Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking

 ,
, 
 and
and 
Abstract
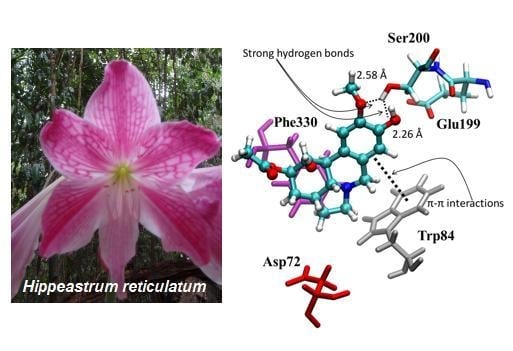
1. Introduction
2. Results and Discussion
2.1. Alkaloid Identification by GC-MS
2.2. Structural Elucidation by NMR
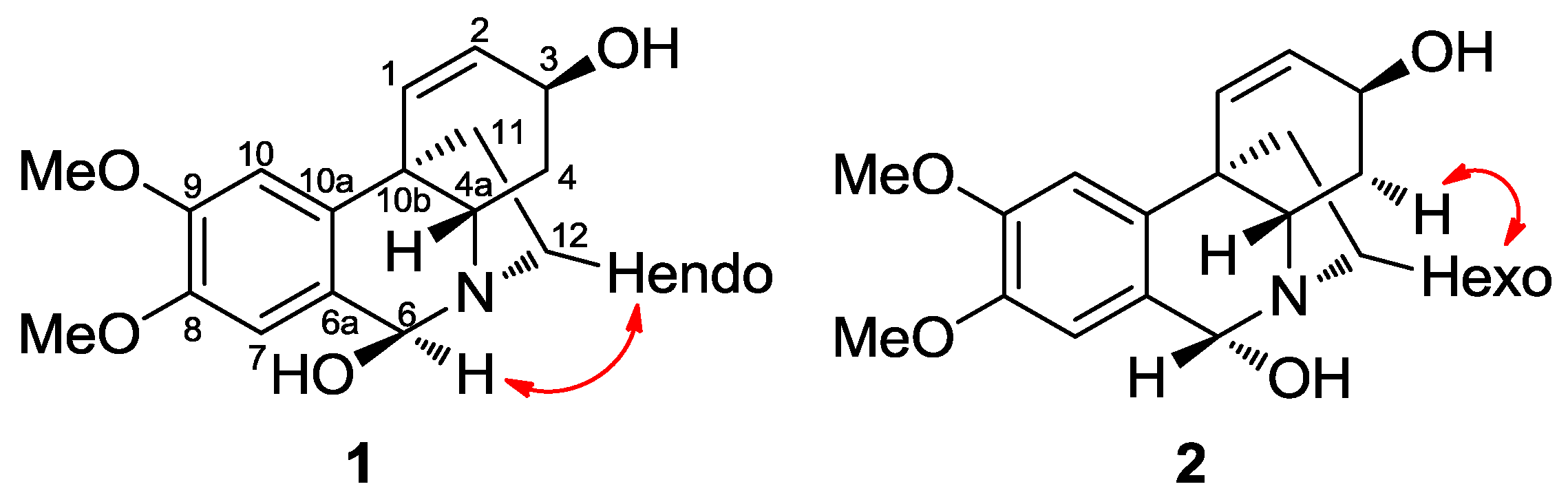
2.2.1. 6β-Hydroxymaritidine (1) and 6α-hydroxymaritidine (2)
2.2.2. Reticulinine (3) and Isoreticulinine (4)
2.3. Biological Activity
2.3.1. Antiprotozoal Activity
2.3.2. Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities
2.4. Molecular Docking
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Characterization of Compounds
3.5. Biological Activity
3.5.1. Antiprotozoal Activity
3.5.2. Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities
3.6. Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, M.-L.; Schneider, G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: application in the design of natural product- based combinatorial libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A. Strategies for discovering drugs from previously unexplored natural products. Drug Discov. Today 2000, 5, 294–300. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Stratton, C.F.; Newman, D.J.; Tan, D.S. Cheminformatic comparision of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 2015, 25, 4802–4807. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- APG III. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef]
- Chase, M.W.; Reveal, J.L.; Fay, M.F. A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot. J. Linn. Soc. 2009, 161, 132–136. [Google Scholar] [CrossRef]
- The Plant List. A Working List of All Plant Species. Available online: http://www.theplantlist.org/1.1/browse/A/Amaryllidaceae/ (accessed on 27 April 2017).
