Conserved Binding Regions Provide the Clue for Peptide-Based Vaccine Development: A Chemical Perspective

Abstract
1. Introduction
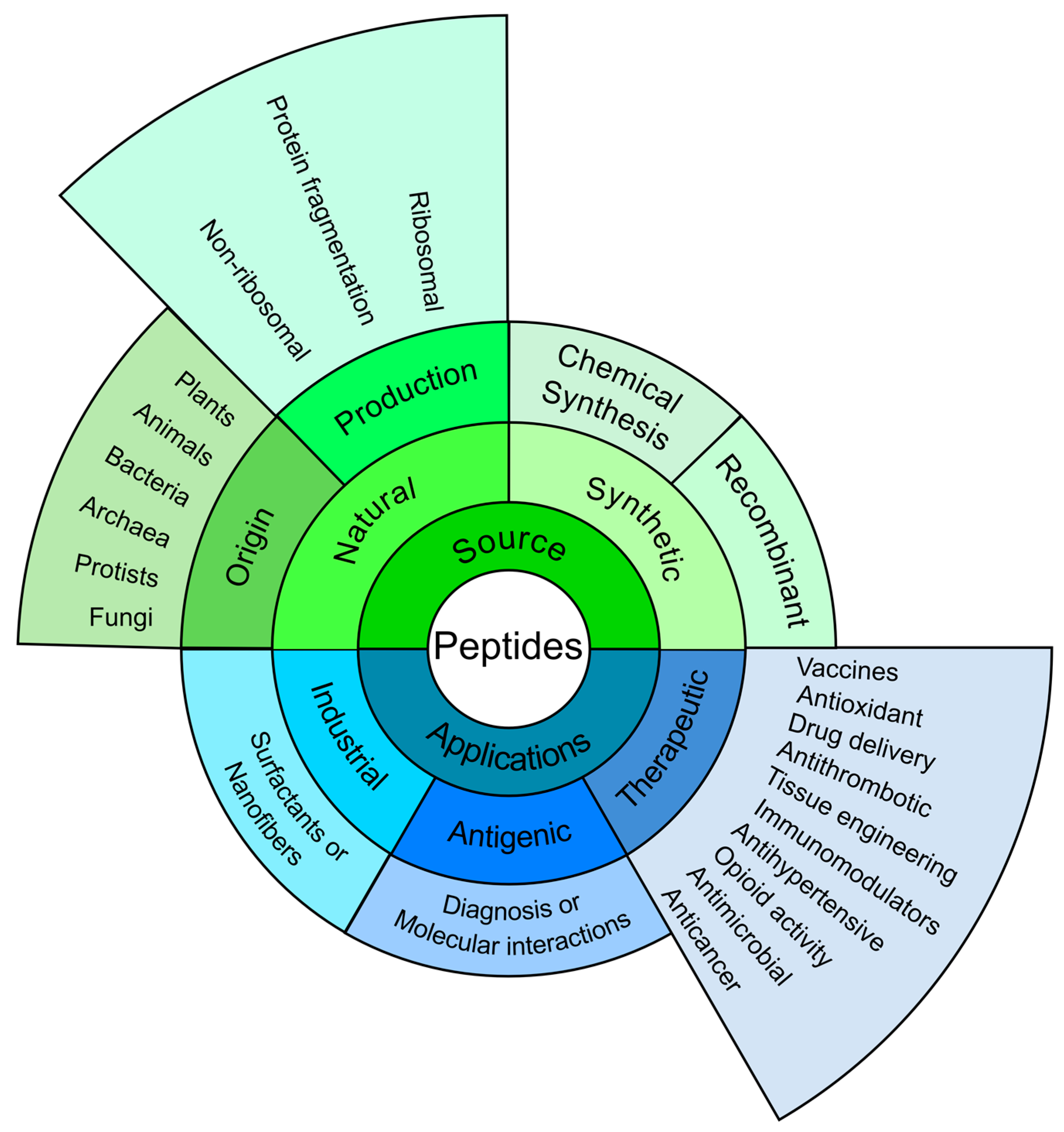
2. A Synthetic Peptide-Based Vaccine Is Feasible
3. Adopting a Functional Approach for Vaccine Development
4. Subunit Component Selection
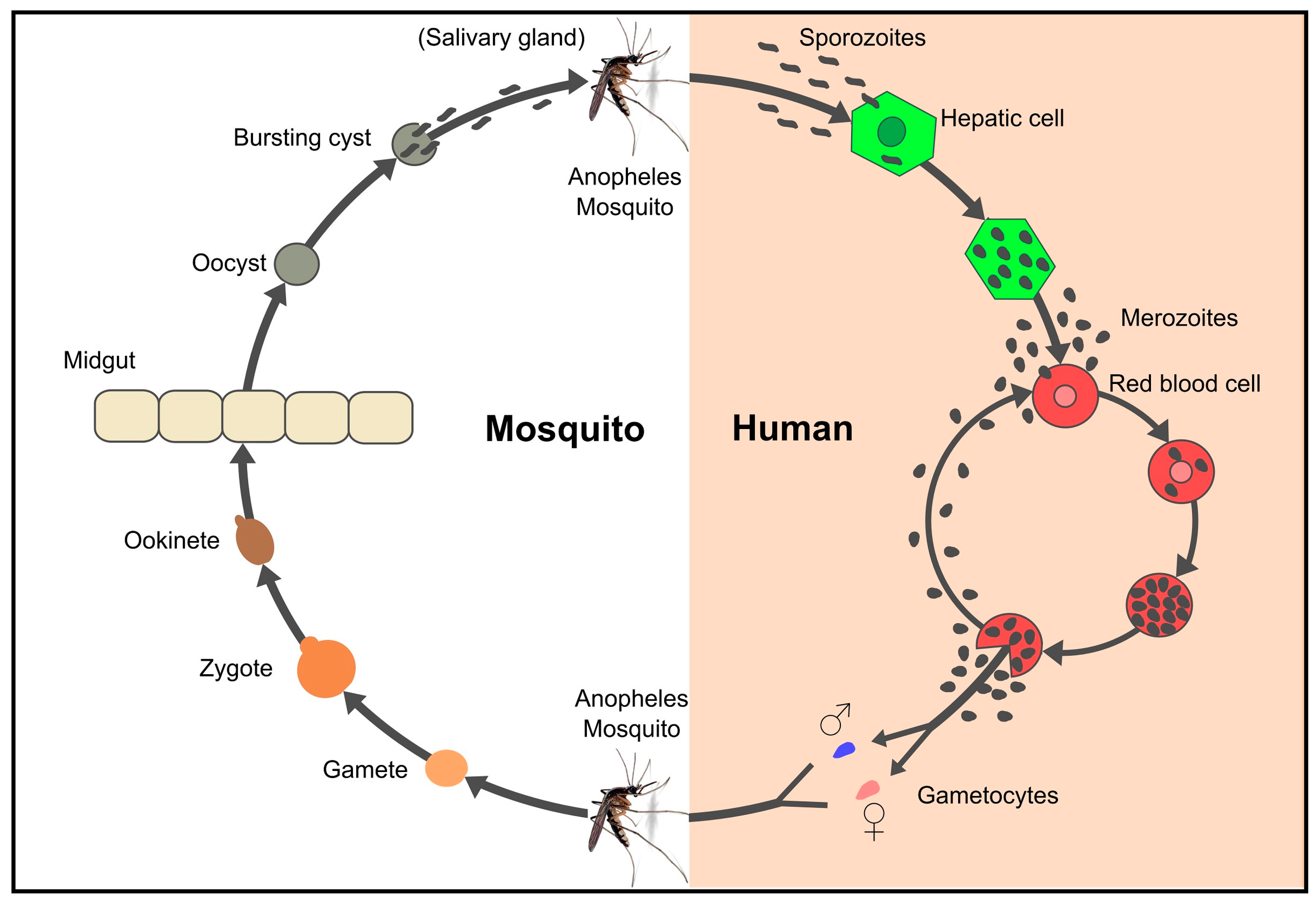
5. Breaking the Parasite’s Immunological Code of Silence
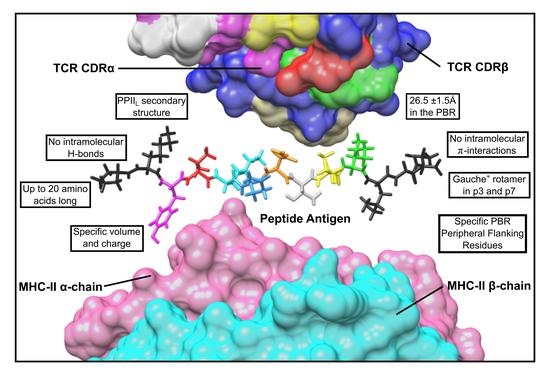
6. Chemical Features to Be Taken into Account in Vaccine Development
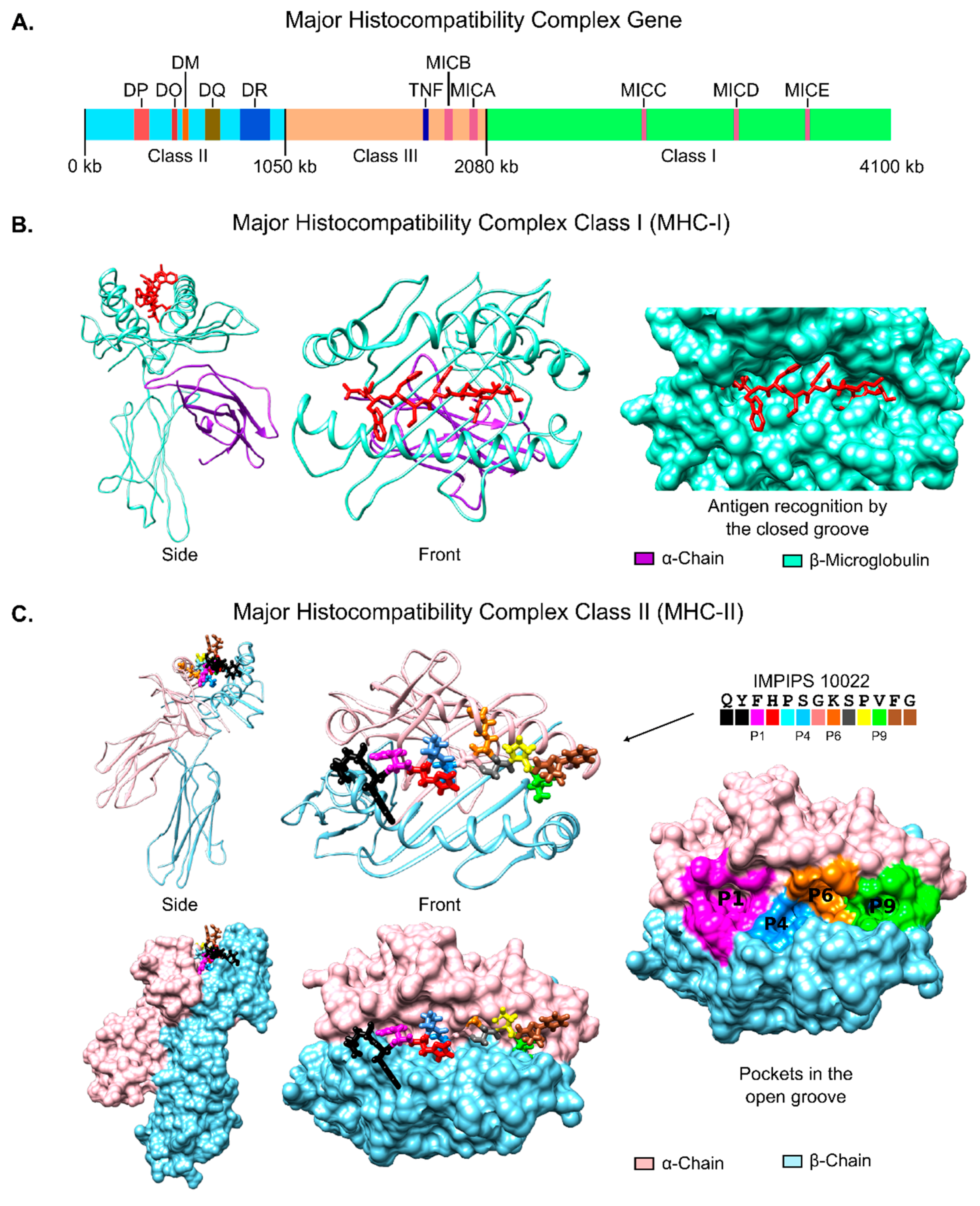
6.1. Immune System Molecules’ 3D Structure
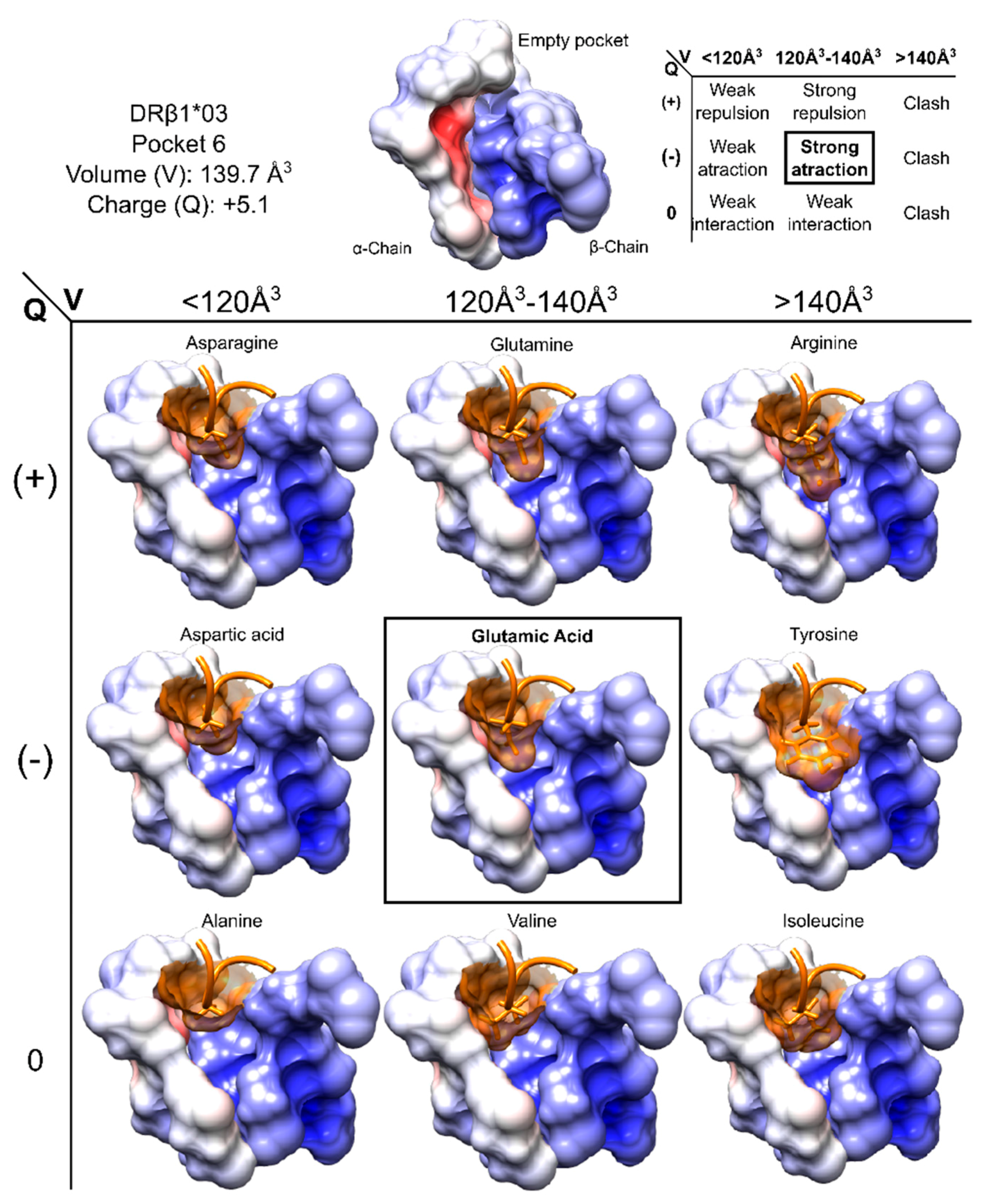
6.2. Charge and Volume
6.3. Peptide–Antigen Length
6.4. Polyproline as Preferred Secondary Structure
6.5. Amino Acid Side chain Orientation
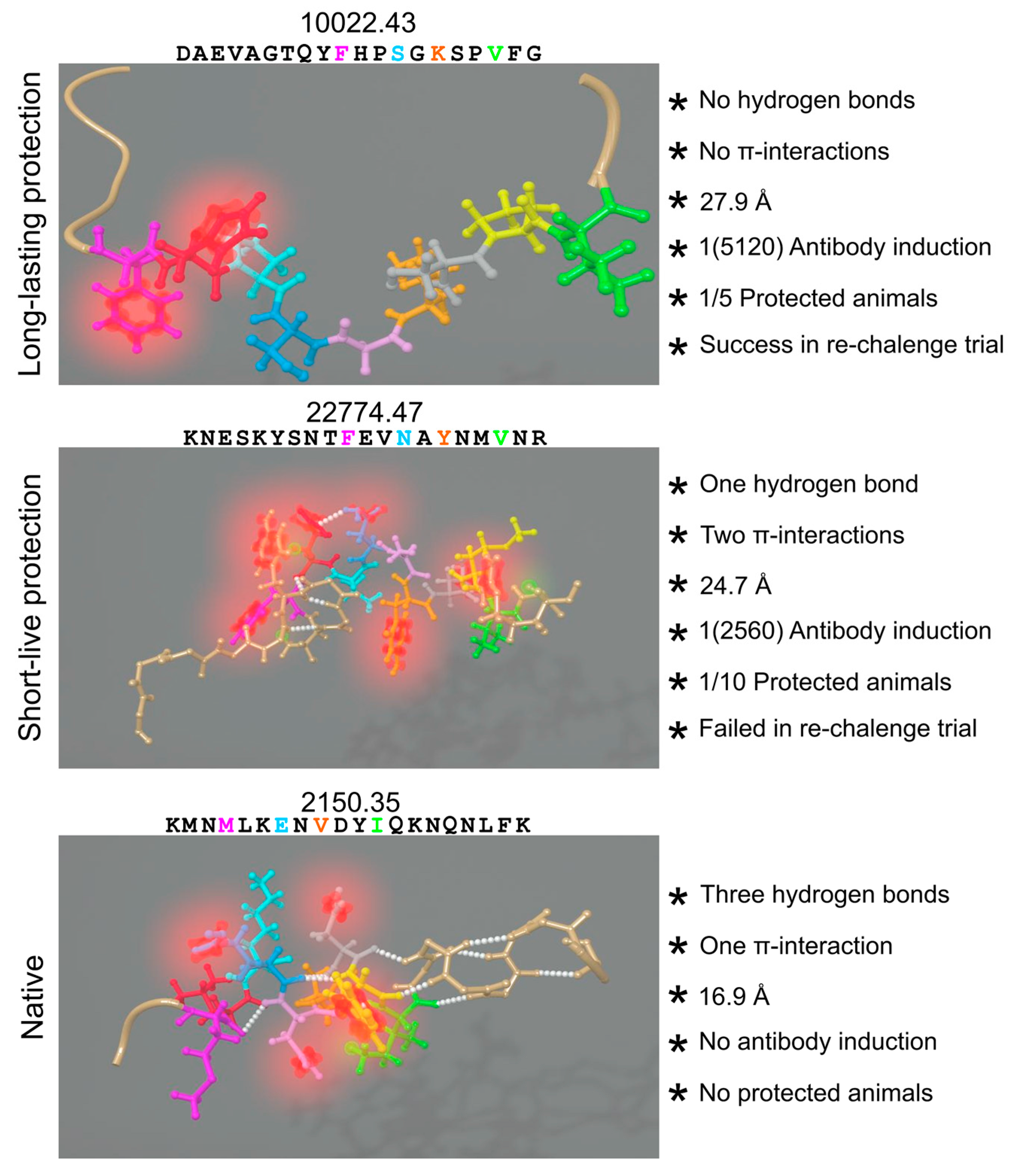
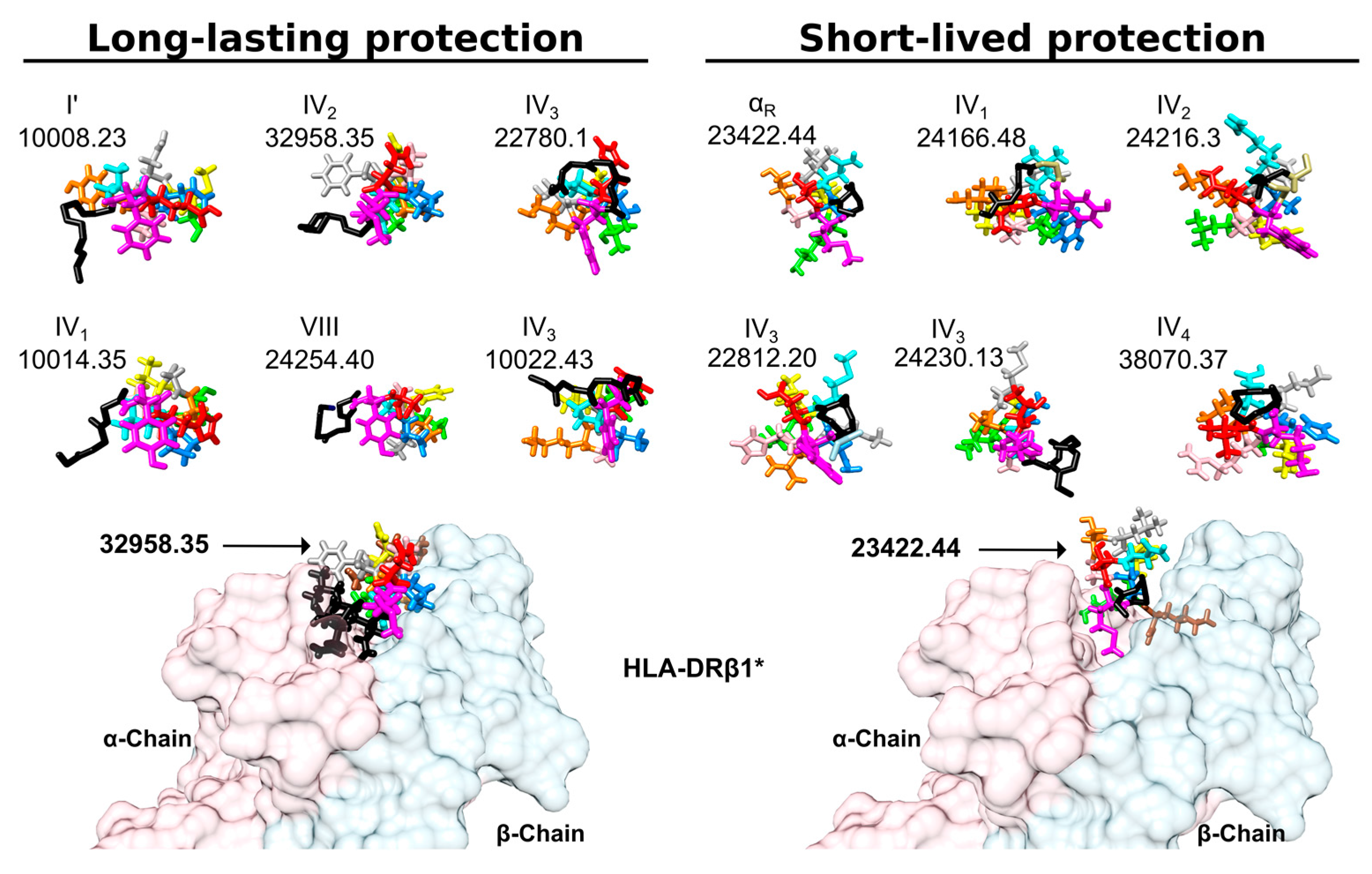
6.6. Hydrogen Bonds and Intramolecular π-Interactions
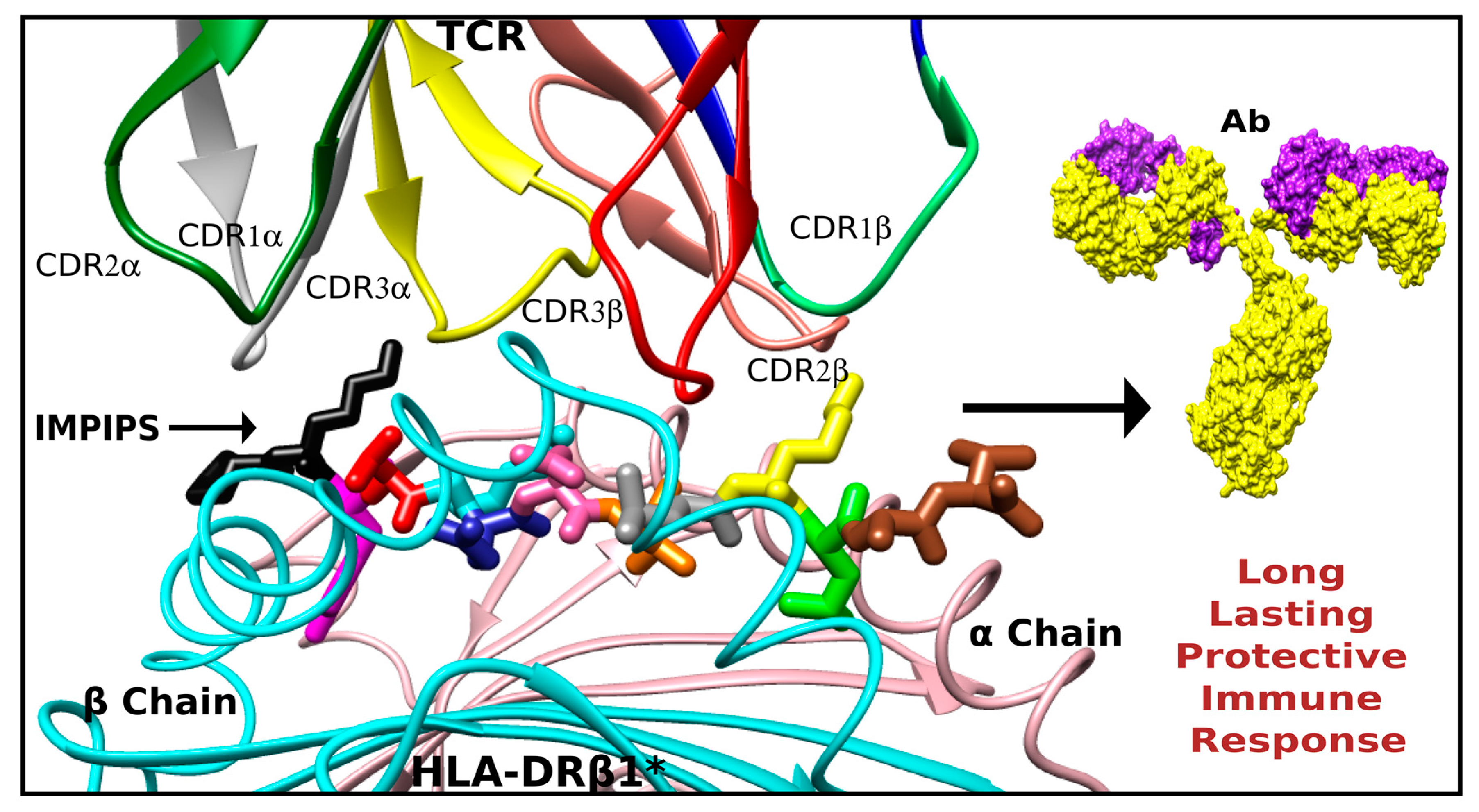
7. Peripheral Flanking Residues (PFR)
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bull, P.C.; Marsh, K. The role of antibodies to Plasmodium falciparum-infected-erythrocyte surface antigens in naturally acquired immunity to malaria. Trends Microbiol. 2002, 10, 55–58. [Google Scholar] [CrossRef]
- Bouharoun-Tayoun, H.; Attanath, P.; Sabchareon, A.; Chongsuphajaisiddhi, T.; Druilhe, P. Antibodies that protect humans against Plasmodium falciparum blood stages do not on their own inhibit parasite growth and invasion in vitro, but act in cooperation with monocytes. J. Exp. Med. 1990, 172, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- McGregor, I.A. Studies in the Acquisition of Immunity of Plasmodium Falciparum Infections in Africa. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 80–92. [Google Scholar] [CrossRef]
- Miyahira, Y.; Garcia-Sastre, A.; Rodriguez, D.; Rodriguez, J.R.; Murata, K.; Tsuji, M.; Palese, P.; Esteban, M.; Zavala, F.; Nussenzweig, R.S. Recombinant viruses expressing a human malaria antigen can elicit potentially protective immune CD8+ responses in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3954–3959. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Zavala, F.; Nussenzweig, R.S.; Wilson, J.M.; Tsuji, M. Efficient induction of protective anti-malaria immunity by recombinant adenovirus. Vaccine 1998, 16, 1812–1817. [Google Scholar] [CrossRef]
- Nussenzweig, R.; Vanderberg, J.; Most, H. Protective immunity produced by the injection of x-irradiated sporozoites of Plasmodium berghei. IV. Dose response, specificity and humoral immunity. Mil. Med. 1969, 134, 1176–1182. [Google Scholar] [PubMed]
- Weiss, W.R.; Sedegah, M.; Beaudoin, R.L.; Miller, L.H.; Good, M.F. CD8+ T cells (cytotoxic/suppressors) are required for protection in mice immunized with malaria sporozoites. Proc. Natl. Acad. Sci. USA 1988, 85, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Gwadz, R.W.; Carter, R.; Green, I. Gamete vaccines and transmission-blocking immunity in malaria. Bull. World Health Organ. 1979, 57 (Suppl. 1), 175–180. [Google Scholar] [PubMed]
- Hoffman, S.L.; Goh, L.M.; Luke, T.C.; Schneider, I.; Le, T.P.; Doolan, D.L.; Sacci, J.; de la Vega, P.; Dowler, M.; Paul, C.; et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J. Infect. Dis. 2002, 185, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, A.S.; Lyke, K.E.; DeZure, A.; Berry, A.A.; Richie, T.L.; Mendoza, F.H.; Enama, M.E.; Gordon, I.J.; Chang, L.J.; Sarwar, U.N.; et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat. Med. 2016, 22, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Amador, R.; Clavijo, P.; Moreno, A.; Guzman, F.; Romero, P.; Tascon, R.; Franco, A.; Murillo, L.A.; Ponton, G.; et al. A synthetic vaccine protects humans against challenge with asexual blood stages of Plasmodium falciparum malaria. Nature 1988, 332, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Noya, O.; Gabaldon Berti, Y.; Alarcon de Noya, B.; Borges, R.; Zerpa, N.; Urbaez, J.D.; Madonna, A.; Garrido, E.; Jimenez, M.A.; Borges, R.E.; et al. A population-based clinical trial with the SPf66 synthetic Plasmodium falciparum malaria vaccine in Venezuela. J. Infect. Dis. 1994, 170, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.F.; Blackman, M.J.; Kaslow, D.C. Vaccine efficacy of recombinant Plasmodium falciparum merozoite surface protein 1 in malaria-naive, -exposed, and/or-rechallenged Aotus vociferans monkeys. Infect. Immun. 2000, 68, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.D.; Baldeviano, G.C.; Lucas, C.M.; Lugo-Roman, L.A.; Crosnier, C.; Bartholdson, S.J.; Diouf, A.; Miura, K.; Lambert, L.E.; Ventocilla, J.A.; et al. A PfRH5-based vaccine is efficacious against heterologous strain blood-stage Plasmodium falciparum infection in Aotus monkeys. Cell Host Microbe 2015, 17, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Collins, W.E.; Anders, R.F.; Pappaioanou, M.; Campbell, G.H.; Brown, G.V.; Kemp, D.J.; Coppel, R.L.; Skinner, J.C.; Andrysiak, P.M.; Favaloro, J.M.; et al. Immunization of Aotus monkeys with recombinant proteins of an erythrocyte surface antigen of Plasmodium falciparum. Nature 1986, 323, 259–262. [Google Scholar] [CrossRef] [PubMed]
- WHO. World malaria report. In WHO Global Malaria Programme 2016; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Anderer, F.A. Preparation and properties of an artificial antigen immunologically related to tobacco mosaic virus. Biochim. Biophys. Acta 1963, 71, 246–248. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Collier, J.H.; Segura, T. Evolving the use of peptides as components of biomaterials. Biomaterials 2011, 32, 4198–4204. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Imondi, A.R.; Stradley, R.P.; Wolgemuth, R. Synthetic peptides in the diagnosis of exocrine pancreatic insufficiency in animals. Gut 1972, 13, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Fargeas, C.; Hommel, M.; Maingon, R.; Dourado, C.; Monsigny, M.; Mayer, R. Synthetic peptide-based enzyme-linked immunosorbent assay for serodiagnosis of visceral leishmaniasis. J. Clin. Microbiol. 1996, 34, 241–248. [Google Scholar] [PubMed]
- Volkmer, R.; Kretzschmar, I.; Tapia, V. Mapping receptor-ligand interactions with synthetic peptide arrays: Exploring the structure and function of membrane receptors. Eur. J. Cell Biol. 2012, 91, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.E.; Puentes, A.; Patarroyo, M.E. Developmental biology of sporozoite-host interactions in Plasmodium falciparum malaria: Implications for vaccine design. Clin. Microbiol. Rev. 2006, 19, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.E.; Urquiza, M.; Ocampo, M.; Suarez, J.; Curtidor, H.; Guzman, F.; Vargas, L.E.; Trivinos, M.; Rosas, M.; Patarroyo, M.E. Plasmodium falciparum EBA-175 kDa protein peptides which bind to human red blood cells. Parasitology 2000, 120 Pt 3, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.P.; Ocampo, M.; Varela, Y.; Curtidor, H.; Patarroyo, M.A.; Patarroyo, M.E. Identifying and characterising PPE7 (Rvo354c) high activity binding peptides and their role in inhibiting cell invasion. Mol. Cell. Biochem. 2017, 430, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Holmes, T.C. Novel peptide-based biomaterial scaffolds for tissue engineering. Trends Biotechnol. 2002, 20, 16–21. [Google Scholar] [CrossRef]
- Boman, H.G.; Wade, D.; Boman, I.A.; Wahlin, B.; Merrifield, R.B. Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett. 1989, 259, 103–106. [Google Scholar] [CrossRef]
- Ge, Y.; MacDonald, D.L.; Holroyd, K.J.; Thornsberry, C.; Wexler, H.; Zasloff, M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999, 43, 782–788. [Google Scholar] [PubMed]
- Mather, R.; Karenchak, L.M.; Romanowski, E.G.; Kowalski, R.P. Fourth generation fluoroquinolones: New weapons in the arsenal of ophthalmic antibiotics. Am. J. Ophthalmol. 2002, 133, 463–466. [Google Scholar] [CrossRef]
