperature in an ice bath. The reaction mixture was quenched with water (2 mL) and loaded in Sep-Pak C18 Plus light Cartridge. The cartridge was washed with 5 mL water and the product was eluted with 1 mL EtOH. EtOH was removed under reduced pressure (600 mBar) using a stream of air at 120 °C within 5 min. 48% HBr (1 mL) was added and the reaction mixture was stirred at 130 °C for 10 min. Hydrolysis of the protected [18F]OMFD was carried out using 38% HCl at 100 °C for 10 min. The reaction mixture was cooled down, diluted with H2O (3 mL) and analyzed by HPLC. RCC was calculated from amount of 18F− loaded onto QMA cartridge, radioactivity amount in the reaction vial after hydrolysis step and HPLC chromatogram.
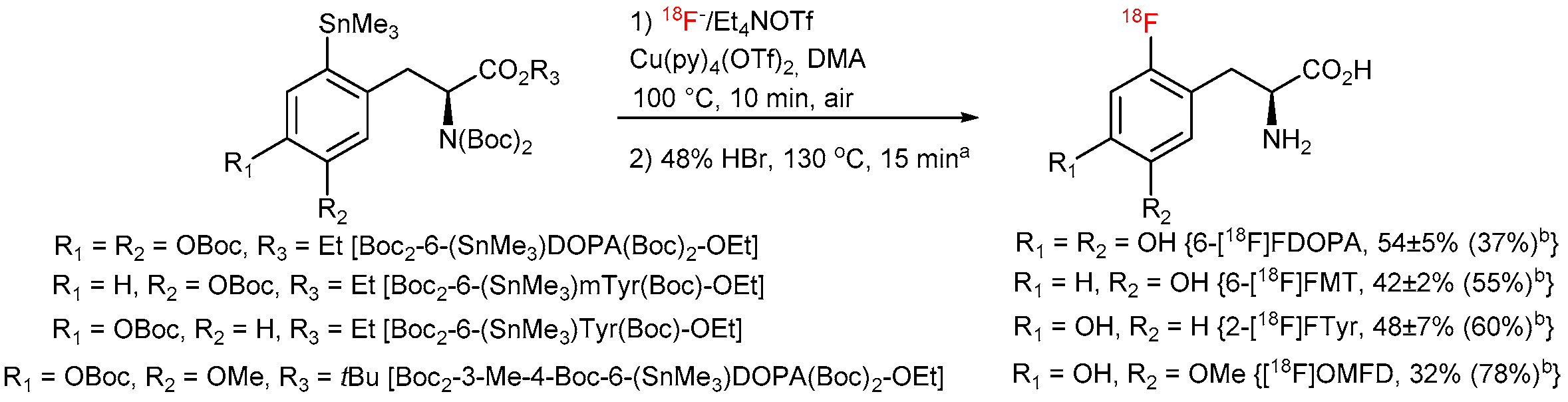
3.5.19. Automated Synthesis of Radiofluorinated Amino Acids—General Procedure 8 (GP8)
Trapping of [18F]fluoride on an QMA ion exchange cartridge;
Washing of the QMA with MeOH;
Closing air valve (50), and system venting;
Elution of [18F]fluoride from the ion exchange cartridge with a methanolic solution of Et4NOTf into RV 1;
Open air valve (50) to completely transfer methanolic solution from QMA to RV 1;
Evaporation of MeOH in RV1 at 100 °C for 3 min using a flow of synthetic air under reduced pressure;
Addition of a solution of the radiolabeling precursor (30 µmol) and Cu(OTf)2(py)4 (40 mg) in DMA (1 mL);
Heating of the reaction mixture in RV1 at 100 °C for 10 min;
Cooling of RV1 down to 50 °C;
Addition of water (1 mL) → in the case of [18F]OMFD: precipitation of precursor;
Loading of the mixture onto a SPE cartridge (C18);
Rinsing of the SPE cartridge with H2O (9 mL);
Elution of the radiolabeled intermediate into RV2 using CH2Cl2 (2.0 mL);
Evaporation of CH2Cl2 at 100 °C within 3 min using a flow of He under reduced pressure;
Addition of 48% HBr (1 mL) and heating at 130 °C for 10 min; in the case of [18F]OMFD: addition of 38% HCl (1 mL) and heating at 100 °C for 10 min;
Cooling of RV2 to 55 °C and addition of a solution consisting of 45% NaOH (300 µL) and 25 mM Na phosphate buffer (3 mL, pH 4.5);
Loading of the mixture onto the HPLC loop for injection;
Injection of the loop content onto the HPLC column and elution with 25 mM sodium phosphate buffer (pH 4.5) at 8 mL/min;
Manual collection of the product fraction in a collection vial (CV1);
Transfer the product solution from CV1 into a sterile, filter-vented final product vial via a 0.22 μm sterile membrane filter using a flow of He.
3.5.20. Molar Activity Calculation
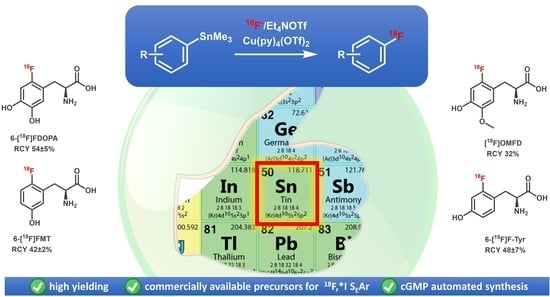
The molar activities (GBq/µmol) were calculated by dividing the radioactivity of the 18F-labeled product by the amount of the unlabeled tracer determined from the peak area in a UV-HPLC chromatograms (λ = 225 or 230 nm). The amounts of unlabeled compounds were determined from the UV absorbance/concentration calibration curves. The molar activities of 6-[18F]FDOPA (7.4 GBq), 6-[18F]FMT (2.5 GBq), 2-[18F]FTyr (1.7 GBq) and [18F]OMFD (1 GBq) were determined to 57, 39, 50 and 27 GBq/µmol, respectively.
3.5.21. Determination of Sn and Cu Content
The contents of tin and copper were determined by Agilent 7900 ICP-MS. Solutions of amino acid derivatives obtained after HPLC purification were concentrated under reduced pressure and the residues were taken up in high purity H2O (1 mL). The measured samples were diluted 1:100. The higher metal content in 6-[18F]FDOPA and 2-[18F]FTyr is explained by the application of 5 mL instead of 9 mL H2O for Step 12 (GP8) in the respective productions.

 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}