- Candido, R.S.; da Silva Fourny, A.C.; Gonçalves-Esteves, V.; Lopes, R.C. Hippeastrum species in areas of resting in the state of Rio de Janeiro, Brazil: pollen characters. Acta Bot. Bras. 2013, 27, 661–668. [Google Scholar] [CrossRef]
- Bastida, J.; Lavilla, R.; Viladomat, F. Chemical and biological aspects of Narcissus Alkaloids. In The Alkaloids: Chemistry and Physiology; Cordell, G.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 63, pp. 87–179. [Google Scholar]
- Maelicke, A.; Samochocki, M.; Jostock, R.; Fehrenbacher, A.; Ludwig, J.; Albuquerque, E.X.; Zerlin, M. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer’s disease. Biol. Psychiatry 2001, 49, 279–288. [Google Scholar] [CrossRef]
- Alzheimer´s Disease International. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf (accessed on 13 October 2017).
- Querfurth, H.W.; LaFerla, F.M. Alzheimer´s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Konrath, E.L.; Passos, C.S.; Klein-Júnior, L.C.; Henriques, A.T. Alkaloids as source of potential anticholinesterase inhibitors for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1701–1725. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Neglected Tropical Diseases. Available online: http://www.who.int/neglected_diseases/diseases/en/ (accessed on 1 September 2017).
- Klug, D.M.; Gelb, M.H.; Pollastri, M.P. Repursposing strategies for tropical disease drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Meerow, A.W.; Snijman, D.A. Amaryllidaceae. In The families and genera of vascular plants. Flowering Plants, Monocotyledons Lilianae. (except Orchidaceae.); Kubitzki, K., Ed.; Springer: Berlin, Germany, 1998; Volume 3, pp. 83–110. [Google Scholar]
- Dutilh, J.H.A. Amaryllidaceae. In Catálogo de Plantas e Fungos do Brasil; Jakobsson, A., Ed.; Sindicato Nacional de Editores de Livros: Rio de Janeiro, Brazil, 2010; Volume 1, pp. 596–599. [Google Scholar]
- Deepa, C.P.; Kuriakose, B.B. Pharmacognostic and phytochemical evaluation of the bulbs of Hippeastrum. puniceum (Lam.) Voss. IJPPR 2014, 6, 399–404. [Google Scholar]
- Da Silva, A.F.S.; de Andrade, J.P.; Bevilaqua, L.R.M.; de Souza, M.M.; Izquierdo, I.; Henriques, A.T.; Zuanazzi, J.A.S. Anxiolytic-, antidepressant- and anticonvulsant-like effects of the alkaloid montanine isolated from Hippeastrum. vittatum. Pharmacol. Biochem. Behav. 2006, 85, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Giordani, R.B.; Junior, C.O.R.; de Andrade, J.P.; Bastida, J.; Zuanazzi, J.A.S.; Tasca, T.; Almeida, M.V. Lycorine derivatives against Trichomonas vaginalis. Chem. Biol. Drug. Des. 2012, 80, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.R.; Silva, A.B.; Torres, M.C.M.; Pinto, F.C.L.P.; Guimaraes, L.A.; Rocha, D.D.; Silveira, E.R.; Costa-Lotufo, L.V.; Braz-Filho, R.; Pessoa, O.D.L. Cytotoxic Alkaloids from Hippeastrum. solandriflorum Lindl. J. Braz. Chem. Soc. 2015, 26, 1976–1980. [Google Scholar] [CrossRef]
- Silva, A.F.S.; de Andrade, J.P.; Machado, K.R.B.; Rocha, A.B.; Apel, M.A.; Sobral, M.E.G.; Henriques, A.T.; Zuanazzi, J.A. Screening for cytotoxic activity of extracts and isolated alkaloids from bulbs of Hippeastrum. vittatum. Phytomedicine 2008, 15, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Nitteranon, V.; Kittiwongwattana, C.; Vuttipongchaikij, S.; Sakulkoo, J.; Srijakkoat, M.; Chokratin, P.; Harinasut, P.; Suputtitada, S.; Apisitwanich, S. Evaluations of the mutagenicity of a pigment extract from bulb culture of Hippeastrum. reticulatum. Food Chem. Toxicol. 2014, 69, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Clippel, J.K.; Carmo, H.N.C.; Nascimento, L.C.Z.; Cuzzuol, G.R.F. Análise química em órgãos de reserva de algumas herbáceas e arbóreas ocorrentes na flora do Espírito Santo, Brasil. Acta Bot. Bras. 2008, 22, 1057–1067. [Google Scholar] [CrossRef]