- Ma, Y.; Ai, G.; Zhang, C.; Zhao, M.; Dong, X.; Han, Z.; Wang, Z.; Zhang, M.; Liu, Y.; Gao, W.; et al. Novel Linear Peptides with High Affinity to αvβ3 Integrin for Precise Tumor Identification. Theranostics 2017, 7, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Koliakos, G.; Trontzos, C.; Kouzi-Koliakos, K.; Kanellaki, M.; Grammaticos, P. Lung carcinoma imaging using a synthetic laminin derivative radioiodinated peptide YIGSR. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1997, 38, 1940–1944. [Google Scholar]
- Lee, T.Y.; Wu, H.C.; Tseng, Y.L.; Lin, C.T. A novel peptide specifically binding to nasopharyngeal carcinoma for targeted drug delivery. Cancer Res. 2004, 64, 8002–8008. [Google Scholar] [CrossRef] [PubMed]
- Garanger, E.; Boturyn, D.; Jin, Z.; Dumy, P.; Favrot, M.C.; Coll, J.L. New multifunctional molecular conjugate vector for targeting, imaging, and therapy of tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.R.; Gotch, F.M.; Davey, J. Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell 1985, 42, 457–467. [Google Scholar] [CrossRef]
- Shinnick, T.M.; Sutcliffe, J.G.; Green, N.; Lerner, R.A. Synthetic peptide immunogens as vaccines. Annu. Rev. Microbiol. 1983, 37, 425–446. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, H.O.; Sela, M. Genetic control of the antibody response. II. Further analysis of the specificity of determinant-specific control, and genetic analysis of the response to (H,G)-A--L in CBA and C57 mice. J. Exp. Med. 1967, 126, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Zavala, F.; Tam, J.P.; Barr, P.J.; Romero, P.J.; Ley, V.; Nussenzweig, R.S.; Nussenzweig, V. Synthetic peptide vaccine confers protection against murine malaria. J. Exp. Med. 1987, 166, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Romero, P.; Torres, M.L.; Clavijo, P.; Moreno, A.; Martinez, A.; Rodriguez, R.; Guzman, F.; Cabezas, E. Induction of protective immunity against experimental infection with malaria using synthetic peptides. Nature 1987, 328, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Arnon, R. Chemically defined antiviral vaccines. Annu. Rev. Microbiol. 1980, 34, 593–618. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.H.; Brandling-Bennett, A.D.; Roberts, J.M.; Collins, F.H.; Kaseje, D.C.; Barber, A.M.; Turner, A. Detection of antibodies in human sera to the repeating epitope of the circumsporozoite protein of Plasmodium falciparum using the synthetic peptide (NANP)3 in an enzyme-linked immunosorbent assay (ELISA). Am. J. Trop. Med. Hyg. 1987, 37, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Zavala, F.; Tam, J.P.; Hollingdale, M.R.; Cochrane, A.H.; Quakyi, I.; Nussenzweig, R.S.; Nussenzweig, V. Rationale for development of a synthetic vaccine against Plasmodium falciparum malaria. Science 1985, 228, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Moreno, A.; Guzman, F.; Calvo, M.; Patarroyo, M.E. Studies in owl monkeys leading to the development of a synthetic vaccine against the asexual blood stages of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 1990, 43, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Audibert, F.; Jolivet, M.; Chedid, L.; Arnon, R.; Sela, M. Successful immunization with a totally synthetic diphtheria vaccine. Proc. Natl. Acad. Sci. USA 1982, 79, 5042–5046. [Google Scholar] [CrossRef] [PubMed]
- Bittle, J.L.; Houghten, R.A.; Alexander, H.; Shinnick, T.M.; Sutcliffe, J.G.; Lerner, R.A.; Rowlands, D.J.; Brown, F. Protection against foot-and-mouth disease by immunization with a chemically synthesized peptide predicted from the viral nucleotide sequence. Nature 1982, 298, 30–33. [Google Scholar] [CrossRef] [PubMed]
- DiMarchi, R.; Brooke, G.; Gale, C.; Cracknell, V.; Doel, T.; Mowat, N. Protection of cattle against foot-and-mouth disease by a synthetic peptide. Science 1986, 232, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Streckert, H.J.; Brussow, H.; Werchau, H. A synthetic peptide corresponding to the cleavage region of VP3 from rotavirus SA11 induces neutralizing antibodies. J. Virol. 1988, 62, 4265–4269. [Google Scholar] [PubMed]
- Chanh, T.C.; Dreesman, G.R.; Kanda, P.; Linette, G.P.; Sparrow, J.T.; Ho, D.D.; Kennedy, R.C. Induction of anti-HIV neutralizing antibodies by synthetic peptides. EMBO J. 1986, 5, 3065–3071. [Google Scholar] [PubMed]
- Amador, R.; Moreno, A.; Murillo, L.A.; Sierra, O.; Saavedra, D.; Rojas, M.; Mora, A.L.; Rocha, C.L.; Alvarado, F.; Falla, J.C.; et al. Safety and immunogenicity of the synthetic malaria vaccine SPf66 in a large field trial. J. Infect. Dis. 1992, 166, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Amador, R.; Moreno, A.; Valero, V.; Murillo, L.; Mora, A.L.; Rojas, M.; Rocha, C.; Salcedo, M.; Guzman, F.; Espejo, F.; et al. The first field trials of the chemically synthesized malaria vaccine SPf66: Safety, immunogenicity and protectivity. Vaccine 1992, 10, 179–184. [Google Scholar] [CrossRef]
- Patarroyo, G.; Franco, L.; Amador, R.; Murillo, L.A.; Rocha, C.L.; Rojas, M.; Patarroyo, M.E. Study of the safety and immunogenicity of the synthetic malaria SPf66 vaccine in children aged 1–14 years. Vaccine 1992, 10, 175–178. [Google Scholar] [CrossRef]
- Alonso, P.L.; Smith, T.; Schellenberg, J.R.; Masanja, H.; Mwankusye, S.; Urassa, H.; Bastos de Azevedo, I.; Chongela, J.; Kobero, S.; Menendez, C.; et al. Randomised trial of efficacy of SPf66 vaccine against Plasmodium falciparum malaria in children in southern Tanzania. Lancet 1994, 344, 1175–1181. [Google Scholar] [CrossRef]
- Sempertegui, F.; Estrella, B.; Moscoso, J.; Piedrahita, L.; Hernandez, D.; Gaybor, J.; Naranjo, P.; Mancero, O.; Arias, S.; Bernal, R.; et al. Safety, immunogenicity and protective effect of the SPf66 malaria synthetic vaccine against Plasmodium falciparum infection in a randomized double-blind placebo-controlled field trial in an endemic area of Ecuador. Vaccine 1994, 12, 337–342. [Google Scholar] [CrossRef]
- Valero, M.V.; Amador, R.; Aponte, J.J.; Narvaez, A.; Galindo, C.; Silva, Y.; Rosas, J.; Guzman, F.; Patarroyo, M.E. Evaluation of SPf66 malaria vaccine during a 22-month follow-up field trial in the Pacific coast of Colombia. Vaccine 1996, 14, 1466–1470. [Google Scholar] [CrossRef]
- Langeveld, J.P.; Casal, J.I.; Osterhaus, A.D.; Cortes, E.; de Swart, R.; Vela, C.; Dalsgaard, K.; Puijk, W.C.; Schaaper, W.M.; Meloen, R.H. First peptide vaccine providing protection against viral infection in the target animal: studies of canine parvovirus in dogs. J. Virol. 1994, 68, 4506–4513. [Google Scholar] [PubMed]
- Snewin, V.A.; Premawansa, S.; Kapilananda, G.M.; Ratnayaka, L.; Udagama, P.V.; Mattei, D.M.; Khouri, E.; Del Giudice, G.; Peiris, J.S.; Mendis, K.N.; et al. Transmission blocking immunity in Plasmodium vivax malaria: antibodies raised against a peptide block parasite development in the mosquito vector. J. Exp. Med. 1995, 181, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Oscherwitz, J.; Yu, F.; Cease, K.B. A synthetic peptide vaccine directed against the 2ss2-2ss3 loop of domain 2 of protective antigen protects rabbits from inhalation anthrax. J. Immunol. 2010, 185, 3661–3668. [Google Scholar] [CrossRef] [PubMed]
- Stanekova, Z.; Kiraly, J.; Stropkovska, A.; Mikuskova, T.; Mucha, V.; Kostolansky, F.; Vareckova, E. Heterosubtypic protective immunity against influenza A virus induced by fusion peptide of the hemagglutinin in comparison to ectodomain of M2 protein. Acta Virol. 2011, 55, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Monso, M.; Tarradas, J.; de la Torre, B.G.; Sobrino, F.; Ganges, L.; Andreu, D. Peptide vaccine candidates against classical swine fever virus: T cell and neutralizing antibody responses of dendrimers displaying E2 and NS2-3 epitopes. J. Pept. Sci. 2011, 17, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Solares, A.M.; Baladron, I.; Ramos, T.; Valenzuela, C.; Borbon, Z.; Fanjull, S.; Gonzalez, L.; Castillo, D.; Esmir, J.; Granadillo, M.; et al. Safety and Immunogenicity of a Human Papillomavirus Peptide Vaccine (CIGB-228) in Women with High-Grade Cervical Intraepithelial Neoplasia: First-in-Human, Proof-of-Concept Trial. ISRN Obstet. Gynecol. 2011, 2011, 292951–292959. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Bermudez, A.; Alba, M.P.; Vanegas, M.; Moreno-Vranich, A.; Poloche, L.A.; Patarroyo, M.A. IMPIPS: The Immune Protection-Inducing Protein Structure Concept in the Search for Steric-Electron and Topochemical Principles for Complete Fully-Protective Chemically Synthesised Vaccine Development. PLoS ONE 2015, 10, e0123249. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Patarroyo, M.A.; Pabon, L.; Curtidor, H.; Poloche, L.A. Immune protection-inducing protein structures (IMPIPS) against malaria: The weapons needed for beating Odysseus. Vaccine 2015, 33, 7525–7537. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.J.; Galindo, C.M.; Schellenberg, D.; Aponte, J.J.; Kahigwa, E.; Urassa, H.; Schellenberg, J.R.; Masanja, H.; Hayes, R.; Kitua, A.Y.; et al. Evaluation of the SPf66 vaccine for malaria control when delivered through the EPI scheme in Tanzania. Trop. Med. Int. Health 1999, 4, 368–376. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, U.; Leach, A.; Drakeley, C.J.; Bennett, S.; Olaleye, B.O.; Fegan, G.W.; Jawara, M.; Langerock, P.; George, M.O.; Targett, G.A.; et al. Efficacy trial of malaria vaccine SPf66 in Gambian infants. Lancet 1995, 346, 462–467. [Google Scholar] [CrossRef]
- Graves, P.; Gelband, H. Vaccines for preventing malaria (SPf66). Cochrane Database Syst. Rev. 2006, 2, 1–30. [Google Scholar]
- Bozdech, Z.; Llinas, M.; Pulliam, B.L.; Wong, E.D.; Zhu, J.; DeRisi, J.L. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1, E5. [Google Scholar] [CrossRef] [PubMed]
- Bozdech, Z.; Zhu, J.; Joachimiak, M.P.; Cohen, F.E.; Pulliam, B.; DeRisi, J.L. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 2003, 4, R9. [Google Scholar] [CrossRef] [PubMed]
- Kappe, S.H.; Gardner, M.J.; Brown, S.M.; Ross, J.; Matuschewski, K.; Ribeiro, J.M.; Adams, J.H.; Quackenbush, J.; Cho, J.; Carucci, D.J.; et al. Exploring the transcriptome of the malaria sporozoite stage. Proc. Natl. Acad. Sci. USA 2001, 98, 9895–9900. [Google Scholar] [CrossRef] [PubMed]
- Amino, R.; Thiberge, S.; Martin, B.; Celli, S.; Shorte, S.; Frischknecht, F.; Menard, R. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat. Med. 2006, 12, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Ejigiri, I.; Sinnis, P. Plasmodium sporozoite-host interactions from the dermis to the hepatocyte. Curr. Opin. Microbiol. 2009, 12, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Bannister, L.; Mitchell, G. The ins, outs and roundabouts of malaria. Trends Parasitol. 2003, 19, 209–213. [Google Scholar] [CrossRef]
- Sinden, R.E. Plasmodium differentiation in the mosquito. Parassitologia 1999, 41, 139–148. [Google Scholar] [PubMed]
- Pereira da Silva, L. Genetic aspects of malaria parasite infection and the host immune response in relation to parasite evasion. Annal. Parasitol. Hum. Comp. 1990, 65 (Suppl. 1), 15–17. [Google Scholar] [CrossRef] [PubMed]
- Casares, S.; Richie, T.L. Immune evasion by malaria parasites: A challenge for vaccine development. Curr. Opin. Immunol. 2009, 21, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hisaeda, H.; Yasutomo, K.; Himeno, K. Malaria: Immune evasion by parasites. Int. J. Biochem. Cell Biol. 2005, 37, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Bermudez, A.; Patarroyo, M.A. Structural and immunological principles leading to chemically synthesized, multiantigenic, multistage, minimal subunit-based vaccine development. Chem. Rev. 2011, 111, 3459–3507. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, F.M.; Lem, L.; Solache, A.; Bennett, E.M. Human pathogen subversion of antigen presentation. Immunol. Rev. 1999, 168, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Aza-Conde, J.; Moreno-Vranich, A.; Pabon, L.; Varela, Y.; Patarroyo, M.A. Far from the Madding Crowd: The Molecular Basis for Immunological Escape of Plasmodium falciparum. Curr. Issues Mol. Biol. 2017, 22, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Daubenberger, C.A.; Nickel, B.; Ciatto, C.; Grutter, M.G.; Poltl-Frank, F.; Rossi, L.; Siegler, U.; Robinson, J.; Kashala, O.; Patarroyo, M.E.; et al. Amino acid dimorphism and parasite immune evasion: Cellular immune responses to a promiscuous epitope of Plasmodium falciparum merozoite surface protein 1 displaying dimorphic amino acid polymorphism are highly constrained. Eur. J. Immunol. 2002, 32, 3667–3677. [Google Scholar] [CrossRef]
- Schofield, L. The circumsporozoite protein of Plasmodium: A mechanism of immune evasion by the malaria parasite? Bull. World Health Organ. 1990, 68, 66–73. [Google Scholar] [PubMed]
- Kemp, D.J.; Cowman, A.F.; Walliker, D. Genetic diversity in Plasmodium falciparum. Adv. Parasitol. 1990, 29, 75–149. [Google Scholar] [PubMed]
- Valero, M.V.; Amador, L.R.; Galindo, C.; Figueroa, J.; Bello, M.S.; Murillo, L.A.; Mora, A.L.; Patarroyo, G.; Rocha, C.L.; Rojas, M.; et al. Vaccination with SPf66, a chemically synthesised vaccine, against Plasmodium falciparum malaria in Colombia. Lancet 1993, 341, 705–710. [Google Scholar] [CrossRef]
- Siddiqui, W.A. An effective immunization of experimental monkeys against a human malaria parasite, Plasmodium falciparum. Science 1977, 197, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Playfair, J.H.; De Souza, J.B.; Cottrell, B.J. Protection of mice against malaria by a killed vaccine: Differences in effectiveness against P. yoelii and P. berghei. Immunology 1977, 33, 507–515. [Google Scholar] [PubMed]
- Nussenzweig, R.S.; Vanderberg, J.; Most, H.; Orton, C. Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei. Nature 1967, 216, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, R.S.; Vanderberg, J.; Spitalny, G.L.; Rivera, C.I.; Orton, C.; Most, H. Sporozoite-induced immunity in mammalian malaria. A review. Am. J. Trop. Med. Hyg. 1972, 21, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.F.; Mohan, B.N. The Immunization of Fowls against Mosquito-Borne Plasmodium Gallinaceum by Injections of Serum and of Inactivated Homologous Sporozoites. J. Exp. Med. 1942, 76, 477–495. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.H.; Dayal, R.; Rieder, H. Characterization of antigens from erythrocytic stages of Plasmodium falciparum reacting with human immune sera. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 163–165. [Google Scholar] [CrossRef]
- Perrin, L.H.; Ramirez, E.; Lambert, P.H.; Miescher, P.A. Inhibition of P. falciparum growth in human erythrocytes by monoclonal antibodies. Nature 1981, 289, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.C.; Yamaga, K.M.; Kramer, K.J.; Case, S.E.; Siddiqui, W.A. Plasmodium falciparum: Protein antigens identified by analysis of serum samples from vaccinated Aotus monkeys. Infect. Immun. 1984, 43, 276–282. [Google Scholar] [PubMed]
- Villard, V.; Agak, G.W.; Frank, G.; Jafarshad, A.; Servis, C.; Nebie, I.; Sirima, S.B.; Felger, I.; Arevalo-Herrera, M.; Herrera, S.; et al. Rapid identification of malaria vaccine candidates based on α-helical coiled coil protein motif. PLoS ONE 2007, 2, e645. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.H.; Simitsek, P.; Fasani-Ghersa, P. Asexual blood stage vaccines: From merozoites to peptides. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 53–56. [Google Scholar] [CrossRef]
- Perlmann, H.; Berzins, K.; Wahlgren, M.; Carlsson, J.; Bjorkman, A.; Patarroyo, M.E.; Perlmann, P. Antibodies in malarial sera to parasite antigens in the membrane of erythrocytes infected with early asexual stages of Plasmodium falciparum. J. Exp. Med. 1984, 159, 1686–1704. [Google Scholar] [CrossRef] [PubMed]
- Wahlin, B.; Wahlgren, M.; Perlmann, H.; Berzins, K.; Bjorkman, A.; Patarroyo, M.E.; Perlmann, P. Human antibodies to a Mr 155,000 Plasmodium falciparum antigen efficiently inhibit merozoite invasion. Proc. Natl. Acad. Sci. USA 1984, 81, 7912–7916. [Google Scholar] [CrossRef] [PubMed]