- Hang, T.T.M.; Phuong, P.T.M.; Dung, T.T.M.; Miyajima, I. Preliminary chromosome studies on Hippeastrum. species from Vietnam. J. Fac. Agric. Kyushu Univ. 2015, 60, 51–55. [Google Scholar]
- Bastida, J.; Codina, C.; Francesc, V. Narcissus alkaloids, XIII. Complete assignment of the NMR spectra of papyramine and 6-epi-papyramine by two-dimensional NMR spectroscopy. J. Nat. Prod. 1990, 53, 1456–1462. [Google Scholar] [CrossRef]
- Herrera, M.R.; Machocho, A.K.; Nair, J.J.; Campbell, W.E.; Brun, R.; Viladomat, F.; Codina, C.; Bastida, J. Alkaloids from Cyrtanthus. elatus. Fitoterapia 2001, 72, 444–448. [Google Scholar] [CrossRef]
- Evidente, A.; Iasiello, I.; Randazzo, G. Isolation of sternbergine, a new alkaloid from bulbs of Sternbergia. lutea. J. Nat. Prod. 1984, 47, 1003–1008. [Google Scholar] [CrossRef]
- Tallini, L.R.; Andrade, J.P.; Kaiser, M.; Viladomat, F.; Nair, J.J.; Zuanazzi, J.A.S.; Bastida, J. Alkaloid constituents of the Amaryllidaceae plant Amaryllis belladonna L. Molecules 2017, 22, 1437. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Lahiri, D.K.; Sambamurti, K. Butyrylcholinesterase: An important new target in Alzheimer's disease therapy. Int. Psychogeriatr. 2002, 14, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.G. Advances in the treatment of Alzheimer’s disease: benefits of dual cholinesterase inhibition. Eur. Neurol. 2002, 47, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Coll, J.C.; Bowden, B.F. The application of vacuum liquid chromatography to the separation of terpene mixtures. J. Nat. Prod. 1986, 49, 934–936. [Google Scholar] [CrossRef]
- Munier, R.; Macheboeuf, M. Paper partion microchromatography of alkaloids and various biological nitrogenous bases. III. Examples of the separation of various alkaloids by the acid solvent phase technic (atropine, cocaine, nicotine, sparteine, strychnine and corynanthine families). Boll. Soc. Chim. Biol. (Paris) 1951, 33, 846–856. [Google Scholar]
- Jatzkewitz, H. A clinical method for determination of basic addiction drugs in urine. Hoppe-Seyler’s Z. Physiol. Chem. 1953, 295, 94–100. [Google Scholar] [CrossRef]
- Bessa, C.D.P.B.; Andrade, J.P.; Oliveira, R.S.; Domingos, E.; Santos, H.; Romão, W.; Bastida, J.; Borges, W.S. Identification of alkaloids from Hippeastrum. aulicum (Ker Gawl.) Herb. (Amaryllidaceae) using CGC-MS and ambient ionization mass spectrometry (PS-MS and LS-MS). J. Braz. Chem. Soc. 2017, 28, 819–830. [Google Scholar] [CrossRef]
- Orhan, I.; Şener, B.; Kaiser, M.; Brun, R.; Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 2010, 8, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- López, S.; Bastida, J.; Viladomat, F.; Codina, C. Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts. Life Sci. 2002, 71, 2521–2529. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 Å resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements os specificity for anti-Alzheimer´s grugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Bushteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- ChemCraft. Available online: http://www.chemcraftprog.com/citation.htmL (accessed on 12 September 2017).