- Carucci, D.J.; Gardner, M.J.; Tettelin, H.; Cummings, L.M.; Smith, H.O.; Adams, M.D.; Venter, J.C.; Hoffman, S.L. Sequencing the genome of Plasmodium falciparum. Curr. Opin. Infect. Dis. 1998, 11, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Tettelin, H.; Carucci, D.J.; Cummings, L.M.; Aravind, L.; Koonin, E.V.; Shallom, S.; Mason, T.; Yu, K.; Fujii, C.; et al. Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science 1998, 282, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Florens, L.; Washburn, M.P.; Raine, J.D.; Anthony, R.M.; Grainger, M.; Haynes, J.D.; Moch, J.K.; Muster, N.; Sacci, J.B.; Tabb, D.L.; et al. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002, 419, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Khan, F.; Mishra, B.N. Computational characterization of Plasmodium falciparum proteomic data for screening of potential vaccine candidates. Hum. Immunol. 2010, 71, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Tuju, J.; Kamuyu, G.; Murungi, L.M.; Osier, F.H.A. Vaccine candidate discovery for the next generation of malaria vaccines. Immunology 2017, 152, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Vinasco, J.; Amador, R.; Espejo, F.; Silva, Y.; Moreno, A.; Rojas, M.; Mora, A.L.; Salcedo, M.; Valero, V.; et al. Genetic control of the immune response to a synthetic vaccine against Plasmodium falciparum. Parasite Immunol. 1991, 13, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, M.; Barreto, L.; Rojas, M.; Moya, R.; Cote, J.; Patarroyo, M.E. Studies on the humoral immune response to a synthetic vaccine against Plasmodium falciparum malaria. Clin. Exp. Immunol. 1991, 84, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Van Schravendijk, M.R.; Rock, E.P.; Marsh, K.; Ito, Y.; Aikawa, M.; Neequaye, J.; Ofori-Adjei, D.; Rodriguez, R.; Patarroyo, M.E.; Howard, R.J. Characterization and localization of Plasmodium falciparum surface antigens on infected erythrocytes from west African patients. Blood 1991, 78, 226–236. [Google Scholar] [PubMed]
- Wahlgren, M.; Bejarano, M.T.; Troye-Blomberg, M.; Perlmann, P.; Riley, E.; Greenwood, B.M.; Patarroyo, M.E.; Gonzales, C.I.; Martinez, A. Epitopes of the Plasmodium falciparum clustered-asparagine-rich protein (CARP) recognized by human T-cells and antibodies. Parasite Immunol. 1991, 13, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Guzman, F.; Perez, E.; Segura, C.H.; Molano, A.; Patarroyo, M.E. Specific interactions of synthetic peptides derived from P. falciparum merozoite proteins with human red blood cells. Pept. Res. 1991, 4, 324–333. [Google Scholar] [PubMed]
- Weiland, G.A.; Molinoff, P.B. Quantitative analysis of drug-receptor interactions: I. Determination of kinetic and equilibrium properties. Life Sci. 1981, 29, 313–330. [Google Scholar] [CrossRef]
- Urquiza, M.; Rodriguez, L.E.; Suarez, J.E.; Guzman, F.; Ocampo, M.; Curtidor, H.; Segura, C.; Trujillo, E.; Patarroyo, M.E. Identification of Plasmodium falciparum MSP-1 peptides able to bind to human red blood cells. Parasite Immunol. 1996, 18, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Curtidor, H.; Patino, L.C.; Arevalo-Pinzon, G.; Patarroyo, M.E.; Patarroyo, M.A. Identification of the Plasmodium falciparum rhoptry neck protein 5 (PfRON5). Gene 2011, 474, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Curtidor, H.; Urquiza, M.; Suarez, J.E.; Rodriguez, L.E.; Ocampo, M.; Puentes, A.; Garcia, J.E.; Vera, R.; Lopez, R.; Ramirez, L.E.; et al. Plasmodium falciparum acid basic repeat antigen (ABRA) peptides: Erythrocyte binding and biological activity. Vaccine 2001, 19, 4496–4504. [Google Scholar] [CrossRef]
- Curtidor, H.; Vanegas, M.; Alba, M.P.; Patarroyo, M.E. Functional, immunological and three-dimensional analysis of chemically synthesised sporozoite peptides as components of a fully-effective antimalarial vaccine. Curr. Med. Chem. 2011, 18, 4470–4502. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.E.; Curtidor, H.; Urquiza, M.; Cifuentes, G.; Reyes, C.; Patarroyo, M.E. Intimate molecular interactions of P. falciparum merozoite proteins involved in invasion of red blood cells and their implications for vaccine design. Chem. Rev. 2008, 108, 3656–3705. [Google Scholar] [CrossRef] [PubMed]
- Curtidor, H.; Rodriguez, L.E.; Ocampo, M.; Lopez, R.; Garcia, J.E.; Valbuena, J.; Vera, R.; Puentes, A.; Vanegas, M.; Patarroyo, M.E. Specific erythrocyte binding capacity and biological activity of Plasmodium falciparum erythrocyte binding ligand 1 (EBL-1)-derived peptides. Protein Sci. Publ. Protein Soc. 2005, 14, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Arevalo-Pinzon, G.; Reyes, C.; Moreno-Vranich, A.; Patarroyo, M.A. Malaria Parasite Survival Depends on Conserved Binding Peptides’ Critical Biological Functions. Curr. Issues Mol. Biol. 2016, 18, 57–78. [Google Scholar] [PubMed]
- Alba, M.P.; Salazar, L.M.; Puentes, A.; Pinto, M.; Torres, E.; Patarroyo, M.E. 6746 SERA peptide analogues immunogenicity and protective efficacy against malaria is associated with short α helix formation: Malaria protection associated with peptides α helix shortening. Peptides 2003, 24, 999–1006. [Google Scholar] [CrossRef]
- Espejo, F.; Cubillos, M.; Salazar, L.M.; Guzman, F.; Urquiza, M.; Ocampo, M.; Silva, Y.; Rodriguez, R.; Lioy, E.; Patarroyo, M.E. Structure, Immunogenicity, and Protectivity Relationship for the 1585 Malarial Peptide and Its Substitution Analogues. Angew. Chem. Int. Ed. Engl. 2001, 40, 4654–4657. [Google Scholar] [CrossRef]
- Guzman, F.; Jaramillo, K.; Salazar, L.M.; Torres, A.; Rivera, A.; Patarroyo, M.E. 1H-NMR structures of the Plasmodium falciparum 1758 erythrocyte binding peptide analogues and protection against malaria. Life Sci. 2002, 71, 2773–2785. [Google Scholar] [CrossRef]
- Purmova, J.; Salazar, L.M.; Espejo, F.; Torres, M.H.; Cubillos, M.; Torres, E.; Lopez, Y.; Rodriguez, R.; Patarroyo, M.E. NMR structure of Plasmodium falciparum malaria peptide correlates with protective immunity. Biochim. Biophys. Acta 2002, 1571, 27–33. [Google Scholar] [CrossRef]
- Herrington, D.A.; Clyde, D.F.; Losonsky, G.; Cortesia, M.; Murphy, J.R.; Davis, J.; Baqar, S.; Felix, A.M.; Heimer, E.P.; Gillessen, D.; et al. Safety and immunogenicity in man of a synthetic peptide malaria vaccine against Plasmodium falciparum sporozoites. Nature 1987, 328, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Takala, S.L.; Plowe, C.V. Genetic diversity and malaria vaccine design, testing and efficacy: Preventing and overcoming ‘vaccine resistant malaria’. Parasite Immunol. 2009, 31, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Escalante, A.A.; Lal, A.A.; Ayala, F.J. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics 1998, 149, 189–202. [Google Scholar] [PubMed]
- Ahouidi, A.D.; Bei, A.K.; Neafsey, D.E.; Sarr, O.; Volkman, S.; Milner, D.; Cox-Singh, J.; Ferreira, M.U.; Ndir, O.; Premji, Z.; et al. Population genetic analysis of large sequence polymorphisms in Plasmodium falciparum blood-stage antigens. Infect. Genet. Evol. 2010, 10, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Patthy, L. Protein Evolution, 2nd ed.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Patarroyo, M.E.; Alba, M.P.; Reyes, C.; Rojas-Luna, R.; Patarroyo, M.A. The Malaria Parasite’s Achilles’ Heel: Functionally-relevant Invasion Structures. Curr. Issues Mol. Biol. 2016, 18, 11–19. [Google Scholar] [PubMed]
- Patarroyo, M.E.; Alba, M.P.; Rojas-Luna, R.; Bermudez, A.; Aza-Conde, J. Functionally relevant proteins in Plasmodium falciparum host cell invasion. Immunotherapy 2017, 9, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, G.; Bermudez, A.; Rodriguez, R.; Patarroyo, M.A.; Patarroyo, M.E. Shifting the polarity of some critical residues in malarial peptides’ binding to host cells is a key factor in breaking conserved antigens’ code of silence. Med. Chem. 2008, 4, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Espejo, F.; Bermudez, A.; Torres, E.; Urquiza, M.; Rodriguez, R.; Lopez, Y.; Patarroyo, M.E. Shortening and modifying the 1513 MSP-1 peptide’s α-helical region induces protection against malaria. Biochem. Biophys. Res. Commun. 2004, 315, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993, 364, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. The discovery of MHC restriction. Immunol. Today 1997, 18, 14–17. [Google Scholar] [CrossRef]
- Rammensee, H.G.; Friede, T.; Stevanoviic, S. MHC ligands and peptide motifs: First listing. Immunogenetics 1995, 41, 178–228. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.; Daubenberger, C.A.; Zalac, T.; Rodriguez, R.; Patarroyo, M.E. Sequence and expression of MHC-DPB1 molecules of the New World monkey Aotus nancymaae, a primate model for Plasmodium falciparum. Immunogenetics 2002, 54, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Diaz, O.L.; Daubenberger, C.A.; Rodriguez, R.; Naegeli, M.; Moreno, A.; Patarroyo, M.E.; Pluschke, G. Immunoglobulin kappa light-chain V, J, and C gene sequences of the owl monkey Aotus nancymaae. Immunogenetics 2000, 51, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.F.; Patarroyo, M.E.; Trujillo, E.; Estupinan, M.; Baquero, J.E.; Parra, C.; Rodriguez, R. Owl monkey MHC-DRB exon 2 reveals high similarity with several HLA-DRB lineages. Immunogenetics 2006, 58, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Porter, J.A., Jr.; Johnson, C.M. Plasmodium vivax transmitted from man to monkey to man. Science 1966, 153, 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Moncada, C.A.; Guerrero, E.; Cardenas, P.; Suarez, C.F.; Patarroyo, M.E.; Patarroyo, M.A. The T-cell receptor in primates: Identifying and sequencing new owl monkey TRBV gene sub-groups. Immunogenetics 2005, 57, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Van Dyke, K.; Rossan, R.N. Effective treatment with a tetrandrine/chloroquine combination for chloroquine-resistant falciparum malaria in Aotus monkeys. Malar. J. 2013, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Powles, L.; Xiang, S.D.; Selomulya, C.; Plebanski, M. The Use of Synthetic Carriers in Malaria Vaccine Design. Vaccines 2015, 3, 894–929. [Google Scholar] [CrossRef] [PubMed]
- Rosas, J.E.; Hernandez, R.M.; Gascon, A.R.; Igartua, M.; Guzman, F.; Patarroyo, M.E.; Pedraz, J.L. Biodegradable PLGA microspheres as a delivery system for malaria synthetic peptide SPf66. Vaccine 2001, 19, 4445–4451. [Google Scholar] [CrossRef]
- Alexander, J.; del Guercio, M.F.; Maewal, A.; Qiao, L.; Fikes, J.; Chesnut, R.W.; Paulson, J.; Bundle, D.R.; DeFrees, S.; Sette, A. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J. Immunol. 2000, 164, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, W.A.; Taylor, D.W.; Kan, S.C.; Kramer, K.; Richmond-Crum, S.M.; Kotani, S.; Shiba, T.; Kasumoto, S. Immunization of experimental monkeys against Plasmodium falciparum: Use of synthetic adjuvants. Bull. World Health Organ. 1979, 57 (Suppl. 1), 199–203. [Google Scholar] [PubMed]
- Good, M.F.; Yanow, S.K. Cryptic epitope for antibodies should not be forgotten in vaccine design. Expert Rev. Vaccines 2016, 15, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, B.; Berzofsky, J.A.; Boykins, R.A.; Majam, V.; Zheng, H.; Chattopadhyay, R.; de la Vega, P.; Moch, J.K.; Haynes, J.D.; Belyakov, I.M.; et al. Multiple antigen peptide vaccines against Plasmodium falciparum malaria. Infect. Immun. 2010, 78, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Flower, D.R.; Feighery, C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res. 2008, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Zhang, G.L.; Tongchusak, S.; Reinherz, E.L.; Brusic, V. Evaluation of MHC-II peptide binding prediction servers: Applications for vaccine research. BMC Bioinform. 2008, 9 (Suppl. 12), S22. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Ahlers, J.D.; Belyakov, I.M. Strategies for designing and optimizing new generation vaccines. Nat. Rev. Immunol. 2001, 1, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.P.; Salazar, L.M.; Vargas, L.E.; Trujillo, M.; Lopez, Y.; Patarroyo, M.E. Modifying RESA protein peptide 6671 to fit into HLA-DRβ1* pockets induces protection against malaria. Biochem. Biophys. Res. Commun. 2004, 315, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, G.; Guzman, F.; Alba, M.P.; Salazar, L.M.; Patarroyo, M.E. Analysis of a Plasmodium falciparum EBA-175 peptide with high binding capacity to erythrocytes and their analogues using 1H-NMR. J. Struct. Biol. 2003, 141, 115–121. [Google Scholar] [CrossRef]
- Getzoff, E.D.; Tainer, J.A.; Lerner, R.A.; Geysen, H.M. The chemistry and mechanism of antibody binding to protein antigens. Adv. Immunol. 1988, 43, 1–98. [Google Scholar] [PubMed]
- Allen, P.M.; Matsueda, G.R.; Adams, S.; Freeman, J.; Roof, R.W.; Lambert, L.; Unanue, E.R. Enhanced immunogenicity of a T cell immunogenic peptide by modifications of its N and C termini. Int. Immunol. 1989, 1, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Alba, M.P.; Vargas, L.E.; Silva, Y.; Rosas, J.; Rodriguez, R. Peptides inducing short-lived antibody responses against Plasmodium falciparum malaria have shorter structures and are read in a different MHC II functional register. Biochemistry 2005, 44, 6745–6754. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Patarroyo, M.A. Emerging rules for subunit-based, multiantigenic, multistage chemically synthesized vaccines. Acc. Chem. Res. 2008, 41, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.M.; Alba, M.P.; Curtidor, H.; Bermudez, A.; Luis, E.V.; Rivera, Z.J.; Patarroyo, M.E. Changing ABRA protein peptide to fit into the HLA-DRβ1*0301 molecule renders it protection-inducing. Biochem. Biophys. Res. Commun. 2004, 322, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.M.; Alba, M.P.; Torres, M.H.; Pinto, M.; Cortes, X.; Torres, L.; Patarroyo, M.E. Protection against experimental malaria associated with AMA-1 peptide analogue structures. FEBS Lett. 2002, 527, 95–100. [Google Scholar] [CrossRef]
- Patarroyo, M.E.; Bermudez, A.; Moreno-Vranich, A. Towards the development of a fully protective Plasmodium falciparum antimalarial vaccine. Expert Rev. Vaccines 2012, 11, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Cifuentes, G.; Vargas, L.E.; Rosas, J. Structural modifications enable conserved peptides to fit into MHC molecules thus inducing protection against malaria. Chembiochem Eur. J. Chem. Biol. 2004, 5, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.A.; Bermudez, A.; Lopez, C.; Yepes, G.; Patarroyo, M.E. 3D analysis of the TCR/pMHCII complex formation in monkeys vaccinated with the first peptide inducing sterilizing immunity against human malaria. PLoS ONE 2010, 5, e9771. [Google Scholar] [CrossRef] [PubMed]
- Strominger, J.L. Human histocompatibility proteins. Immunol. Rev. 2002, 185, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Vandiedonck, C.; Knight, J.C. The human Major Histocompatibility Complex as a paradigm in genomics research. Brief. Funct. Genom. Proteom. 2009, 8, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.; Wilming, L.; Rand, V.; Lovering, R.C.; Bruford, E.A.; Khodiyar, V.K.; Lush, M.J.; Povey, S.; Talbot, C.C., Jr.; Wright, M.W.; et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004, 5, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Sato, A. The HLA system. Second of two parts. N. Engl. J. Med. 2000, 343, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Sato, A. The HLA system. First of two parts. N. Engl. J. Med. 2000, 343, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Albring, J.; Koopmann, J.O.; Hammerling, G.J.; Momburg, F. Retrotranslocation of MHC class I heavy chain from the endoplasmic reticulum to the cytosol is dependent on ATP supply to the ER lumen. Mol. Immunol. 2004, 40, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.K.; van den Berg, H.A.; Lloyd, A.; Crowther, M.D.; Beck, K.; Ekeruche-Makinde, J.; Miles, J.J.; Bulek, A.M.; Dolton, G.; Schauenburg, A.J.; et al. Structural Mechanism Underpinning Cross-reactivity of a CD8+ T-cell Clone That Recognizes a Peptide Derived from Human Telomerase Reverse Transcriptase. J. Biol. Chem. 2017, 292, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Amaya, M.; Mellins, E.; Wiley, D.C. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 1995, 378, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Hennecke, J.; Wiley, D.C. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J. Exp. Med. 2002, 195, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Zhong, G.; Germain, R.N. Antigen presentation by MHC class II molecules: Invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum. Immunol. 1997, 54, 159–169. [Google Scholar] [CrossRef]