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110:13, 6158–6170. [Google Scholar] [CrossRef]
- Petersson, G.A.; Benett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revis. E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Moris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| alkaloid | RI | M+ | MS |
|---|---|---|---|
| galanthamine (11) | 2410.8 | 287 (84) | 288 (20), 286 (100), 270 (15), 244 (27), 230 (15), 216 (37), 174 (32), 159 (11), 115 (16) |
| sanguinine (12) | 2430.5 | 273 (100) | 274 (16), 272 (77), 256 (23), 202 (31), 197 (15), 165 (14) 160 (45), 152 (15), 115 (19) |
| 6β- and 6α-hydroxymaritidine (1) and (2) | 2495.5 | 303 (20) | 286 (9), 274 (12), 260 (21), 259 (100), 258 (12), 256 (20), 241 (30) 128 (16), 115 (20) |
| 8-O-demethylmaritidine (6) | 2522.4 | 273 (100) | 274 (18), 230 (24), 203 (19), 202 (25), 201(89), 189 (55), 175 (23), 115 (19), 56 (20) |
| maritidine (5) | 2528.5 | 287 (100) | 288 (20), 244 (35), 217 (20) 216 (25), 215 (95), 203 (55), 189 (18), 128 (17), 115 (20) |
| 11,12-dehydroanhydrolycorine (10) | 2629.3 | 249 (59) | 248 (100), 218 (1),190 (26), 163 (8), 137 (1), 123 (5), 95 (13) |
| m/z 264 | 2693.6 | 265 (76) | 264 (100), 248 (18), 220 (12), 191 (14),178 (18) |
| 11-hydroxyvittatine (7) | 2732.5 | 287 (5) | 259 (16), 258 (100), 242 (9), 214 (9), 212 (8), 211 (13), 186 (13), 181 (14), 128 (11) |
| lycorine (9) | 2771.8 | 287 (18) | 286 (10), 268 (19), 250 (16), 238 (7), 227 (78), 226 (100), 211 (6), 147 (5), 119 (3) |
| isoreticulinine (4) | 2829.1 | 333 (<1) | 291 (37), 290 (100), 274 (11), 272 (6), 256 (5), 228 (5), 147 (13) |
| reticulinine (3) | 2133.4 | 333 (42) | 332 (100), 316 (5) 290 (6), 272 (56), 256 (31), 244 (14), 216 (14), 147 (26) |
| m/z 294 | 2950.3 | 295 (87) | 294 (100), 278 (10), 264 (3), 250 (7), 235 (3), 221 (5), 207 (3), 194 (5) |
| m/z 280 | 2978.6 | 281 (75) | 280 (100), 264 (12), 250 (3), 236 (11), 219 (4), 207 (4), 194 (8), 178 (4), 167 (3) |
| 2-methoxypratosine (8) | 3071.1 | 309 (100) | 310 (20), 294 (18), 284 (7), 266 (24), 251 (14), 236 (6), 164 (4), 152 (6) |
| 1 | 2 | |||
|---|---|---|---|---|
| No. | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 | 130.7, d | 6.59, d (10.0) | 129.2, d | 6.51, d (10.1) |
| 2 | 127.9, d | 5.98, dd (5.3 and 10.4) | 128.4, d | 6.02, dd (5.1 and 10.0) |
| 3 | 63.2, d | 4.32, m | 62.1, d | 4.37, m |
| 4α | 31.0, t | 1.72, dt (4.0 and 13.7) | 30.5, t | 1.85, dt (4.0 and 13.4) |
| 4β | 31.0, t | 2.14, dd (4.1 and 13.3) | 30.5, t | 2.29, dd (4.2 and 13.8) |
| 4a | 56.4, d | 4.15, dd (4.1 and 13.6) | 61.7, d | 3.89, m |
| 6a | 125.2, s | - | 123.4, s | - |
| 6 | 88.8, d | 5.35, s | 86.6, d | 6.12, s |
| 7 | 111.8, d | 6.89, s | 110.7, d | 7.03, s |
| 8 | 148.0, s | - | 148.4, s | - |
| 9 | 148.9, s | - | 149.2, s | - |
| 10 | 105.3, d | 6.83, s | 105.1, d | 6.78, s |
| 10a | 136.0, s | - | 133.7, s | - |
| 10b | 44.2, s | - | 44.8, s | - |
| 11exo | 39.9, t | 2.03, m | 39.8, t | 2.03 |
| 11endo | 39.9, t | 2.03, m | 39.8, t | 2.03 |
| 12exo | 47.4, t | 3.37, ddd (3.8; 10.0 and 13.6) | 41.2, t | 3.19, ddd (3.5; 10.4 and 13.6) |
| 12endo | 47.4, t | 2.87, ddd (6.5; 9.0 and 13.1) | 41.2, t | 3.94, m |
| OMe | 56.1, q | 3.90, s | 56.1, q | 3.90, s |