- Beck, H.P.; Felger, I.; Barker, M.; Bugawan, T.; Genton, B.; Alexander, N.; Jazwinska, E.; Erlich, H.; Alpers, M. Evidence of HLA class II association with antibody response against the malaria vaccine SPF66 in a naturally exposed population. Am. J. Trop. Med. Hyg. 1995, 53, 284–288. [Google Scholar] [PubMed]
- Draheim, M.; Wlodarczyk, M.F.; Crozat, K.; Saliou, J.M.; Alayi, T.D.; Tomavo, S.; Hassan, A.; Salvioni, A.; Demarta-Gatsi, C.; Sidney, J.; et al. Profiling MHC II immunopeptidome of blood-stage malaria reveals that cDC1 control the functionality of parasite-specific CD4 T cells. EMBO Mol. Med. 2017, 9, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Ndungu, F.M.; Sanni, L.; Urban, B.; Stephens, R.; Newbold, C.I.; Marsh, K.; Langhorne, J. CD4 T cells from malaria-nonexposed individuals respond to the CD36-Binding Domain of Plasmodium falciparum erythrocyte membrane protein-1 via an MHC class II-TCR-independent pathway. J. Immunol. 2006, 176, 5504–5512. [Google Scholar] [CrossRef] [PubMed]
- Cigel, F.; Batchelder, J.; Burns, J.M., Jr.; Yanez, D.; van der Heyde, H.; Manning, D.D.; Weidanz, W.P. Immunity to blood-stage murine malarial parasites is MHC class II dependent. Immunol. Lett. 2003, 89, 243–249. [Google Scholar] [CrossRef]
- Rammensee, H.G. Chemistry of peptides associated with MHC class I and class II molecules. Curr. Opin. Immunol. 1995, 7, 85–96. [Google Scholar] [CrossRef]
- Stern, L.J.; Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 1994, 368, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Moreno-Vranich, A.; Bermudez, A. Phi (Phi) and psi (Psi) angles involved in malarial peptide bonds determine sterile protective immunity. Biochem. Biophys. Res. Commun. 2012, 429, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Urquiza, M.; Suarez, J.E.; Cardenas, C.; Lopez, R.; Puentes, A.; Chavez, F.; Calvo, J.C.; Patarroyo, M.E. Plasmodium falciparum AMA-1 erythrocyte binding peptides implicate AMA-1 as erythrocyte binding protein. Vaccine 2000, 19, 508–513. [Google Scholar] [CrossRef]
- Bermudez, A.; Calderon, D.; Moreno-Vranich, A.; Almonacid, H.; Patarroyo, M.A.; Poloche, A.; Patarroyo, M.E. Gauche(+) side chain orientation as a key factor in the search for an immunogenic peptide mixture leading to a complete fully protective vaccine. Vaccine 2014, 32, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, W.A.; Galindo, J.F.; Patarroyo, M.E. Electrostatic potential as a tool to understand interactions between malaria vaccine candidate peptides and MHC II molecules. Biochem. Biophys. Res. Commun. 2011, 410, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Biro, J.C. Amino acid size, charge, hydropathy indices and matrices for protein structure analysis. Theor. Biol. Med. Model. 2006, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Dagliyan, O.; Proctor, E.A.; D’Auria, K.M.; Ding, F.; Dokholyan, N.V. Structural and dynamic determinants of protein-peptide recognition. Structure 2011, 19, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Tsao, D.; Nie, H.; Dokholyan, N.V. Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.J.; Popelier, P.L.A. Where does charge reside in amino acids? The effect of side chain protonation state on the atomic charges of Asp, Glu, Lys, His and Arg. Comput. Theor. Chem. 2015, 1053, 298–304. [Google Scholar] [CrossRef]
- Yuan, Y.; Mills, M.J.; Popelier, P.L.; Jensen, F. Comprehensive analysis of energy minima of the 20 natural amino acids. J. Phys. Chem. A 2014, 118, 7876–7891. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Takeshita, T.; Pendleton, C.D.; Houghten, R.A.; Sadegh-Nasseri, S.; Racioppi, L.; Berzofsky, J.A.; Germain, R.N. The importance of dominant negative effects of amino acid side chain substitution in peptide-MHC molecule interactions and T cell recognition. J. Immunol. 1993, 150, 331–341. [Google Scholar] [PubMed]
- Kim, Y.; Sidney, J.; Pinilla, C.; Sette, A.; Peters, B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinform. 2009, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.F.; Pabon, L.; Barrera, A.; Aza-Conde, J.; Patarroyo, M.A.; Patarroyo, M.E. Structural analysis of owl monkey MHC-DR shows that fully-protective malaria vaccine components can be readily used in humans. Biochem. Biophys. Res. Commun. 2017, 491, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Espejo, F.; Bermudez, A.; Vanegas, M.; Rivera, Z.; Torres, E.; Salazar, L.M.; Patarroyo, M.E. Elongating modified conserved peptides eliminates their immunogenicity and protective efficacy against P. falciparum malaria. J. Struct. Biol. 2005, 150, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polyproline II-like conformation for bound peptides. Proc. Natl. Acad. Sci. USA 1996, 93, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Arnott, S.; Dover, S.D. The structure of poly-L-proline II. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1968, 24, 599–601. [Google Scholar] [CrossRef]
- Stapley, B.J.; Creamer, T.P. A survey of left-handed polyproline II helices. Protein Sci. Publ. Protein Soc. 1999, 8, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Poly(pro)II helices in globular proteins: Identification and circular dichroic analysis. Biochemistry 1994, 33, 10022–10025. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Sternberg, M.J. Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 1993, 229, 472–493. [Google Scholar] [CrossRef] [PubMed]
- Rucker, A.L.; Pager, C.T.; Campbell, M.N.; Qualls, J.E.; Creamer, T.P. Host-guest scale of left-handed polyproline II helix formation. Proteins 2003, 53, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.F.; Siligardi, G.; Gibbons, W.A. Reassessment of the electronic circular dichroism criteria for random coil conformations of poly(l-lysine) and the implications for protein folding and denaturation studies. Biophys. Chem. 1988, 31, 143–146. [Google Scholar] [CrossRef]
- Venugopal, M.G.; Ramshaw, J.A.; Braswell, E.; Zhu, D.; Brodsky, B. Electrostatic interactions in collagen-like triple-helical peptides. Biochemistry 1994, 33, 7948–7956. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Davidson, A.R.; Deber, C.M. The structure of “unstructured” regions in peptides and proteins: Role of the polyproline II helix in protein folding and recognition. Biopolymers 2005, 80, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Sternberg, M.J.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Curtidor, H.; Patarroyo, M.E.; Patarroyo, M.A. Recent advances in the development of a chemically synthesised anti-malarial vaccine. Expert Opin. Biol. Ther. 2015, 15, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, S.; Srinivasan, S. Sequence-dependent conformational sampling using a database of phi(i)+1 and psi(i) angles for predicting polypeptide backbone conformations. Protein Eng. 1997, 10, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Karpen, M.E.; de Haseth, P.L.; Neet, K.E. Comparing short protein substructures by a method based on backbone torsion angles. Proteins 1989, 6, 155–167. [Google Scholar] [CrossRef] [PubMed]