| OMe | 55.9, q | 3.87, s | 55.9, q | 3.87, s |
| 3 | 4 | |||
|---|---|---|---|---|
| No. | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 | 70.6 d | 6.01, t (3.9) | 66.9 d | 4.77, t (1.5) |
| 2 | 68.4 d | 4.15, ddd (2.8; 7.1 and 9.8) | 72.1 d | 5.12, ddd (2.5; 5.4 and 11.6) |
| 3α | 25.8 t | 2.02, m | 26.8 t | 2.23, ddd (6.2; 11.8 and 13.4) |
| 3β | 25.8 t | 2.02, m | 26.8 t | 1.95, m |
| 4 | 36.7 d | 2.68, m | 36.4 d | 2.68, m |
| 4a | 58.0 d | 3.11, dd (5.9 and 10.4) | 57.4 d | 3.22, dd (5.9 and 10.7) |
| 6α | 51.4 t | 4.28, d (16.7) | 51.6 t | 4.29, d (16.4) |
| 6β | 51.4 t | 3.73, d (16.7) | 51.6 t | 3.74, d (16.9) |
| 6a | 127.7 s | - | 127.7 s | - |
| 7 | 112.7 d | 6.67, s | 112.9 d | 6.68, s |
| 8 | 144.0 s | - | 144.0 s | - |
| 9 | 145.4 s | - | 145.4 s | - |
| 10 | 106.4 d | 6.73, s | 106.4 d | 6.82, s |
| 10a | 126.7 s | - | 126.7 s | - |
| 10b | 33.1 d | 2.68, m | 34.3 d | 2.59, d (10.5) |
| 11α | 29.7 t | 1.85, m | 29.9 t | 2.04, m |
| 11β | 29.7 t | 1.77, m | 29.9 t | 1.85, m |
| 12α | 52.7 t | 3.39, m | 52.9 t | 3.39, m |
| 12β | 52.7 t | 2.81, m | 52.9 t | 2.81, m |
| OMe | 56.0 q | 3.82, s | 56.1 q | 3.89, s |
| MeCOO | 172.2 q | 2.06, s | 170.4 q | 2.16, s |
| MeCOO | 21.1 q | 2.06, s | 21.3 q | 2.16, s |
| Parasite | T. b. rhodesiense | T. cruzi | L. donovani | P. falciparum | Cytotoxicity |
|---|---|---|---|---|---|
| Stage | Trypomastigotes | Amastigotes | Amastigotes | IEF (intraerythrocytic) | |
| Strain | STIB 900 (IC50 ) | Tulahuen C4 (IC50 ) | MHOM-ET-67/L82 (IC50 ) | NF54 (IC50 ) | L6 (IC50 ) |
| melarsoprol | 0.0010 | ||||
| benznidazole | 1.080 | ||||
| miltefosine | 0.091 | ||||
| chloroquine | 0.002 | ||||
| podophyllotoxin | 0.007 | ||||
| compounds 1 and 2 | 30.68 | 66.11 | >100 | 32.86 | >100 |
| Alkaloids | 1DX6 a (kcal mol−1) | 4EY7 b (kcal mol−1) | 4BDS c (kcal mol−1) |
|---|---|---|---|
| 6β-hydroxymaritidine (1) | −8.49 | −9.44 | −8.95 |
| 6α-hydroxymaritidine (2) | −8.25 | −8.75 | −8.72 |
| reticulinine (3) | −8.88 | −8.87 | −8.10 |
| isoreticulinine (4) | −9.02 | −9.80 | −8.12 |
| galanthamine (11) | −9.36 | −10.10 | −8.74 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallini, L.R.; Osorio, E.H.; Santos, V.D.d.; Borges, W.D.S.; Kaiser, M.; Viladomat, F.; Zuanazzi, J.A.S.; Bastida, J. Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking. Molecules 2017, 22, 2191. https://doi.org/10.3390/molecules22122191
Tallini LR, Osorio EH, Santos VDd, Borges WDS, Kaiser M, Viladomat F, Zuanazzi JAS, Bastida J. Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking. Molecules. 2017; 22(12):2191. https://doi.org/10.3390/molecules22122191
Chicago/Turabian StyleTallini, Luciana R., Edison H. Osorio, Vanessa Dias dos Santos, Warley De Souza Borges, Marcel Kaiser, Francesc Viladomat, José Angelo S. Zuanazzi, and Jaume Bastida. 2017. "Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking" Molecules 22, no. 12: 2191. https://doi.org/10.3390/molecules22122191
APA StyleTallini, L. R., Osorio, E. H., Santos, V. D. d., Borges, W. D. S., Kaiser, M., Viladomat, F., Zuanazzi, J. A. S., & Bastida, J. (2017). Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking. Molecules, 22(12), 2191. https://doi.org/10.3390/molecules22122191