- McGregor, M.J.; Islam, S.A.; Sternberg, M.J. Analysis of the relationship between side chain conformation and secondary structure in globular proteins. J. Mol. Biol. 1987, 198, 295–310. [Google Scholar] [CrossRef]
- Schrauber, H.; Eisenhaber, F.; Argos, P. Rotamers: To be or not to be? An analysis of amino acid side chain conformations in globular proteins. J. Mol. Biol. 1993, 230, 592–612. [Google Scholar] [CrossRef] [PubMed]
- Pickett, S.D.; Sternberg, M.J. Empirical scale of side chain conformational entropy in protein folding. J. Mol. Biol. 1993, 231, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Sternberg, M.J. Side chain conformational entropy in protein folding. Protein Sci. Publ. Protein Soc. 1995, 4, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Searle, M.S.; Williams, D.H. The cost of conformational order: Entropy changes in molecular associations. J. Am. Chem. Soc. 1992, 114, 10690–10697. [Google Scholar] [CrossRef]
- Nicholls, A.; Sharp, K.A.; Honig, B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 1991, 11, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Mandel-Gutfreund, Y.; Zaremba, S.M.; Gregoret, L.M. Contributions of residue pairing to β-sheet formation: Conservation and covariation of amino acid residue pairs on antiparallel β-strands. J. Mol. Biol. 2001, 305, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Narwani, T.J.; Santuz, H.; Shinada, N.; Melarkode Vattekatte, A.; Ghouzam, Y.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Recent advances on polyproline II. Amino Acids 2017, 49, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.W.; Derewenda, Z.S. The name is bond—H bond. Nat. Struct. Biol. 1999, 6, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. Publ. Protein Soc. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.M.; Neal, B.L.; Lenhoff, A.M. Van der Waals interactions involving proteins. Biophys. J. 1996, 70, 977–987. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Shirley, B.A.; Hendricks, M.M.; Iimura, S.; Gajiwala, K.; Scholtz, J.M.; et al. Contribution of hydrophobic interactions to protein stability. J. Mol. Biol. 2011, 408, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Suhnel, J.; Hilgenfeld, R. C-H…pi-interactions in proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. The cation-pi interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Honig, B.; Yang, A.S. Free energy balance in protein folding. Adv. Protein Chem. 1995, 46, 27–58. [Google Scholar] [PubMed]
- Yang, A.S.; Honig, B. Free energy determinants of secondary structure formation: II. Antiparallel β-sheets. J. Mol. Biol. 1995, 252, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. Energetics of protein folding. J. Mol. Biol. 2007, 371, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef] [PubMed]
- Deechongkit, S.; Nguyen, H.; Powers, E.T.; Dawson, P.E.; Gruebele, M.; Kelly, J.W. Context-dependent contributions of backbone hydrogen bonding to β-sheet folding energetics. Nature 2004, 430, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Nick Pace, C.; Scholtz, J.M.; Grimsley, G.R. Forces stabilizing proteins. FEBS Lett. 2014, 588, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Herve, M.; Maillere, B.; Mourier, G.; Texier, C.; Leroy, S.; Menez, A. On the immunogenic properties of retro-inverso peptides. Total retro-inversion of T-cell epitopes causes a loss of binding to MHC II molecules. Mol. Immunol. 1997, 34, 157–163. [Google Scholar] [CrossRef]
- Lozano, J.M.; Espejo, F.; Ocampo, M.; Salazar, L.M.; Tovar, D.; Barrera, N.; Guzman, F.; Patarroyo, M.E. Mapping the anatomy of a Plasmodium falciparum MSP-1 epitope using pseudopeptide-induced mono- and polyclonal antibodies and CD and NMR conformation analysis. J. Struct. Biol. 2004, 148, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Lozano, J.M.; Patarroyo, M.E.; Pluschke, G.; Daubenberger, C.A. Characterization of a reduced peptide bond analogue of a promiscuous CD4 T cell epitope derived from the Plasmodium falciparum malaria vaccine candidate merozoite surface protein 1. Mol. Immunol. 2004, 41, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Lozano, J.M.; Lesmes, L.P.; Carreno, L.F.; Gallego, G.M.; Patarroyo, M.E. Development of designed site-directed pseudopeptide-peptido-mimetic immunogens as novel minimal subunit-vaccine candidates for malaria. Molecules 2010, 15, 8856–8889. [Google Scholar] [CrossRef] [PubMed]
- Lozano, J.M.; Espejo, F.; Vera, R.; Vargas, L.E.; Rosas, J.; Lesmes, L.; Torres, E.; Cortes, J.; Silva, Y.; Patarroyo, M.E. Protection against malaria induced by chirally modified Plasmodium falciparum’s MSP-1 42 pseudopeptides. Biochem. Biophys. Res. Commun. 2005, 329, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Cifuentes, G.; Martinez, N.L.; Patarroyo, M.A. Atomic fidelity of subunit-based chemically-synthesized antimalarial vaccine components. Prog. Biophys. Mol. Biol. 2010, 102, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Cifuentes, G.; Pirajan, C.; Moreno-Vranich, A.; Vanegas, M. Atomic evidence that modification of H-bonds established with amino acids critical for host-cell binding induces sterile immunity against malaria. Biochem. Biophys. Res. Commun. 2010, 394, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Tolia, N.H.; Enemark, E.J.; Sim, B.K.; Joshua-Tor, L. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell 2005, 122, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.H.; Salazar, L.M.; Vanegas, M.; Guzman, F.; Rodriguez, R.; Silva, Y.; Rosas, J.; Patarroyo, M.E. Modified merozoite surface protein-1 peptides with short alpha helical regions are associated with inducing protection against malaria. Eur. J. Biochem. FEBS 2003, 270, 3946–3952. [Google Scholar] [CrossRef]
- Cifuentes, G.; Patarroyo, M.E.; Urquiza, M.; Ramirez, L.E.; Reyes, C.; Rodriguez, R. Distorting malaria peptide backbone structure to enable fitting into MHC class II molecules renders modified peptides immunogenic and protective. J. Med. Chem. 2003, 46, 2250–2253. [Google Scholar] [CrossRef] [PubMed]
- Suarez, J.E.; Urquiza, M.; Puentes, A.; Garcia, J.E.; Curtidor, H.; Ocampo, M.; Lopez, R.; Rodriguez, L.E.; Vera, R.; Cubillos, M.; et al. Plasmodium falciparum circumsporozoite (CS) protein peptides specifically bind to HepG2 cells. Vaccine 2001, 19, 4487–4495. [Google Scholar] [CrossRef]
- Bermudez, A.; Vanegas, M.; Patarroyo, M.E. Structural and immunological analysis of circumsporozoite protein peptides: A further step in the identification of potential components of a minimal subunit-based, chemically synthesised antimalarial vaccine. Vaccine 2008, 26, 6908–6918. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, S.; Soteras, I.; Gelpi, J.L.; Dehez, F.; Chipot, C.; Luque, F.J.; Curutchet, C. Structural and energetic study of cation-pi-cation interactions in proteins. Phys. Chem. Chem. Phys. PCCP 2017, 19, 9849–9861. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The Cationminus signpi Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. Engl. 2011, 50, 4808–4842. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.L. Aromatic interactions in peptides: Impact on structure and function. Biopolymers 2004, 76, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Sunner, J.; Nishizawa, K.; Kebarle, P. Ion-solvent molecule interactions in the Gas Phase. The potassium ion and benzene. J. Phys. Chem. 1981, 85, 1814–1820. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation-pi interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Petsko, G.A. Aromatic-aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Petsko, G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986, 203, 139–143. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Dimerization energetics of benzene and aromatic amino acid side chains. J. Am. Chem. Soc. 1986, 108, 7995–8001. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Weakly polar interactions in proteins. Adv. Protein Chem. 1988, 39, 125–189. [Google Scholar] [PubMed]
- Jackson, M.R.; Beahm, R.; Duvvuru, S.; Narasimhan, C.; Wu, J.; Wang, H.N.; Philip, V.M.; Hinde, R.J.; Howell, E.E. A preference for edgewise interactions between aromatic rings and carboxylate anions: The biological relevance of anion-quadrupole interactions. J. Phys. Chem. B 2007, 111, 8242–8249. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting anion-pi interactions into perspective. Angew. Chem. Int. Ed. Engl. 2011, 50, 9564–9583. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F. The chemistry of biological molecular recognition—The role of aromatic rings as hydrogen-bond acceptors in molecular recognition. Philos. Trans. R. Soc. Phys. Eng. Sci. 1993, 345, 105–112. [Google Scholar] [CrossRef]
- Nishio, M. CH/π hydrogen bonds in crystals. CristEngComm 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Zondlo, N.J. Aromatic-proline interactions: Electronically tunable CH/π interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Balaji, P.V. C-H…pi interactions in proteins: Prevalence, pattern of occurrence, residue propensities, location, and contribution to protein stability. J. Mol. Model. 2014, 20, 2136. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Noncovalent interactions in biochemistry. Comp. Mol. Sci. 2011, 1, 3–17. [Google Scholar] [CrossRef]
- Patarroyo, M.E.; Cifuentes, G.; Rodriguez, R. Structural characterisation of sporozoite components for a multistage, multi-epitope, anti-malarial vaccine. Int. J. Biochem. Cell Biol. 2008, 40, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.; Moreno-Vranich, A.; Patarroyo, M.E. The role of pi-interactions and hydrogen bonds in fully protective synthetic malaria vaccine development. Biochem. Biophys. Res. Commun. 2017, 484, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.J.; Cole, D.K.; Godkin, A. Re-Directing CD4(+) T Cell Responses with the Flanking Residues of MHC Class II-Bound Peptides: The Core is Not Enough. Front. Immunol. 2013, 4, 172. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.Y.; La Gruta, N.L.; Miller, T.; Vignali, K.M.; Adams, P.S.; Woodland, D.L.; Vignali, D.A. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J. Immunol. 2002, 169, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Conant, S.B.; Swanborg, R.H. MHC class II peptide flanking residues of exogenous antigens influence recognition by autoreactive T cells. Autoimmun. Rev. 2003, 2, 8–12. [Google Scholar] [CrossRef]
- Nelson, C.A.; Petzold, S.J.; Unanue, E.R. Identification of two distinct properties of class II major histocompatibility complex-associated peptides. Proc. Natl. Acad. Sci. USA 1993, 90, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Petzold, S.J.; Unanue, E.R. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature 1994, 371, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Roof, R.W.; McCourt, D.W.; Unanue, E.R. Identification of the naturally processed form of hen egg white lysozyme bound to the murine major histocompatibility complex class II molecule I-Ak. Proc. Natl. Acad. Sci. USA 1992, 89, 7380–7383. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Viner, N.J.; Young, S.P.; Petzold, S.J.; Unanue, E.R. A negatively charged anchor residue promotes high affinity binding to the MHC class II molecule I-Ak. J. Immunol. 1996, 157, 755–762. [Google Scholar] [PubMed]
- Carson, R.T.; Vignali, K.M.; Woodland, D.L.; Vignali, D.A. T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity 1997, 7, 387–399. [Google Scholar] [CrossRef]
- Moudgil, K.D.; Sercarz, E.E.; Grewal, I.S. Modulation of the immunogenicity of antigenic determinants by their flanking residues. Immunol. Today 1998, 19, 217–220. [Google Scholar] [CrossRef]
- Godkin, A.J.; Smith, K.J.; Willis, A.; Tejada-Simon, M.V.; Zhang, J.; Elliott, T.; Hill, A.V. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J. Immunol. 2001, 166, 6720–6727. [Google Scholar] [CrossRef] [PubMed]
- Lovitch, S.B.; Pu, Z.; Unanue, E.R. Amino-terminal flanking residues determine the conformation of a peptide-class II MHC complex. J. Immunol. 2006, 176, 2958–2968. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.K.; Gallagher, K.; Lemercier, B.; Holland, C.J.; Junaid, S.; Hindley, J.P.; Wynn, K.K.; Gostick, E.; Sewell, A.K.; Gallimore, A.M.; et al. Modification of the carboxy-terminal flanking region of a universal influenza epitope alters CD4(+) T-cell repertoire selection. Nat. Commun. 2012, 3, 665. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.; Rojas-Luna, R.; Aza-Conde, J.; Tabares, L.; Patarroyo, M.A.; Patarroyo, M.E. Critical role of HLA-DRβ* binding peptides’ peripheral flanking residues in fully-protective malaria vaccine development. Biochem. Biophys. Res. Commun. 2017, 489, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Hashimoto, C.; Sticht, H.; Eichler, J. Synthetic Peptides as Protein Mimics. Front. Bioeng. Biotechnol. 2015, 3, 211. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds cHABPs and mHABPs are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Disease | Agent | Model & Outcome | Ref. |
|---|---|---|---|---|
| Diphtheria | Corynebacterium diphtheriae | Guinea pigs became protected | [45] | |
| Foot-and-mouth disease | Foot-and-mouth disease virus | Neutralising antibodies induced in rabbits and guinea pigs | [46] | |
| Foot-and-mouth disease | Foot-and-mouth disease virus | Cattle became protected | [47] | |
| Malaria | Plasmodium berghei | Mice became protected | [38] | |
| Gastroenteritis | Simian rotavirus | Neutralising antibodies induced in rabbits | [48] | |
| Acquired immune deficiency syndrome | Human immunodeficiency virus | Neutralising antibodies induced in rabbits | [49] | |
| First multi-epitope synthetic antimalarial vaccine | Malaria | Plasmodium falciparum | Aotus monkeys became protected | [39] |
| SPf66 | Malaria | Plasmodium falciparum | Humans became protected | [11] |
| SPf66 | Malaria | Plasmodium falciparum | Safe and highly-immunogenic in humans | [50,51,52] |
| SPf66 | Malaria | Plasmodium falciparum | Protective efficacy (30–40%) in semi-immune populations | [12,53,54,55] |
| Haemorrhagic enteritis, Myocarditis | Canine parvovirus | Dogs became protected | [56] | |
| Malaria | Plasmodium vivax | Parasite development in the mosquito was blocked | [57] | |
| Anthrax | Bacillus anthracis | Rabbit became protected | [58] | |
| Fusion peptide | Influenza | Influenza A virus | Mice became protected | [59]. |
| Swine fever | Classical swine fever virus | Neutralising antibodies induced in pigs | [60] | |
| CIGB-228 | Cervical intraepithelial neoplasia | Human papilloma virus | Lesion regression and HPV clearance | [61]. |
| IMPIPS | Malaria | Plasmodium falciparum | Aotus monkeys became protected | [62,63] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curtidor, H.; Reyes, C.; Bermúdez, A.; Vanegas, M.; Varela, Y.; Patarroyo, M.E. Conserved Binding Regions Provide the Clue for Peptide-Based Vaccine Development: A Chemical Perspective. Molecules 2017, 22, 2199. https://doi.org/10.3390/molecules22122199
Curtidor H, Reyes C, Bermúdez A, Vanegas M, Varela Y, Patarroyo ME. Conserved Binding Regions Provide the Clue for Peptide-Based Vaccine Development: A Chemical Perspective. Molecules. 2017; 22(12):2199. https://doi.org/10.3390/molecules22122199
Chicago/Turabian StyleCurtidor, Hernando, César Reyes, Adriana Bermúdez, Magnolia Vanegas, Yahson Varela, and Manuel E. Patarroyo. 2017. "Conserved Binding Regions Provide the Clue for Peptide-Based Vaccine Development: A Chemical Perspective" Molecules 22, no. 12: 2199. https://doi.org/10.3390/molecules22122199
APA StyleCurtidor, H., Reyes, C., Bermúdez, A., Vanegas, M., Varela, Y., & Patarroyo, M. E. (2017). Conserved Binding Regions Provide the Clue for Peptide-Based Vaccine Development: A Chemical Perspective. Molecules, 22(12), 2199. https://doi.org/10.3390/molecules22122199